
The SSR Null Allele Problem, and Its Consequences in Pedigree Reconstruction and Population Genetic Studies in Viticulture

 ,
,  ,
, 
Abstract
:1. The Null Allele Problem
- The primers used in the PCR reaction fail to bind to the DNA because the DNA sequence is different from the conservative reference sequence on which the detection is based [3]. This problem can also be caused by inappropriate primer design.
- Amplification of alleles of different sizes may differ, with “longer” alleles sometimes not amplified [4].
2. Methods for the Estimation of Null Allele Frequencies
2.1. Chakraborty’s Method
2.2. Brookfield’s Method
2.3. The EM (Expectation-Maximization) Algorithm and Its Use for Estimating Null Allele Frequencies
3. The Null Allele Problem in Pedigree Reconstruction in Viticulture
3.1. The Importance of Pedigree Reconstruction from the Grape Breeder’s Point of View
3.2. Consequences of the Presence of Null Alleles in Pedigree Studies-Some Examples
3.3. Solutions for the Correct Pedigree Reconstruction
4. The Null Allele Problem in Population Genetic Studies in Viticulture
4.1. Importance of Population Genetic Studies in Wild and Cultivated Grapevines
4.2. Consequences of the Presence of Null Alleles in Population Genetic Studies
4.3. Solutions for Minimizing the Error of the Estimates
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dakin, E.E.; Avise, J.C. Microsatellite Null Alleles in Parentage Analysis. Heredity 2004, 93, 504–509. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L. Maximum Likelihood Estimation of the Frequency of Null Alleles at Microsatellite Loci. Conserv. Genet. 2006, 7, 991–995. [Google Scholar] [CrossRef]
- Kwok, S.; Kellogg, D.E.; Mckinney, N.; Spasic, D.; Goda, L.; Levenson, C.; Sninsky, J.J. Effects of Primer-Template Mismatches on the Polymerase Chain Reaction: Human Immunodeficiency Virus Type 1 Model Studies. Nucleic Acids Res. 1990, 18, 999. [Google Scholar] [CrossRef] [PubMed]
- Wattier, R.; Engel, C.R.; Saumitou-Laprade, P.; Valero, M. Short Allele Dominance as a Source of Heterozygote Deficiency at Microsatellite Loci: Experimental Evidence at the Dinucleotide Locus Gv1CT in Gracilaria Gracilis (Rhodophyta). Mol. Ecol. 1998, 7, 1569–1573. [Google Scholar] [CrossRef]
- Garcia de Leon, F.J.; Canonne, M.; Quillet, E.; Bonhomme, F.; Chatain, B. The Application of Microsatellite Markers to Breeding Programmes in the Sea Bass, Dicentrarchus Labrax. Aquaculture 1998, 159, 303–316. [Google Scholar] [CrossRef]
- Gagneux, P.; Boesch, C.; Woodruff, D.S. Microsatellite Scoring Errors Associated with Noninvasive Genotyping Based on Nuclear DNA Amplified from Shed Hair. Mol. Ecol. 1997, 6, 861–868. [Google Scholar] [CrossRef]
- Wagner, A.P.; Creel, S.; Kalinowski, S.T. Estimating Relatedness and Relationships Using Microsatellite Loci with Null Alleles. Heredity 2006, 97, 336–345. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Slatkin, M. Maximum-Likelihood Estimation of Molecular Haplotype Frequencies in a Diploid Population. Mol. Biol. Evol. 1995, 12, 921–927. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, R.; Jin, L. Heterozygote Deficiency, Population Substructure and Their Implications in DNA Fingerprinting. Hum. Genet. 1992, 88, 267–272. [Google Scholar] [CrossRef]
- Chakraborty, R.; de Andrade, M.; Daiger, S.P.; Budowle, B. Apparent Heterozygote Deficiencies Observed in DNA Typing Data and Their Implications in Forensic Applications. Ann. Hum. Genet. 1992, 56, 45–57. [Google Scholar] [CrossRef]
- Summers, K.; Amos, W. Behavioral, Ecological, and Molecular Genetic Analyses of Reproductive Strategies in the Amazonian Dart-Poison Frog, Dendrobates Ventrimaculatus. Behav. Ecol. 1997, 8, 260–267. [Google Scholar] [CrossRef] [Green Version]
- Dempster, A.P.; Laird, N.M.; Rubin, D.B. Maximum Likelihood Estimation from Incomplete Data via the EM Algorithm. J. R. Stat. Soc. B 1977, 39, 1–38. [Google Scholar]
- Ceppellini, R.; Siniscalco, M.; Smith, C.A. The Estimation of Gene Frequencies in a Randomly Mating Population. Ann. Hum. Genet. 1955, 20, 97–115. [Google Scholar] [CrossRef]
- Kalinowski, S. ML-Null Freq-Steven Kalinowski|Montana State University. Available online: https://www.montana.edu/kalinowski/software/null-freq.html (accessed on 12 May 2022).
