

Genome-Wide Association Mapping of Oil Content and Seed-Related Traits in Shea Tree (Vitellaria paradoxa subsp. nilotica) Populations


 , ,
, ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Leaf Sampling for DNA Extraction
2.1.1. Shea Oil Extraction Procedure
2.1.2. DNA Extraction and SNP Discovery by DArTseq™ Technology
2.2. Data Analysis
2.2.1. Seed Trait Data Analysis
2.2.2. Genome-Wide Association Analysis and Gene Annotation Identification
- Y = the vector of the phenotypic observations estimated for the traits studied;
- X = the SNP markers (fixed effect) matrix;
- Z = the random kinship (co-ancestry) matrix;
- b = a vector representing the estimated SNP effects;
- u = a vector representing random additive genetic effects, and
- e = the vector for random residual errors.
3. Results
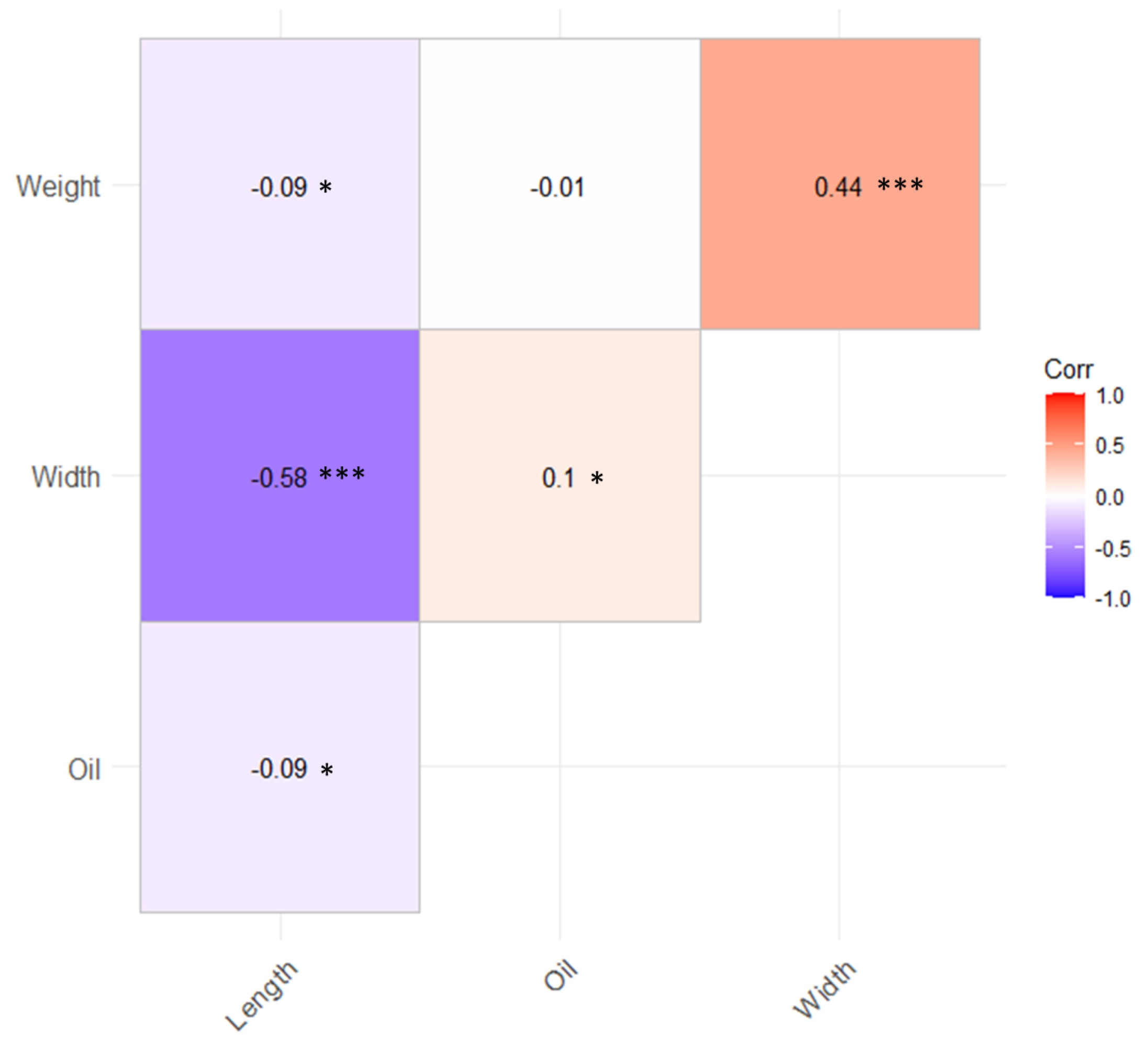
3.1. Phenotypic Variation for the Shea Tree Traits
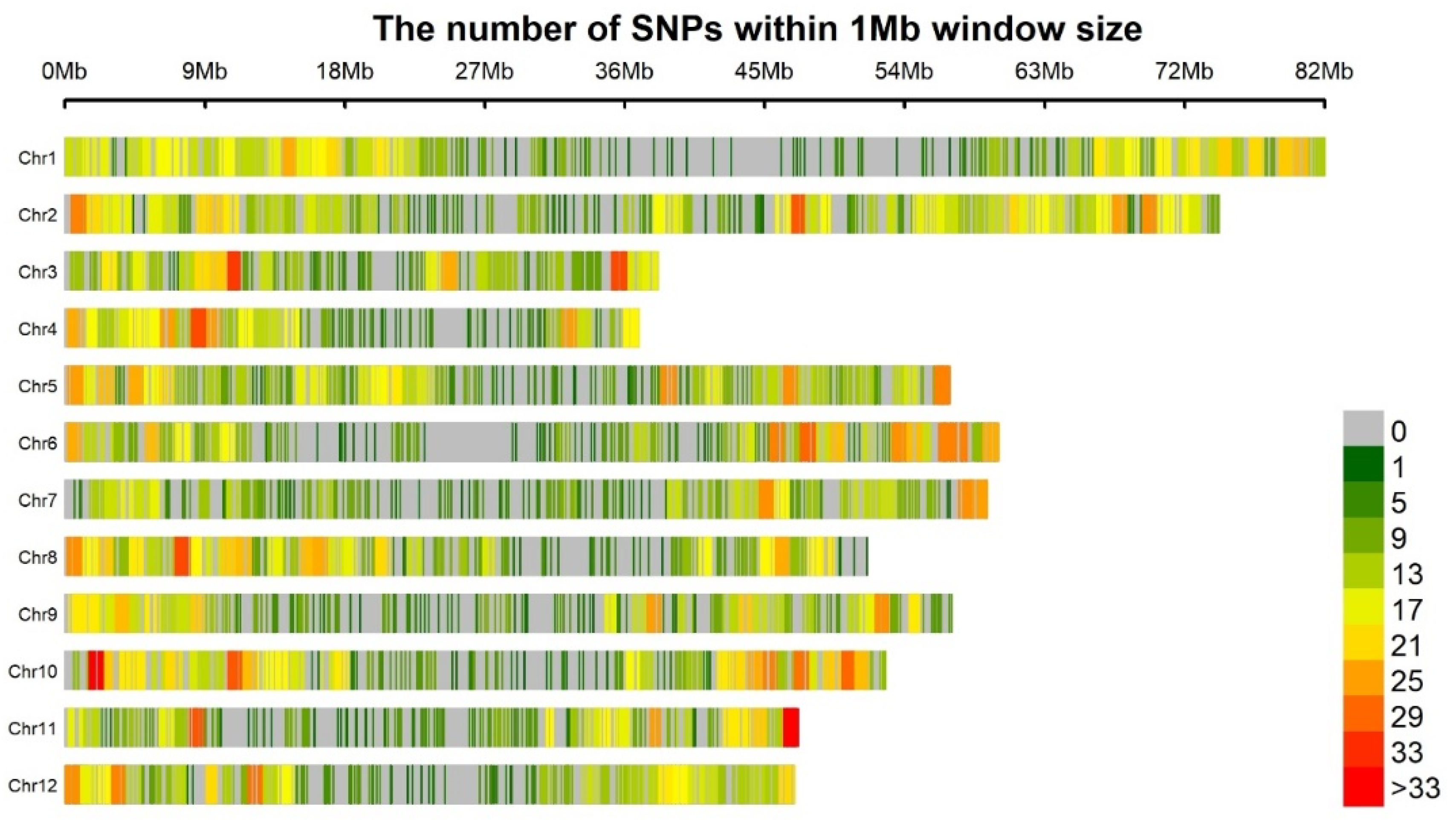
3.2. Marker Coverage and SNP Distribution
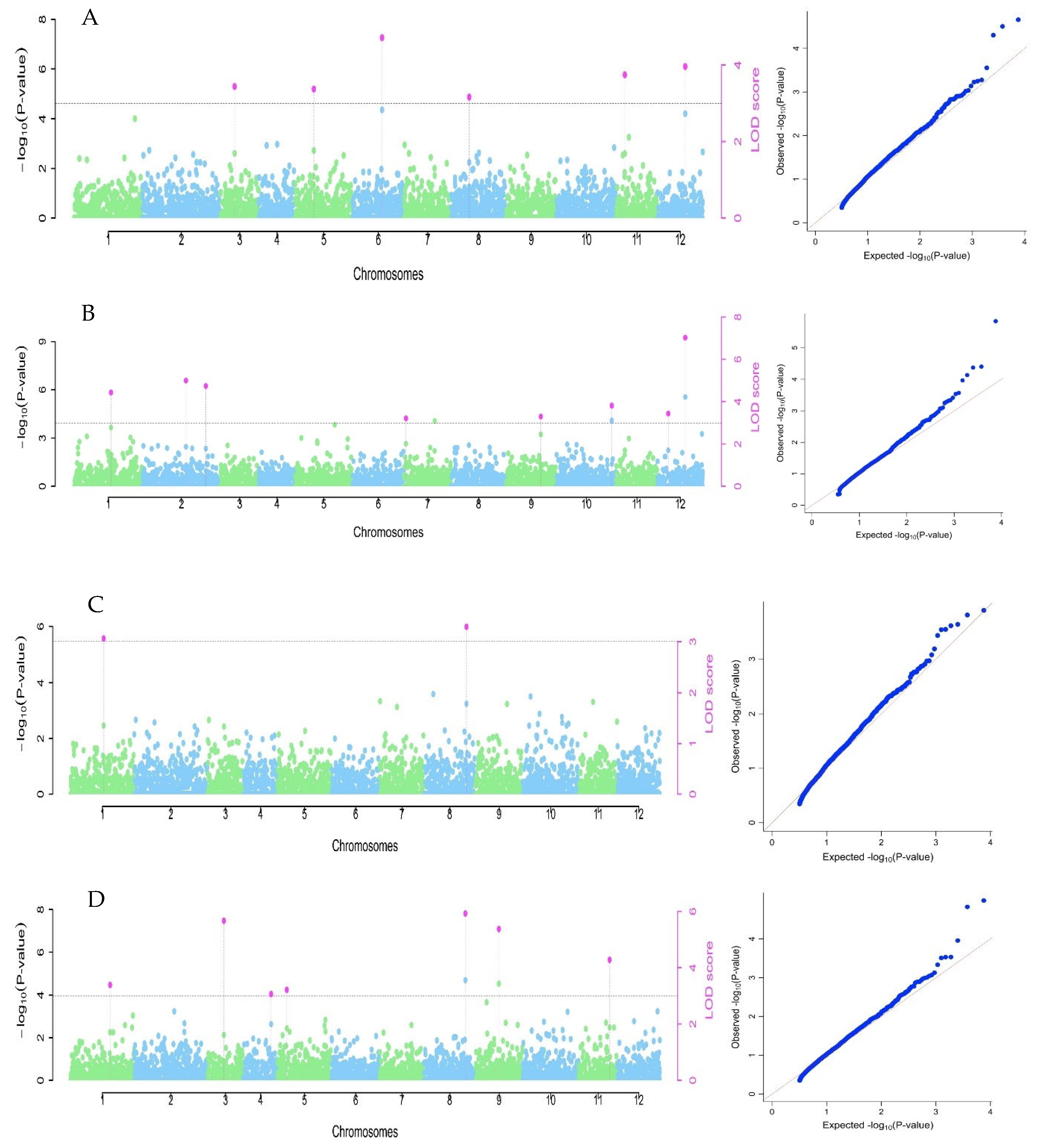
3.3. Marker Association for the Studied Traits
3.4. Potential Candidate Genes
3.5. Linkage Disequilibrium (LD)
4. Discussion
4.1. Phenotypic Data
4.2. Candidate Gene Scan in the Oil Content Traits
4.3. Candidate Gene Scan within the Seed Related Traits
4.4. Linkage Disequilibrium (LD)
4.5. Marker Assisted Selection in Shea Tree
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wei, Y.; Ji, B.; Siewers, V.; Xu, D.; Halkier, B.A.; Nielsen, J. Identification of genes involved in shea butter biosynthesis from Vitellaria paradoxa fruits through transcriptomics and functional heterologous expression. Appl. Microbiol. Biotechnol. 2019, 103, 3727–3736. [Google Scholar] [CrossRef] [PubMed]
