
Recent Advances in DNA Methylation and Their Potential Breeding Applications in Plants


 ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. DNA 5mC in Plants
2.1. Distribution of 5mC in Plant Genome
2.2. Derivatives of 5mC
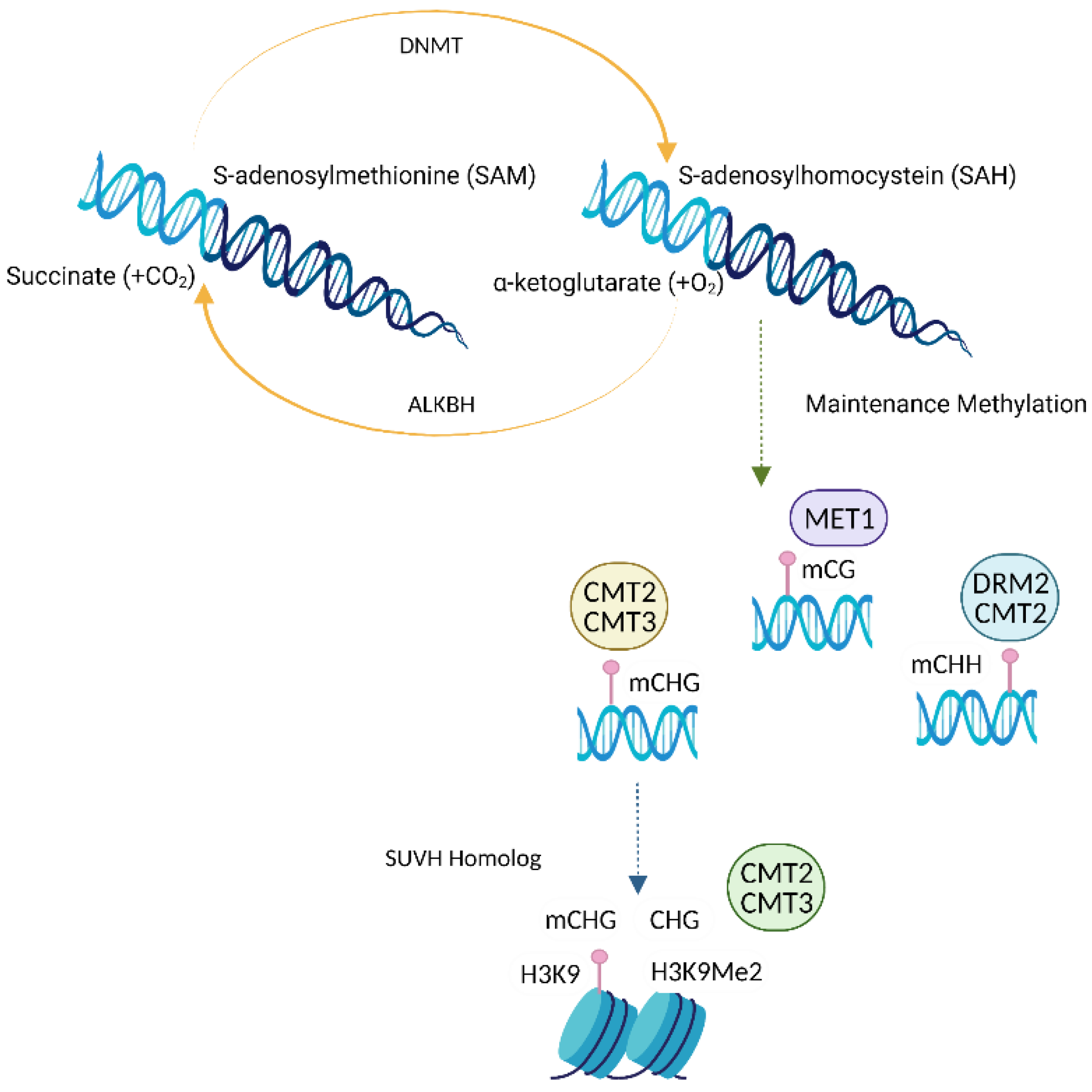
2.3. Writers, Erasers and Readers of DNA Methylation
3. DNA Adenine Methylation 6mA in Plants
4. Molecular and Biological Function of DNA Methylation
4.1. Molecular Functions of DNA Methylation
4.2. Dynamic DNA Methylation in Plant Development
4.3. DNA Methylation Heritability in Plants
4.4. DNA Methylation in Response to Stresses
5. Epigenetics Prospective for Plant Breeding
5.1. Applications of DNA Methylation in Plant Breeding
5.2. Potential Applications of RdDM in Crop Improvement
5.3. DNA Methylation and Heterosis
5.4. Challenges and Opportunities for DNA Methylation in Breeding Applications
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Correction Statement
References
- Varotto, S.; Tani, E.; Abraham, E.; Krugman, T.; Kapazoglou, A.; Melzer, R.; Radanović, A.; Miladinović, D. Epigenetics: Possible Applications in Climate-Smart Crop Breeding. J. Exp. Bot. 2020, 71, 5223–5236. [Google Scholar] [CrossRef]
- Waddington, C.H. The Epigenotype. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar] [CrossRef]
- Kumar, S.; Singh, A. Epigenetic Regulation of Abiotic Stress Tolerance in Plants. Adv. Plants Agric. Res. 2016, 5, 00179. [Google Scholar] [CrossRef]
- Saze, H. Epigenetic Memory Transmission through Mitosis and Meiosis in Plants. Semin. Cell Dev. Biol. 2008, 19, 527–536. [Google Scholar] [CrossRef]
- Thiebaut, F.; Hemerly, A.S.; Ferreira, P.C.G. A Role for Epigenetic Regulation in the Adaptation and Stress Responses of Non-Model Plants. Front. Plant. Sci. 2019, 10, 246. [Google Scholar] [CrossRef]
- Henderson, I.R.; Jacobsen, S.E. Epigenetic Inheritance in Plants. Nature 2007, 447, 418–424. [Google Scholar] [CrossRef]
- Stassen, J.H.M.; López, A.; Jain, R.; Pascual-Pardo, D.; Luna, E.; Smith, L.M.; Ton, J. The Relationship between Transgenerational Acquired Resistance and Global DNA Methylation in Arabidopsis. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Russo, V.E.A.; Martienssen, R.A.; Riggs, A.D. Epigenetic Mechanisms of Gene Regulation; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1996; ISBN 0879694904. [Google Scholar]
- Slotkin, R.K. Plant epigenetics: From genotype to phenotype and back again. Genome Biol. 2016, 17, 57. [Google Scholar] [CrossRef]
- Liu, B.; Wendel, J.F. Epigenetic Phenomena and the Evolution of Plant Allopolyploids. Mol. Phylogenet Evol. 2003, 29, 365–379. [Google Scholar] [CrossRef]
- Song, Q.; Zhang, T.; Stelly, D.M.; Chen, Z.J. Epigenomic and Functional Analyses Reveal Roles of Epialleles in the Loss of Photoperiod Sensitivity during Domestication of Allotetraploid Cottons. Genome Biol. 2017, 18, 1–14. [Google Scholar] [CrossRef]