- Jahnke, G.; Smidla, J. MolMarker-Molekuláris Markerekkel Kapott Kutatási Eredmények Értékelését Segítő Felhasználóbarát Szoftver (MolMarker-User-Friendly Software to Help Evaluate the Research Results Obtained by Molecular Markers); Georgikon Napok Keszthely, M., Kis, L.B., Lukács, G., Nagy, B., Tóth, G., Eds.; Pannon Egyetem Georgikon Mezőgazdaságtudományi Kar: Keszthely, Hungary, 2014; pp. 135–143. Available online: https://napok.georgikon.hu/hu/cikkadatbazis/cikkek-2012/doc_download/203-gyorffyne-jahnke-gizella-smidla-jozsef-molmarker-molekularis-markerekkel-kapott-kutatasi-eredmenyek-ertekeleset-segito-felhasznalobarat-szoftver (accessed on 28 June 2022).
- Jahnke, G.; Smidla, J.; Poczai, P. MolMarker: A Simple Tool for DNA Fingerprinting Studies and Polymorphic Information Content Calculation. Diversity 2022, 14, 497. [Google Scholar] [CrossRef]
- Kozma, P. A Szőlő Teljesítőképessége Növelésének Lehetőségei Keresztezéses Nemesítéssel—Possibilities of Increasing the Productivity of the Vine by Cross-Breeding-Academic Inaugural Lecture; Hungarian Academy of Sciences: Budapest, Hungary, 1973. [Google Scholar]
- Kozma, P. A Szőlő Nemesítése. (Breeding of the Vine); Mezőgazdasági Kiadó: Budapest, Hungary, 1951. [Google Scholar]
- Hajdu, E. A Kertészeti Növények Nemesítése a Szőlő Példáján. (The Horticultural Plant Breeding on the Example of Grape. Gradus 2016, 3, 378–383. [Google Scholar]
- Hajdu, E. 6—Grapevine breeding in Hungary. In Woodhead Publishing Series in Food Science, Technology and Nutrition, Grapevine Breeding Programs for the Wine Industry; Reynolds, A., Ed.; Woodhead Publishing: Cambridge, UK, 2015; pp. 103–134. ISBN 9781782420750. [Google Scholar] [CrossRef]
- Negrul, A.M. Questions of Origin and Breeding of the Grape Vine on a Genetical Basis. Genetika 1968, 4, 84–97. [Google Scholar]
- Negrul’, A.M. Questions of the Origin and Breeding of Vines on a Genetical Basis. NI Vavilov Agric. Sci. 1969, 323–339. [Google Scholar]
- Kozma, P.I. A Szőlőrezisztencia-Nemesítés Szempontjai Es Módszerei Magyarországon. Int. J. Hortic. Sci. 2002, 8, 43–48. [Google Scholar] [CrossRef]
- Korbuly, J. A Vitis Amurensis (Rupp.) Értékei a Szőlő Rezisztencia Nemesítés Számára. Int. J. Hortic. Sci. 2002, 8, 53–58. [Google Scholar] [CrossRef]
- Gorodea, G.; Neagu, M.I. Combining Ability of the Cultivar “Bicane”; INRA: Paris, France, 1978. [Google Scholar]
- Roytchev, V. Combining Ability and Genotype-Environmental Variability of Cluster Weight in F-1 Crosses between Seeded and Seedless Grape-Vine (Vitis vinifera L.) Cultivars. Genet. Breed. 2006, 35, 29–38. [Google Scholar]
- Maul, E.; Eibach, R.; Zyprian, E.; Töpfer, R. The Prolific Grape Variety (Vitis vinifera L.) ‘Heunisch Weiss’ (= ‘Gouais Blanc’): Bud Mutants, “Colored” Homonyms and Further Offspring. VITIS J. Grapevine Res. 2015, 54, 79–86. [Google Scholar] [CrossRef]
- Boursiquot, J.-M.; Lacombe, T.; Bowers, J.E.; Meredith, C.P. Le Gouais, Un Cépage Clé Du Patrimoine Viticole Européen. Bull. L’O.I.V. 2004, 77, 5–19. [Google Scholar]
- Vouillamoz, J.F.; Grando, M.S. Genealogy of Wine Grape Cultivars: ‘Pinot’ Is Related to ‘Syrah’. Heredity 2006, 97, 102–110. [Google Scholar] [CrossRef] [Green Version]
- This, P.; Lacombe, T.; Thomas, M.R. Historical Origins and Genetic Diversity of Wine Grapes. Trends Genet. 2006, 22, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Bowers, J.; Boursiquot, J.M.; This, P.; Chu, K.; Johansson, H.; Meredith, C. Historical Genetics: The Parentage of Chardonnay, Gamay, and Other Wine Grapes of Northeastern France. Science 1999, 285, 1562–1565. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.G.; Small, C.M.; Paczolt, K.A.; Ratterman, N.L. A Practical Guide to Methods of Parentage Analysis. Mol. Ecol. Resour. 2010, 10, 6–30. [Google Scholar] [CrossRef]