- Naughton, C.C.; Lovett, P.N.; Mihelcic, J. Land suitability modeling of shea (Vitellaria paradoxa) distribution across sub-Saharan Africa. Appl. Geogr. 2015, 58, 217–227. [Google Scholar] [CrossRef]
- Lovett, P.; Haq, N. Evidence for anthropic selection of the Sheanut tree (Vitellaria paradoxa). Agrofor. Syst. 2000, 48, 273–288. [Google Scholar] [CrossRef]
- Choungo Nguekeng, P.B.; Hendre, P.; Tchoundjeu, Z.; Kalousová, M.; TchanouTchapda, A.V.; Kyereh, D.; Masters, E.; Lojka, B. The Current State of Knowledge of Shea Butter Tree (Vitellaria paradoxa C.F. Gaertner.) for Nutritional Value and Tree Improvement in West and CentralAfrica. Forests 2021, 12, 1740. [Google Scholar] [CrossRef]
- Hemsley, J.H. Sapotaceae. In Flora of Tropical East Africa; Milne, E., Polhill, R.M., Eds.; Crown Agents for Overseas Governments and Administrations: London, UK, 1968; pp. 47–50. [Google Scholar]
- Issaka, A.; Konstantin, V.K.; Reiner, F. Morphological and genetic diversity of shea tree (Vitellaria paradoxa) in the savannah regions of Ghana. Genet. Resour. Crop Evol. 2017, 64, 1253–1268. [Google Scholar]
- Allal, F.; Piombo, G.; Kelly, B.A.; Okullo, J.B.L.; Thiam, M.; Diallo, O.B.; Nyarko, G.; Davrieux, F.; Lovett, P.N.; Bouvet, J.-M. Fatty acid and tocopherol patterns of variation within the natural range of the shea tree (Vitellaria paradoxa). Agrofor. Syst. 2013, 87, 1065–1082. [Google Scholar] [CrossRef]
- Hale, I.; Ma, X.; Melo, A.T.O.; Padi, F.K.; Hendre, P.S.; Kingan, S.B.; Sullivan, S.T.; Chen, S.; Boffa, J.-M.; Muchugi, A.; et al. Genomic Resources to Guide Improvement of the Shea Tree. Front. Plant Sci. 2021, 12, 720670. [Google Scholar] [CrossRef]
- Cardi, C.; Vaillant, A.; Sanou, H.; Bokary Kelly, A.; Bouvet, J.-M. Characterization of microsatellite markers in the shea tree (Vitellaria paradoxa C. F Gaertn) in Mali. Mol. Resour. 2005, 5, 524–526. [Google Scholar] [CrossRef]