- Zhang, Y.; Fischer, M.; Colot, V.; Bossdorf, O. Epigenetic Variation Creates Potential for Evolution of Plant Phenotypic Plasticity. New Phytol. 2013, 197, 314–322. [Google Scholar] [CrossRef]
- Stroud, H.; Ding, B.; Simon, S.A.; Feng, S.; Bellizzi, M.; Pellegrini, M.; Wang, G.-L.; Meyers, B.C.; Jacobsen, S.E. Plants Regenerated from Tissue Culture Contain Stable Epigenome Changes in Rice. elife 2013, 2, e00354. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Xiao, X.; Zhang, H.; Li, Y.; Liu, S.; Sun, W.; Zhang, X.; Wu, Q. Whole-Genome Bisulfite Sequencing Reveals a Role for Dna Methylation in Variants from Callus Culture of Pineapple (Ananas comosus L.). Genes 2019, 10, 877. [Google Scholar] [CrossRef] [PubMed]
- Stelpflug, S.C.; Eichten, S.R.; Hermanson, P.J.; Springer, N.M.; Kaeppler, S.M. Consistent and Heritable Alterations of DNA Methylation Are Induced by Tissue Culture in Maize. Genetics 2014, 198, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Crisp, P.A.; Stelpflug, S.; Kaeppler, S.M.; Li, Q.; Springer, N.M. Heritable Epigenomic Changes to the Maize Methylome Resulting from Tissue Culture. Genetics 2018, 209, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, Z.; Wang, N.; Gao, Y.; Liu, Y.; Wu, Y.; Bai, Y.; Zhang, Z.; Lin, X.; Dong, Y. Tissue Culture-Induced Heritable Genomic Variation in Rice, and Their Phenotypic Implications. PLoS ONE 2014, 9, e96879. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA Methylation Landscapes: Provocative Insights from Epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Mazzio, E.A.; Soliman, K.F.A. Basic Concepts of Epigenetics: Impact of Environmental Signals on Gene Expression. Epigenetics 2012, 7, 119–130. [Google Scholar] [CrossRef]
- García-Aguilar, M.; Gillmor, C.S. Zygotic Genome Activation and Imprinting: Parent-of-Origin Gene Regulation in Plant Embryogenesis. Curr. Opin. Plant. Biol. 2015, 27, 29–35. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, Maintaining and Modifying DNA Methylation Patterns in Plants and Animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- He, G.; Elling, A.A.; Deng, X.W. The Epigenome and Plant Development. Annu. Rev. Plant Biol. 2011, 62, 411–435. [Google Scholar] [CrossRef]
- He, X.-J.; Chen, T.; Zhu, J.-K. Regulation and Function of DNA Methylation in Plants and Animals. Cell Res. 2011, 21, 442–465. [Google Scholar] [CrossRef]
- Li, Q.; Xu, W.; Cui, Y.; Ma, L.; Richards, J.; Li, W.; Ma, Y.; Fu, G.; Bythwood, T.; Wang, Y. A Preliminary Exploration on DNA Methylation of Transgene across Generations in Transgenic Rats. Sci. Rep. 2015, 5, 8292. [Google Scholar] [CrossRef] [PubMed]
- Hackett, J.A.; Surani, M.A. DNA Methylation Dynamics during the Mammalian Life Cycle. Phil. Trans. R. Soc. B 2013, 368, 20110328. [Google Scholar] [CrossRef] [PubMed]
- Delcuve, G.P.; Rastegar, M.; Davie, J.R. Epigenetic Control. J. Cell. Physiol. 2009, 219, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Altun, G.; Loring, J.F.; Laurent, L.C. DNA Methylation in Embryonic Stem Cells. J. Cell. Biochem. 2010, 109, 1–6. [Google Scholar] [CrossRef]