- Bautista, J.; Dangl, G.S.; Yang, J.; Reisch, B.; Stover, E. Use of Genetic Markers to Assess Pedigrees of Grape Cultivars and Breeding Program Selections. Am. J. Enol. Vitic. 2008, 59, 248–254. [Google Scholar]
- Crespan, M. The Parentage of Muscat of Hamburg. Vitis 2003, 42, 193–197. [Google Scholar]
- Vouillamoz, J.; Maigre, D.; Meredith, C.P. Microsatellite Analysis of Ancient Alpine Grape Cultivars: Pedigree Reconstruction of Vitis Vinifera L. “Cornalin Du Valais”. Theor. Appl. Genet. 2003, 107, 448–454. [Google Scholar] [CrossRef]
- Crespan, M.; Calo, A.; Costacurta, A.; Milani, N.; Giust, M.; Carraro, R.; di Stefano, R. Ciliegiolo and Aglianicone: Only One Variety in a Direct Relationship with Sangiovese [Vitis vinifera L.; Grapevine]. Riv. Vitic. E Enol. 2002, 55, 3–14. [Google Scholar]
- Vouillamoz, J.F.; Monaco, A.; Costantini, L.; Stefanini, M.; Scienza, A.; Grando, M.S. The Parentage of “Sangiovese”, the Most Important Italian Wine Grape. Vitis 2007, 46, 19–22. [Google Scholar]
- Staraz, M.D.V.; Bandinelli, R.; Boselli, M.; This, P.; Boursiquot, J.M.; Laucou, V.; Lacombe, T.; Varès, D. Genetic Structuring and Parentage Analysis for Evolutionary Studies in Grapevine: Kin Group and Origin of the Cultivar Sangiovese Revealed. J. Am. Soc. Hortic. Sci. 2007, 132, 514–524. [Google Scholar] [CrossRef]
- Cipriani, G.; Spadotto, A.; Jurman, I.; di Gaspero, G.; Crespan, M.; Meneghetti, S.; Frare, E.; Vignani, R.; Cresti, M.; Morgante, M.; et al. The SSR-Based Molecular Profile of 1005 Grapevine (Vitis vinifera L.) Accessions Uncovers New Synonymy and Parentages, and Reveals a Large Admixture amongst Varieties of Different Geographic Origin. Theor. Appl. Genet. 2010, 121, 1569–1585. [Google Scholar] [CrossRef]
- Bergamini, C.; Caputo, A.R.; Gasparro, M.; Perniola, R.; Cardone, M.F.; Antonacci, D. Evidences for an Alternative Genealogy of “Sangiovese”. Mol. Biotechnol. 2013, 53, 278–288. [Google Scholar] [CrossRef]
- Bergamini, C.; Caputo, A.R.; Perniola, R.; Cardone, M.F.; Gasparro, M.; Antonacci, D. Genealogy of “sangiovese”: A new hypothesis. In Proceedings of the 35th World Congress of Vine and Wine, Izmir, Turkey, 18–22 June 2012; OIV: Paris, France, 2012. [Google Scholar]
- Sefc, K.; Pejić, I.; Maletić, E. Microsatellite Markers for Grapevine: Tools for Cultivar Identification & Pedigree Reconstruction. In Grapevine Molecular Physiology & Biotechnology; Springer: Dordrecht, The Netherlands, 2009; pp. 565–596. [Google Scholar]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. Micro-Checker: Software for Identifying and Correcting Genotyping Errors in Microsatellite Data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Cao, S.; Stringer, S.; Gunawan, G.; McGregor, C.; Conner, P.J. Genetic Diversity and Pedigree Analysis of Muscadine Grape Using SSR Markers. J. Am. Soc. Hortic. Sci. 2020, 145, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Mihaljević, M.Ž.; Maletić, E.; Preiner, D.; Zdunić, G.; Bubola, M.; Zyprian, E.; Pejić, I. Genetic Diversity, Population Structure, and Parentage Analysis of Croatian Grapevine Germplasm. Genes 2020, 11, 737. [Google Scholar] [CrossRef]
- Christie, M.R. Parentage in Natural Populations: Novel Methods to Detect Parent-Offspring Pairs in Large Data Sets. Mol. Ecol. Resour. 2010, 10, 115–128. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Wagner, A.P.; Taper, M.L. ML-RELATE: A Computer Program for Maximum Likelihood Estimation of Relatedness and Relationship. Mol. Ecol. Notes 2006, 6, 576–579. [Google Scholar] [CrossRef]
- Ko, A.; Nielsen, R. Joint Estimation of Pedigrees and Effective Population Size Using Markov Chain Monte Carlo. Genetics 2019, 212, 855–868. [Google Scholar] [CrossRef] [Green Version]
- Population Genetics-Latest Research and News|Nature. Available online: https://www.nature.com/subjects/population-genetics (accessed on 7 June 2022).