- Odoi, J.B.; Odong, T.L.; Okia, C.A.; Edema, R.; Muchugi, A.; Gwali, S. Variation in phenotypic traits of high oil yielding and early maturing shea trees (Vitellaria paradoxa) selected using local knowledge. J. Agric. Nat. Resour. Sci. 2020, 7, 34–42. [Google Scholar]
- Global Market Insight. Market Research Report 2021; TechNavio | PRODUCT CODE: 1044351; Global Market Insight: Elmhurst, IL, USA, 2021. [Google Scholar]
- Abdul-Mumeen, I.; Beauty, D.; Adam, A. Shea butter extraction technologies: Current status and future perspective. Afr. J. Biochem. Res. 2019, 13, 9–22. [Google Scholar]
- Global Shea Alliance. Shea Production and Market; Global Shea Alliance: Accra, Ghana, 2021. [Google Scholar]
- Boffa, J.-M. Opportunities and Challenges in the Improvement of the Shea (Vitellaria paradoxa) Resource and Its Management; Occasional Paper 24; World Agroforestry Centre: Nairobi, Kenya, 2015. [Google Scholar]
- Aleza, K.; Villamor, G.B.; Nyarko, B.K.; Wala, K.; Akpagana, K. Shea (Vitellaria paradoxa Gaertn C. F.) fruit yield assessment and management by farm households in the Atacora district of Benin. PLoS ONE 2018, 13, e0190234. [Google Scholar] [CrossRef]
- Yao, S.D.M.; Diarrassouba, N.; Diallo, R.; Koffi, E.-B.Z.; Dago, D.N.; Fofana, I.J. Effects of Sowing Depth and Seed Orientation on the Germination and Seedling Growth in Shea Tree (Vitellaria paradoxa C.F. Gaertn.) for Rootstock Production in Nursery. Res. Plant Sci. 2021, 9, 13–22. [Google Scholar] [CrossRef]
- Chimsah, F.A. Shea Sapling Management and Grafting. In The Way forward to Shea Domestication A Case Research from the University for Development Studies; Tamale: Ghana, West Africa, 2012. [Google Scholar]
- Pilipović, A.; Orlović, S.; Kovačević, B.; Galović, V.; Stojnić, S. Selection and Breeding of Fast-Growing Trees for Multiple Purposes in Serbia. In Forests of Southeast Europe Under a Changing Climate; Advances in Global Change Research; Šijačić-Nikolić, M., Milovanović, J., Nonić, M., Eds.; Springer: Cham, Switzerland, 2019; Volume 65. [Google Scholar] [CrossRef]
- Kilian, A.; Wenzl, P.; Huttner, E.; Carling, J.; Xia, L.; Blois, H.; Caig, V.; Heller-Uszynska, K.; Jaccoud, D.; Hopper, C.; et al. Diversity Arrays Technology: A Generic Genome Profiling Technology on Open Platforms. In Data Production and Analysis in Population Genomics; Methods in Molecular Biology (Methods and Protocols); Pompanon, F., Bonin, A., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 67–89. [Google Scholar]
- Zahid, G.; Aka Kaçar, Y.; Dönmez, D.; Küden, A.; Giordani, T. Perspectives and recent progress of genome-wide association studies (GWAS) in fruits. Mol. Biol. Rep. 2022, 49, 5341–5352. [Google Scholar] [CrossRef] [PubMed]
- AOAC. Association of Official Analytical Chemist, Official Methods of Analysis, 19th ed.; AOAC: Washington, DC, USA, 2012; 130p. [Google Scholar]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 1–15. [Google Scholar]
- Sansaloni, C.; Petroli, C.; Jaccoud, D.; Carling, J.; Detering, F.; Grattapaglia, D.; Kilian, A. Diversity Arrays Technology (DArT) and next-generation sequencing combined: Genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proc. 2011, 5, P54. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Raman, H.; Raman, R.; Kilian, A.; Detering, F.; Carling, J.; Coombes, N.; Diffey, S.; Kadkol, G.; Edwards, D.; McCully, M.; et al. Genome-wide delineation of natural variation for pod shatter resistance in Brassica napus. PLoS ONE 2014, 9, e101673. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 22 March 2022).