- Aravanopoulos, F.A.; Ganopoulos, I.; Tsaftaris, A. Population and Conservation Genomics in Forest and Fruit Trees. Adv. Bot. Res. 2015, 74, 125–155. [Google Scholar] [CrossRef]
- Purugganan, M.D. Evolutionary Insights into the Nature of Plant Domestication. Curr. Biol. 2019, 29, R705–R714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrano, A.; Grzeskowiak, L.; Sanz, P.M.; Lorenzi, S.; Prazzoli, M.L.; Arzumanov, A.; Amanova, M.; Failla, O.; Maghradze, D.; Grando, M.S. Genetic Diversity and Relationships in the Grapevine Germplasm Collection from Central Asia. VITIS J. Grapevine Res. 2015, 54, 233–237. [Google Scholar] [CrossRef]
- Martínez, L.E.; Cavagnaro, P.F.; Masuelli, R.W.; Zúñiga, M. SSR-Based Assessment of Genetic Diversity in South American Vitis Vinifera Varieties. Plant Sci. 2006, 170, 1036–1044. [Google Scholar] [CrossRef]
- Doulati-Baneh, H.; Mohammadi, S.A.; Labra, M. Genetic Structure and Diversity Analysis in Vitis Vinifera L. Cultivars from Iran Using SSR Markers. Sci. Hortic. 2013, 160, 29–36. [Google Scholar] [CrossRef]
- Jahnke, G.; Májer, J.; Lakatos, A.; Molnár, J.G.; Deák, E.; Stefanovits-Bányai, E.; Varga, P. Isoenzyme and Microsatellite Analysis of Vitis Vinifera L. Varieties from the Hungarian Grape Germplasm. Sci. Hortic. 2009, 120, 213–221. [Google Scholar] [CrossRef]
- Halász, G.; Veres, A.; Kozma, P.; Kiss, E.; Balogh, A.; Galli, Z.; Szőke, A.; Hoffmann, S.; Heszky, L. Microsatellite Fingerprinting of Grapevine (Vitis vinifera L.) Varieties of the Carpathian Basin. Vitis 2005, 44, 173. [Google Scholar]
- Scali, M.; Zifferero, A.; Vignani, R. Distribution and Characterization of the Vitis vinifera L. Subsp Sylvestris in Southern Tuscany. Recent Pat. Biotechnol. 2018, 12, 208–220. [Google Scholar] [CrossRef]
- Zdunić, G.; Maul, E.; Dias, J.E.E.; Organero, G.M.; Carka, F.; Maletic, E.; Savvides, S.; Jahnke, G.G.; Nagy, Z.A.; Nikolić, D.; et al. Guiding Principles for Identification, Evaluation and Conservation of Vitis vinifera L. Subsp Sylvestris. Vitis J. Grapevine Res. 2017, 56, 127–131. [Google Scholar] [CrossRef]
- Söylemezoğlu, G.; Ağaoğlu, Y.S.; Uzun, H.I. Ampelographic Characteristics and Isozymic Analysis of Vitis vinifera spp. Sylvestris Gmel. In Southwestern Turkey. Biotechnol. Biotechnol. Equip. 2001, 15, 106–113. [Google Scholar] [CrossRef] [Green Version]
- Bodor, P.; Höhn, M.; Pedryc, A.; Deák, T.; Dücso, I.; Uzun, I.; Cseke, K.; Böhm, É.I.; Bisztray, G.D. Conservation Value of the Native Hungarian Wild Grape (Vitis sylvestris Gmel.) Evaluated by Microsatellite Markers. Vitis J. Grapevine Res. 2010, 49, 23–27. [Google Scholar]
- Jahnke, G.; Nagy, Z.A.; Koltai, G.; Hajdú, E.; Májer, J. Preservation and Characterization of Woodland Grape (Vitis vinifera ssp. Sylvestris GMEL) Genotypes of the Szigetköz, Hungary. In Germplasm: Characteristics, Diversity and Preservation; Walton, M., Ed.; Nova Science Publishers: New York, NY, USA, 2016; pp. 27–45. [Google Scholar]
- Jahnke, G.; Nagy, Z.A.; Koltai, G.; Oláh, R.; Májer, J. Morphological, phenological and molecular diversity of woodland grape (Vitis sylvestris gmel.) genotypes from the szigetköz, hungary. Mitt. Klosterneubg. 2021, 71, 90–98. [Google Scholar]
- Zoghlami, N.; Riahi, L.; Laucou, V.; Mliki, A.; Ghorbel, A.; This, P. Genetic Structure of Endangered Wild Grapevine Vitis vinifera ssp. Sylvestris Populations from Tunisia: Implications for Conservation and Management. For. Ecol. Manag. 2013, 310, 896–902. [Google Scholar] [CrossRef]
- Jahnke, G.; Nagy, Z.; Taller, J.; Májer, J.; Kocsis, L. Application of Isozymes and SSR Markers for the Analysis of the Genetic Background of Some Rootstocks Derived from Teleki’s Seedlings (Teleki 5C, Kober 5BB, SO4). Mitt. Klosterneubg. 2015, 65, 221–236. [Google Scholar]
- Arnold, C.; Schnitzler, A. Ecology and Genetics of Natural Populations of North American Vitis Species Used as Rootstocks in European Grapevine Breeding Programs. Front. Plant Sci. 2020, 11, 866. [Google Scholar] [CrossRef]
- Poczai, P.; Hyvönen, J.; Taller, J.; Jahnke, G.; Kocsis, L. Phylogenetic Analyses of Teleki Grapevine Rootstocks Using Three Chloroplast DNA Markers. Plant Mol. Biol. Report. 2013, 31, 371–386. [Google Scholar] [CrossRef]