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; Buckler, E.S.A. Unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Rosyara, U.R.; De Jong, W.S.; Douches, D.S.; Endelman, J.B. Software for genome-wide association studies in autopolyploids and its application to potato. Plant Genome 2016, 9, 1–10. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Lwaka Tamba, C.; Wen, Y.J.; Li, P.; Ren, W.L.; Ni, Y.L.; Gao, J.; Zhang, Y.M. mrMLM v4.0: An R platform for multi-locus genome-wide association studies. Genom. Proteom. Bioinform. 2020, 18, 481–487. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery Rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Gatarira, C.; Agre, P.; Matsumoto, R.; Edemodu, A.; Adetimirin, V.; Bhattacharjee, R.; Asiedu, R.; Asfaw, A. Genome-Wide Association Analysis for Tuber Dry Matter and Oxidative Browning in Water Yam (Dioscorea alata L.). Plants 2020, 9, 969. [Google Scholar] [CrossRef] [PubMed]
- Verde, I.; Jenkins, J.; Dondini, L.; Micali, S.; Pagliarani, G.; Vendramin, E.; Paris, R.; Aramini, V.; Gazza, L.; Rossini, L.; et al. The peach v2.0 release: High-resolution linkage mapping and deep resequencing improve chromosome-scale assembly and contiguity. BMC Genom. 2017, 18, 225. [Google Scholar] [CrossRef] [PubMed]
- Odoi, J.B.; Adjei, E.A.; Hendre, P.; Nantongo, J.S.; Ozimati, A.A.; Badji, A.; Nakabonge, G.; Edema, R.; Gwali, S.; Odong, T.L. Genetic diversity and population structure among Ugandan shea tree (Vitellariaparadoxa subsp. nilotica) accessions based on DarTSeq markers. Crop Sci. 2023, 1–13. [Google Scholar] [CrossRef]
- Ma, W.; Kong, Q.; Mantyla, J.J.; Yang, Y.; Ohlrogge, J.B.; Benning, C. 14-3-3 protein mediates plant seed oil biosynthesis through interaction with AtWRI1. Plant J. 2016, 88, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kong, Q.; Lim, A.R.Q.; Lu, S.; Zhao, H.; Guo, L.; Yuan, L.; Ma, W. Transcriptional regulation of oil biosynthesis in seed plants: Current understanding, applications, and perspectives. Plant Commun. 2022, 3, 100328. [Google Scholar] [CrossRef]
- Gwali, S.; Nakabonge, G.; Okullo, J.B.L.; Eilu, G.; Forestier-Chironc, N.; Piombod, G.; Davrieux, F. Fat content and fatty acid profiles of shea tree (Vitellaria paradoxa subspecies nilotica) ethno-varieties in Uganda. For. Trees Livelihoods 2012, 21, 267–278. [Google Scholar] [CrossRef]
- Okullo, J.B.L.; Omujai, F.; Agea, J.G.; Vuzi, P.C.; Namutebi, A.; Okello, J.B.; Nyanzi, S.A. Physico-chemical characteristics of Shea butter (Vitellaria Paradoxa C. F. Gaertu) oil from the shea districts of Uganda. AJFAND Afr. J. Food Agric. Nutr. Dev. 2010, 10, 2070–2084. [Google Scholar]
- Mattia, M.R.; Du, D.; Yu, Q.; Kahn, T.; Roose, M.; Hiraoka, Y.; Wang, Y.; Munoz, P.; Gmitter, F.G., Jr. Genome-Wide Association Study of Healthful Flavonoids among Diverse Mandarin Accessions. Plants 2022, 11, 317. [Google Scholar] [CrossRef]
- Hall, D.; Tegstrom, C.; Ingvarsson, P.K. Using association mapping to dissect the genetic basis of complex traits in plants. Brief. Funct. Genom. Proteom. 2010, 9, 157–165. [Google Scholar] [CrossRef]
- Kumar, S.; Chagne, D.; Bink, M.C.A.M.; Volz, R.K.; Whitworth, C.; Carlisle, C. Genomic selection for fruit quality traits in apple (Malus 3 domestica Borkh.). PLoS ONE 2012, 7, e36674. [Google Scholar] [CrossRef]
- Gwali, S.; Vaillant, A.; Nakabonge, G.; Okullo, J.B.L.; Eilu, G.; Muchugi, A.; Jean-Marc Bouvet, J.-M. Genetic diversity in shea tree (Vitellaria paradoxa subspecies nilotica) ethno-varieties in Uganda assessed with microsatellite markers. For. Trees Livelihoods 2014, 24, 163–175. [Google Scholar] [CrossRef]
- Fontaine, C.; Lovett, P.N.; Sanou, H.; Maley, J.; Bouvet, J.-M. Genetic diversity of the shea tree (Vitellaria paradoxa C.F. Gaertn), detected by RAPD and chloroplast microsatellite markers. Heredity 2004, 93, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.D.; Stymne, S.; Ohlrogge, J. Biochemical pathways in seed oil synthesis. Curr. Opin. Plant Biol. 2013, 16, 358–364. [Google Scholar] [CrossRef]