- Lin, H.; Walker, M.A. Identifying Grape Rootstocks With Simple Sequence Repeat (SSR) DNA Markers. Am. J. Enol. Vitic. 1998, 49, 403–407. [Google Scholar]
- Migliaro, D.; de Lorenzis, G.; di Lorenzo, G.S.; de Nardi, B.; Gardiman, M.; Failla, O.; Brancadoro, L.; Crespan, M. Grapevine Non-Vinifera Genetic Diversity Assessed by Simple Sequence Repeat Markers as a Starting Point for New Rootstock Breeding Programs. Am. J. Enol. Vitic. 2019, 70, 390–397. [Google Scholar] [CrossRef]
- Jahnke, G.; Kocsisné Molnár, G.; Májer, J.; Szöke, B.; Tarczal, E.; Varga, P.; Kocsis, L. Analysis of Grape Rootstocks by SSR Markers. J. Int. Des Sci. Vigne Vin 2011, 45, 199–210. [Google Scholar] [CrossRef]
- Băileştianu, N.A.; Mitrea, R. The black rot—A new challenge for vine crops. Ann. Univ. Craiova Agric. Montanol. Cadastre Ser. 2020, 49, 20–25. [Google Scholar]
- Migicovsky, Z.; Sawler, J.; Gardner, K.M.; Aradhya, M.K.; Prins, B.H.; Schwaninger, H.R.; Bustamante, C.D.; Buckler, E.S.; Zhong, G.Y.; Brown, P.J.; et al. Patterns of Genomic and Phenomic Diversity in Wine and Table Grapes. Hortic. Res. 2017, 4, 17035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacilieri, R.; Lacombe, T.; le Cunff, L.; di Vecchi-Staraz, M.; Laucou, V.; Genna, B.; Péros, J.P.; This, P.; Boursiquot, J.M. Genetic Structure in Cultivated Grapevines Is Linked to Geography and Human Selection. BMC Plant Biol. 2013, 13, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battilana, J.; Lorenzi, S.; Moreira, F.M.; Moreno-Sanz, P.; Failla, O.; Emanuelli, F.; Grando, M.S. Linkage Mapping and Molecular Diversity at the Flower Sex Locus in Wild and Cultivated Grapevine Reveal a Prominent Ssr Haplotype in Hermaphrodite Plants. Mol. Biotechnol. 2013, 54, 1031–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riaz, S.; de Lorenzis, G.; Velasco, D.; Koehmstedt, A.; Maghradze, D.; Bobokashvili, Z.; Musayev, M.; Zdunic, G.; Laucou, V.; Andrew Walker, M.; et al. Genetic Diversity Analysis of Cultivated and Wild Grapevine (Vitis vinifera L.) Accessions around the Mediterranean Basin and Central Asia. BMC Plant Biol. 2018, 18, 137. [Google Scholar] [CrossRef] [PubMed]