- Xianghan, L.; Tianxiang, T.; Chao, S.; Libo, S.; Hui, Z.; Chuanli, Z.; Liping, L.; Liangbin, L. Several Key Enzymes in Oil Synthesis of the Brassica napus. J. Chin. Cereals Oils Assoc. 2017, 12, 100–104. [Google Scholar]
- Zhao, H.; Kosma, D.K.; Lü, S. Functional Role of Long-Chain Acyl-CoA Synthetases in Plant Development and Stress Responses. Front. Plant Sci. 2021, 12, 640996. [Google Scholar] [CrossRef]
- Jasinski, S.; Chardon, F.; Nesi, N.; Lécureuil, A.; Guerche, P. Improving seed oil and protein content in Brassicaceae: Some new genetic insights from Arabidopsis thaliana. OCL 2018, 25, D603. [Google Scholar] [CrossRef]
- Osorio-Guarín, J.A.; Garzón-Martínez, G.A.; Delgadillo-Duran, P.; Bastidas, S.; Moreno, L.P.; Enciso-Rodríguez, F.E.; Cornejo, O.E.; Barrero, L.S. Genome-wide association study (GWAS) for morphological and yield-related traits in an oil palm hybrid (Elaeis oleifera x Elaeis guineensis) population. BMC Plant Biol. 2019, 19, 533. [Google Scholar] [CrossRef]
- Raboanatahiry, N.; Wang, B.; Yu, L.; Li, M. Functional and Structural Diversity of Acyl-coA Binding Proteins in Oil Crops. Front. Genet. 2018, 9, 182. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.-S.; Tang, Y.-H.; Sun, J.; Zhao, D.-Q.; Zhang, K.-L.; Tao, J. Identification of genes associated with the biosynthesis of unsaturated fatty acid and oil accumulation in herbaceous peony ‘Hangshao’ (Paeonia lactiflora ‘Hangshao’) seeds based on transcriptome analysis. BMC Genom. 2021, 22, 94. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.-Z.; Xue, H.-W. Arabidopsis b-Ketoacyl-[Acyl Carrier Protein] Synthase I Is Crucial for Fatty Acid Synthesis and Plays a Role in Chloroplast Division and Embryo Development. Plant Cell 2010, 22, 3726–3744. [Google Scholar] [CrossRef] [PubMed]
- Janick, P.; Huleux, M.; Spaniol, B.; Sommer, F.; Neunzig, J.; Schroda, M.; Li-Beisson, Y.; Philippar, K. Fatty acid export (FAX) proteins contribute to oil production in the green microalga Chlamydomonas reinhardtii. Front. Mol. Biosci. 2022, 9, 939834. [Google Scholar] [CrossRef]
- Martins-Noguerol, R.; DeAndres-Gil, C.; Garces, R.; Salas, J.J.; Martínez-Force, E.; Moreno-Perez, A.J. Characterization of the acyl-ACP thioesterases from Koelreuteria paniculata reveals a new type of FatB thioesterase. Heliyon 2020, 6, e05237. [Google Scholar] [CrossRef] [PubMed]
- Hajiahmadi, Z.; Abedi, A.; Wei, H.; Sun, W.; Ruan, H.; Zhuge, Q.; Movahedi, A. Identification, evolution, expression, and docking studies of fatty acid desaturase genes in wheat (Triticum aestivum L.). BMC Genom. 2020, 21, 778. [Google Scholar] [CrossRef] [PubMed]
- Botha, F.; Dennis, D. Phosphoglyceromutase activity and concentration in the endosperm of developing and germinating Ricinus communis seeds. Biol. Chem. 1987. [Google Scholar] [CrossRef]
- Golub, E.E.; Boesze-Battaglia, K. The role of alkaline phosphatase in mineralization. Curr. Opin. Orthop. 2007, 18, 444–448. [Google Scholar] [CrossRef]
- Minamikawa, M.F.; Nonaka, K.; Kaminuma, E.; Kajiya-Kanegae, H.; Onogi, A.; Goto, S.; Yoshioka, T.; Imai, A.; Hamada, H.; Hayashi, T.; et al. Genome-wide association study and genomic prediction in citrus: Potential of genomics-assisted breeding for fruit quality traits. Sci. Rep. 2017, 7, 4721. [Google Scholar] [CrossRef]
- Kim, S.; Plagnol, V.; Hu, T.T.; Toomajian, C.; Clark, R.M.; Ossowski, S.; Ecker, J.R.; Weigel, D.; Nordborg, M. Recombination and linkage disequilibrium in Arabidopsis thaliana. Nat. Genet. 2007, 39, 1151–1155. [Google Scholar] [CrossRef]
- Thomson, M.J.; Ismail, A.M.; McCouch, S.R.; Mackill, D.J. Abiotic Stress Adaptation in Plants; Pareek, A., Sopory, S.K., Bohnert, H.J., Eds.; Springer: Dordrecht, The Netherlands, 2009; pp. 451–469. [Google Scholar]
- Tartarini, S.; Sansavini, S. Advances in the use of molecular markers in Pome fruit breeding. In Proceedings of the XXVIth International Horticultural Conference and Exhibition, Toronto, ON, Canada, 11–17 August 2002; p. 622. [Google Scholar]