- De AndrÉs, M.T.; Benito, A.; PÉrez-Rivera, G.; Ocete, R.; Lopez, M.A.; Gaforio, L.; Muñoz, G.; Cabello, F.; MartÍnez Zapater, J.M.; Arroyo-GarcÍa, R. Genetic Diversity of Wild Grapevine Populations in Spain and Their Genetic Relationships with Cultivated Grapevines. Mol. Ecol. 2012, 21, 800–816. [Google Scholar] [CrossRef] [PubMed]
- Emanuelli, F.; Lorenzi, S.; Grzeskowiak, L.; Catalano, V.; Stefanini, M.; Troggio, M.; Myles, S.; Martinez-Zapater, J.M.; Zyprian, E.; Moreira, F.M.; et al. Genetic Diversity and Population Structure Assessed by SSR and SNP Markers in a Large Germplasm Collection of Grape. BMC Plant Biol. 2013, 13, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maraš, V.; Tello, J.; Gazivoda, A.; Mugoša, M.; Perišić, M.; Raičević, J.; Štajner, N.; Ocete, R.; Božović, V.; Popović, T.; et al. Population Genetic Analysis in Old Montenegrin Vineyards Reveals Ancient Ways Currently Active to Generate Diversity in Vitis vinifera. Sci. Rep. 2020, 10, 15000. [Google Scholar] [CrossRef]
- Oddou-Muratorio, S.; Vendramin, G.G.; Buiteveld, J.; Fady, B. Population Estimators or Progeny Tests: What Is the Best Method to Assess Null Allele Frequencies at SSR Loci? Conserv. Genet. 2009, 10, 1343–1347. [Google Scholar] [CrossRef]
- Carlsson, J. Effects of Microsatellite Null Alleles on Assignment Testing. J. Hered. 2008, 99, 616–623. [Google Scholar] [CrossRef] [Green Version]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring Weak Population Structure with the Assistance of Sample Group Information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [Green Version]
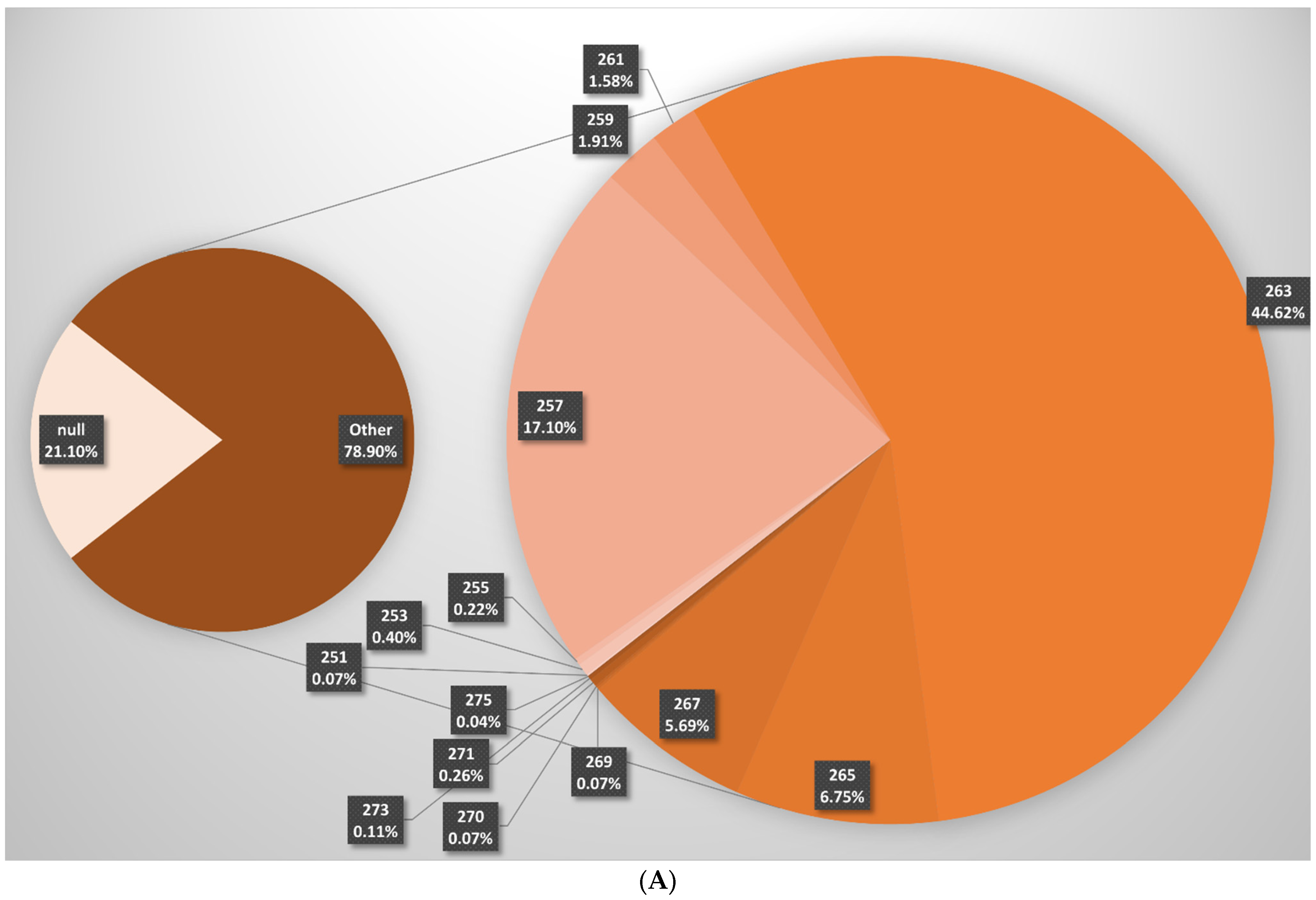
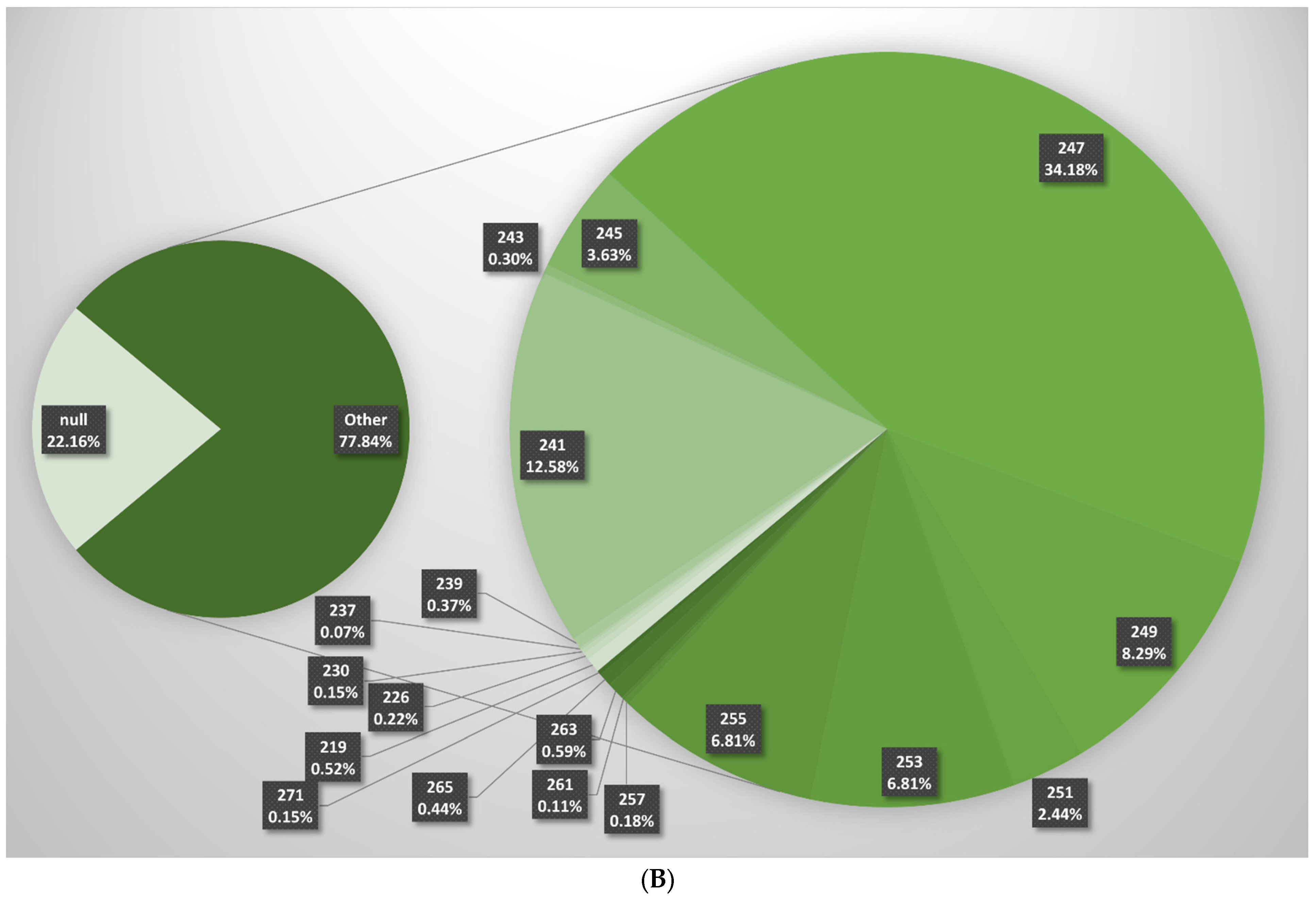


{kind=link}
{kind=link}
{kind=link}
| V. vinifera ssp. sylvestris | V. vinifera ssp. sativa | |||
|---|---|---|---|---|
| Null | No | Null | No | |
| VMC1B11 | 14.84% | 12 | 4.97% | 13 |
| VMC4F3-1 | 10.21% | 14 | 3.62% | 13 |
| VVIB01 | 11.98% | 6 | 13.81% | 6 |
| VVIH54 | 4.17% | 12 | 11.88% | 12 |
| VVIN16 | 8.59% | 6 | 12.78% | 6 |
| VVIN73 | 13.28% | 7 | 26.46% | 7 |
| VVIP31 | 8.65% | 15 | 4.16% | 13 |
| VVIP60 | 12.79% | 9 | 8.56% | 13 |
| VVIQ52 | 9.35% | 5 | 15.47% | 6 |
| VVIV37 | 11.72% | 10 | 4.97% | 12 |
| VVIV67 | 8.33% | 10 | 0.29% | 14 |
| VVMD21 | 18.75% | 7 | 12.50% | 8 |
| VVMD24 | 10.18% | 8 | 8.03% | 8 |
| VVMD25 | 1.30% | 10 | 8.84% | 12 |
| VVMD27 | 11.75% | 10 | 3.88% | 9 |
| VVMD28 | 8.56% | 15 | 7.45% | 14 |
| VVMD32 | 8.31% | 11 | 5.01% | 9 |
| VVMD5 | 7.29% | 10 | 4.43% | 10 |
| VVMD7 | 11.46% | 14 | 10.19% | 10 |
| VVS2 | 12.50% | 14 | 3.59% | 13 |
| ZAG112 | 16.45% | 8 | n.a. | n.a. |
| ZAG29 | 26.82% | 4 | n.a. | n.a. |
| ZAG62 | 6.77% | 8 | n.a. | n.a. |
| ZAG67 | 13.84% | 14 | n.a. | n.a. |
| ZAG83 | 23.38% | 7 | n.a. | n.a. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahnke, G.; Smidla, J.; Deák, T.; Oláh, R.; Szőke, B.Á.; Nyitrainé Sárdy, D.Á. The SSR Null Allele Problem, and Its Consequences in Pedigree Reconstruction and Population Genetic Studies in Viticulture. Horticulturae 2022, 8, 658. https://doi.org/10.3390/horticulturae8070658
Jahnke G, Smidla J, Deák T, Oláh R, Szőke BÁ, Nyitrainé Sárdy DÁ. The SSR Null Allele Problem, and Its Consequences in Pedigree Reconstruction and Population Genetic Studies in Viticulture. Horticulturae. 2022; 8(7):658. https://doi.org/10.3390/horticulturae8070658
Chicago/Turabian StyleJahnke, Gizella, József Smidla, Tamás Deák, Róbert Oláh, Barna Árpád Szőke, and Diána Ágnes Nyitrainé Sárdy. 2022. "The SSR Null Allele Problem, and Its Consequences in Pedigree Reconstruction and Population Genetic Studies in Viticulture" Horticulturae 8, no. 7: 658. https://doi.org/10.3390/horticulturae8070658
APA StyleJahnke, G., Smidla, J., Deák, T., Oláh, R., Szőke, B. Á., & Nyitrainé Sárdy, D. Á. (2022). The SSR Null Allele Problem, and Its Consequences in Pedigree Reconstruction and Population Genetic Studies in Viticulture. Horticulturae, 8(7), 658. https://doi.org/10.3390/horticulturae8070658