- Odoi, J.B.; Muchugi, A.; Okia, C.A.; Gwali, S.; Odong, T.L. Local knowledge, identification and selection of shea tree (Vitellaria paradoxa) ethnovarieties for pre-breeding in Uganda. J. Agric. Nat. Resour. Sci. 2020, 7, 22–33. [Google Scholar]
- Agúndez, D.; Nouhoheflin, T.; Coulibaly, O.; Soliño, M.; Alía, R. Local Preferences for Shea Nut and Butter Production in Northern Benin: Preliminary Results. Forests 2020, 11, 13. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Traits | Mean ± (SD a) | Minimum | Maximum |
|---|---|---|---|
| Kernel dry matter oil content (% b) | 53.53 ± 2.28 | 39.05 | 69.77 |
| Kernel length (cm c) | 3.19 ± 0.34 | 1.90 | 8.43 |
| Kernel width (cm) | 3.61 ± 0.43 | 2.23 | 4.97 |
| Kernel weight (mg d) | 10.30 ± 0.30 | 2.00 | 18.8 |
| Source of Variation | Df a | KOC b | KL c | KW d | KWt e |
|---|---|---|---|---|---|
| Replications | 2 | 4.81 | 0.01307 | 0.0249 | 0.08108 |
| Environment | 4 | 1840.82 *** | 0.694 *** | 0.82403 *** | 0.90574 *** |
| Genotypes | 373 | 60.42 *** | 1.45026 *** | 2.54701 *** | 0.9112 *** |
| Genotype x Environment | 1492 | 35.9 ** | 0.01524 | 20.69 | 0.01666 |
| Residuals | 3738 | 8.61 | 0.0159 | 0.01553 | 0.02156 |
| Chromosomes | All SNPs a | Filtered SNPs | Chr b Size (Mbs) | PIC c | Gene Div d |
|---|---|---|---|---|---|
| 1 | 2893 | 805 | 82 | 0.262 | 0.32 |
| 2 | 3450 | 960 | 74.5 | 0.260 | 0.32 |
| 3 | 1545 | 430 | 38.6 | 0.261 | 0.32 |
| 4 | 1527 | 425 | 37 | 0.258 | 0.31 |
| 5 | 2336 | 650 | 56.5 | 0.261 | 0.32 |
| 6 | 2210 | 615 | 58 | 0.259 | 0.31 |
| 7 | 2088 | 581 | 57.3 | 0.262 | 0.32 |
| 8 | 2318 | 645 | 48 | 0.260 | 0.32 |
| 9 | 2124 | 591 | 56.5 | 0.262 | 0.32 |
| 10 | 2803 | 780 | 50 | 0.265 | 0.32 |
| 11 | 1791 | 498 | 47.1 | 0.265 | 0.32 |
| 12 | 1978 | 550 | 46.9 | 0.269 | 0.33 |
| Total/Mean | 27,063 | 7530 | 652.4 | 0.260 | 0.32 |
| Trait | Pσ a | Marker | Chr b | Position (bp) | Alleles | QTN Effect | LOD Score | −log10 c | r2 d | MAF e |
|---|---|---|---|---|---|---|---|---|---|---|
| Oil content | S1_60237300 | 1 | 60237300 | AA | 0.83 | 3.39 | 4.11 | 6.61 | 0.12 | |
| S3_14843482 | 3 | 14843482 | AA | −1.06 | 5.67 | 6.49 | 11.80 | 0.14 | ||
| S4_32032310 | 4 | 32032310 | AA | 0.74 | 3.07 | 3.77 | 6.76 | 0.19 | ||
| 4.03 | S5_6275145 | 5 | 6275145 | AA | 0.68 | 3.21 | 3.92 | 5.11 | 0.15 | |
| S8_41696703 | 8 | 41696703 | TT | −1.06 | 5.93 | 6.76 | 13.31 | 0.17 | ||
| S9_32689981 | 9 | 32689981 | CC | −1.22 | 5.38 | 6.19 | 11.52 | 0.09 | ||
| S11_43126044 | 11 | 43126044 | CC | 0.81 | 4.28 | 5.05 | 8.18 | 0.31 | ||
| kernel length | S3_11153087 | 3 | 11153087 | TT | −0.13 | 3.44 | 4.16 | 8.19 | 0.12 | |
| S5_15524578 | 5 | 15524578 | AA | 0.10 | 3.37 | 4.09 | 6.51 | 0.32 | ||
| S6_46530240 | 6 | 46530240 | TT | −0.25 | 4.71 | 5.49 | 14.55 | 0.05 | ||
| 0.095 | S8_11121701 | 8 | 11121701 | GG | −0.14 | 3.16 | 3.87 | 9.08 | 0.10 | |
| S11_8320549 | 11 | 8320549 | CC | −0.13 | 3.74 | 4.48 | 7.28 | 0.10 | ||
| S12_32853547 | 12 | 32853547 | CC | −0.18 | 3.96 | 4.71 | 9.31 | 0.06 | ||
| kernel width | S1_32402910 | 1 | 32402910 | CC | −0.19 | 4.42 | 5.20 | 9.75 | 0.12 | |
| 0.169 | S2_47786838 | 2 | 47786838 | CC | 0.16 | 4.99 | 5.79 | 9.01 | 0.26 | |
| S2_64059706 | 2 | 64059706 | AA | 0.17 | 4.73 | 5.52 | 8.28 | 0.13 | ||
| S7_3025298 | 7 | 3025298 | CC | 0.15 | 3.22 | 3.92 | 5.29 | 0.10 | ||
| S9_43700743 | 9 | 43700743 | AA | −0.18 | 3.30 | 4.01 | 7.77 | 0.11 | ||
| S10_50604452 | 10 | 50604452 | GG | 0.19 | 3.81 | 4.55 | 8.69 | 0.10 | ||
| S12_32853547 | 12 | 32853547 | CC | 0.29 | 7.02 | 7.89 | 13.14 | 0.06 | ||
| S12_7613999 | 12 | 7613999 | TT | 0.12 | 3.44 | 4.17 | 4.47 | 0.20 | ||
| kernel weight | S1_30720144 | 1 | 30720144 | CC | −0.08 | 3.06 | 3.76 | 9.20 | 0.22 | |
| 0.061 | S8_43605016 | 8 | 43605016 | CC | −0.11 | 3.29 | 4.00 | 15.70 | 0.18 |
| Traits | Marker | Chr a | Pos b | Gene ID | GO. c | Function |
|---|---|---|---|---|---|---|
| Kernel length | S3_11153087 | 3 | 11153087 | Vitpa03g07900 | IPR006968 | UVB-sensing and in early seedling morphogenesis and development |
| S5_15524578 | 5 | 15524578 | Vitpa05g09840 | GO:0005515 | ion transportation and signal transduction | |
| S6_46530240 | 6 | 46530240 | Vitpa06g28930 | PTHR23155 | Disease resistance (R) | |
| S8_11121701 | 8 | 11121701 | Vitpa08g10570 | GO:0004017 | Predicts residues in protein biosythesis | |
| S11_8320549 | 11 | 8320549 | Vitpa11g07160 | PTHR33052 | Protein translocation | |
| S12_32853547 | 12 | 32853547 | Vitpa12g19540 | GO:0003824 | Protein metabolism and gluconeogenesis | |
| Kernel width | S1_32402910 | 1 | 32402910 | Vitpa01g21080 | GO:0005515 | Consensus disorder prediction |
| S2_47786838 | 2 | 47786838 | Vitpa02g27300 | GO:0043190 | Glutathione synthetase ATP-binding | |
| S2_64059706 | 2 | 64059706 | Vitpa02g39460 | Zinc finger | ||
| S7_3025298 | 7 | 3025298 | Vitpa07g02460 | GO:0005515 | Calcium signaling | |
| S9_43700743 | 9 | 43700743 | Vitpa09g19440 | PTHR14859 | Protein binding | |
| S10_50604452 | 10 | 50604452 | Vitpa10g25960 | GO:0003677 | Chromosome cohesion | |
| S12_32853547 | 12 | 32853547 | Vitpa12g19540 | GO:0003824 | Protein metabolism and gluconeogenesis | |
| S12_7613999 | 12 | 7613999 | Vitpa12g07520 | GO:0055114 | Catalyze the oxidation of alcohols to aldehydes and ketones | |
| Kernel weight | S1_30720144 | 1 | 30720144 | Vitpa01g20620 | GO:0003676 | Hydrolyze ATP |
| S8_43605016 | 8 | 43605016 | Vitpa08g25310 | GO:0004672 | Predict protein residues as disordered |
| Traits | Marker | Chr a | Pos b | Gene ID c | GO. d | Function |
|---|---|---|---|---|---|---|
| Oil content | S1_60237300 | 1 | 62536299 | Vitpa01g27780 (Acyl-ACP Thioesterase Fat B (FATB)) | GO:0004553 | Consensus disorder prediction |
| S3_14843482 | 3 | 14843482 | Vitpa03g10720 (Acyl-CoA-binding protein (ACBP)) | GO:0005515 | Protein binding | |
| S4_32032310 | 4 | 32032310 | Vitpa04g14070 Long Chain Acyl-CoA Synthetase (LACS)) | G3DSA | Oxidoreductase activity | |
| S5_6275145 | 5 | 6275145 | Vitpa05g04280 (Acyl-CoA-binding protein (ACBP)) | GO:0000160 | Transcriptional regulation of oil biosynthesis in seed plants | |
| S8_41696703 | 8 | 41696703 | Vitpa08g23790 (Fatty acid exporter (FAX2)) | GO:0008168 | methyltransferase activity | |
| S9_32689981 | 9 | 32689981 | Vitpa09g14250 (3-ketoacyl-ACP synthase II (KASII)) | GO:0004672 | Early noduling | |
| S11_43126044 | 11 | 43126044 | Vitpa11g24760 (Fatty acid desaturases (FADs)) | abiotic stress reduction |
| Chromosome | Chr1 | Chr2 | Chr3 | Chr4 | Chr5 | Chr6 | Chr7 | Chr8 | Chr9 | Chr10 | Chr11 | Chr12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| # marker pairs | 39,461 | 46,764 | 22,990 | 21,698 | 34,482 | 30,834 | 28,187 | 32,786 | 30,232 | 35,784 | 24,736 | 27,091 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Odoi, J.B.; Adjei, E.A.; Barnor, M.T.; Edema, R.; Gwali, S.; Danquah, A.; Odong, T.L.; Hendre, P. Genome-Wide Association Mapping of Oil Content and Seed-Related Traits in Shea Tree (Vitellaria paradoxa subsp. nilotica) Populations. Horticulturae 2023, 9, 811. https://doi.org/10.3390/horticulturae9070811
Odoi JB, Adjei EA, Barnor MT, Edema R, Gwali S, Danquah A, Odong TL, Hendre P. Genome-Wide Association Mapping of Oil Content and Seed-Related Traits in Shea Tree (Vitellaria paradoxa subsp. nilotica) Populations. Horticulturae. 2023; 9(7):811. https://doi.org/10.3390/horticulturae9070811
Chicago/Turabian StyleOdoi, Juventine Boaz, Emmanuel Amponsah Adjei, Michael Teye Barnor, Richard Edema, Samson Gwali, Agyemang Danquah, Thomas Lapaka Odong, and Prasad Hendre. 2023. "Genome-Wide Association Mapping of Oil Content and Seed-Related Traits in Shea Tree (Vitellaria paradoxa subsp. nilotica) Populations" Horticulturae 9, no. 7: 811. https://doi.org/10.3390/horticulturae9070811
APA StyleOdoi, J. B., Adjei, E. A., Barnor, M. T., Edema, R., Gwali, S., Danquah, A., Odong, T. L., & Hendre, P. (2023). Genome-Wide Association Mapping of Oil Content and Seed-Related Traits in Shea Tree (Vitellaria paradoxa subsp. nilotica) Populations. Horticulturae, 9(7), 811. https://doi.org/10.3390/horticulturae9070811