- Smith, Z.D.; Meissner, A. DNA Methylation: Roles in Mammalian Development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Elhamamsy, A.R. DNA Methylation Dynamics in Plants and Mammals: Overview of Regulation and Dysregulation. Cell Biochem. Funct. 2016, 34, 289–298. [Google Scholar] [CrossRef]
- Goettel, W.; Messing, J. Epiallele Biogenesis in Maize. Gene 2013, 516, 8–23. [Google Scholar] [CrossRef]
- Ito, H. Small RNAs and Regulation of Transposons in Plants. Genes Genet. Syst. 2013, 88, 3–7. [Google Scholar] [CrossRef]
- Shi, J.; Shi, W.; Ni, L.; Xu, X.; Su, X.; Xia, L.; Xu, F.; Chen, J.; Zhu, J. OCT4 Is Epigenetically Regulated by DNA Hypomethylation of Promoter and Exon in Primary Gliomas. Oncol. Rep. 2013, 30, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Ma, W.; Solov’Yov, I.A.; Chipot, C.; Schulten, K. Recognition of Methylated DNA through Methyl-CpG Binding Domain Proteins. Nucleic Acids Res. 2012, 40, 2747–2758. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.; Allshire, R.C. RNA Silencing and Genome Regulation. Trends Cell Biol. 2005, 15, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, F.K.; Heredia, F.; Sarazin, A.; Roudier, F.; Boccara, M.; Ciaudo, C.; Cruaud, C.; Poulain, J.; Berdasco, M.; Fraga, M.F. A Role for RNAi in the Selective Correction of DNA Methylation Defects. Science 2009, 323, 1600–1604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X. The Epigenetic Landscape of Plants. Science 2008, 320, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-Wide Analysis of Arabidopsis thaliana DNA Methylation Uncovers an Interdependence between Methylation and Transcription. Nat. Genet. 2007, 39, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly Integrated Single-Base Resolution Maps of the Epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef]
- Barkan, A.; Martienssen, R.A. Inactivation of Maize Transposon Mu Suppresses a Mutant Phenotype by Activating an Outward-Reading Promoter near the End of Mu1. Proc. Natl. Acad. Sci. USA 1991, 88, 3502–3506. [Google Scholar] [CrossRef]
- Zhang, X.; Shiu, S.; Cal, A.; Borevitz, J.O. Global Analysis of Genetic, Epigenetic and Transcriptional Polymorphisms in Arabidopsis thaliana Using Whole Genome Tiling Arrays. PLoS Genet. 2008, 4, e1000032. [Google Scholar] [CrossRef]
- Kawakatsu, T.; Ecker, J.R. Diversity and Dynamics of DNA Methylation: Epigenomic Resources and Tools for Crop Breeding. Breed. Sci. 2019, 69, 191–204. [Google Scholar] [CrossRef]
- Tirnaz, S.; Batley, J. DNA Methylation: Toward Crop Disease Resistance Improvement. Trends Plant. Sci. 2019, 24, 1137–1150. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Sanchez, R.; Kundariya, H.; Maher, T.; Dopp, I.; Schwegel, R.; Virdi, K.; Axtell, M.J.; Mackenzie, S.A. Segregation of an MSH1 RNAi Transgene Produces Heritable Non-Genetic Memory in Association with Methylome Reprogramming. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Do Kim, K.; El Baidouri, M.; Jackson, S.A. Accessing Epigenetic Variation in the Plant Methylome. Brief. Funct. Genom. 2014, 13, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Duttke, S.H.; Hetzel, J.; Groth, M.; Feng, S.; Gallego-Bartolome, J.; Zhong, Z.; Kuo, H.Y.; Wang, Z.; Zhai, J.; et al. RNA-Directed DNA Methylation Involves Co-Transcriptional Small-RNA-Guided Slicing of Polymerase V Transcripts in Arabidopsis. Nat. Plants 2018, 4, 181–188. [Google Scholar] [CrossRef]
- Matzke, M.A.; Mosher, R.A. RNA-Directed DNA Methylation: An Epigenetic Pathway of Increasing Complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef]
- Zhong, X.; Du, J.; Hale, C.J.; Gallego-Bartolome, J.; Feng, S.; Vashisht, A.A.; Chory, J.; Wohlschlegel, J.A.; Patel, D.J.; Jacobsen, S.E. Molecular Mechanism of Action of Plant DRM de Novo DNA Methyltransferases. Cell 2014, 157, 1050–1060. [Google Scholar] [CrossRef]
- Law, J.A.; Du, J.; Hale, C.J.; Feng, S.; Krajewski, K.; Palanca, A.M.S.; Strahl, B.D.; Patel, D.J.; Jacobsen, S.E. Polymerase IV Occupancy at RNA-Directed DNA Methylation Sites Requires SHH1. Nature 2013, 498, 385–389. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, Z.-Y.; Zeng, L.; Tanaka, K.; Zhang, C.-J.; Ma, J.; Bai, G.; Wang, P.; Zhang, S.-W.; Liu, Z.-W. DTF1 Is a Core Component of RNA-Directed DNA Methylation and May Assist in the Recruitment of Pol IV. Proc. Natl. Acad. Sci. USA 2013, 110, 8290–8295. [Google Scholar] [CrossRef]
- Pikaard, C.S.; Mittelsten Scheid, O. Epigenetic Regulation in Plants. Cold Spring Harb. Perspect. Biol. 2014, 6, a019315. [Google Scholar] [CrossRef]
- Bartels, A.; Han, Q.; Nair, P.; Stacey, L.; Gaynier, H.; Mosley, M.; Huang, Q.Q.; Pearson, J.K.; Hsieh, T.-F.; An, Y.-Q.C.; et al. Dynamic DNA Methylation in Plant Growth and Development. Int. J. Mol. Sci. 2018, 19, 2144. [Google Scholar] [CrossRef]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.-L.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E. Genome-Wide High-Resolution Mapping and Functional Analysis of DNA Methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread Natural Variation of DNA Methylation within Angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef] [PubMed]
- Bouyer, D.; Kramdi, A.; Kassam, M.; Heese, M.; Schnittger, A.; Roudier, F.; Colot, V. DNA Methylation Dynamics during Early Plant Life. Genome Biol. 2017, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.Q.; Ali, I.; Tang, J.; Yang, W.C. New Insights into 5hmC DNA Modification: Generation, Distribution and Function. Front. Genet. 2017, 8, 100. [Google Scholar] [CrossRef]
- Plongthongkum, N.; Diep, D.H.; Zhang, K. Advances in the Profiling of DNA Modifications: Cytosine Methylation and Beyond. Nat. Rev. Genet. 2014, 15, 647–661. [Google Scholar] [CrossRef]
- Rajakumara, E.; Nakarakanti, N.K.; Nivya, M.A.; Satish, M. Mechanistic Insights into the Recognition of 5-Methylcytosine Oxidation Derivatives by the SUVH5 SRA Domain. Sci. Rep. 2016, 6, 20161. [Google Scholar] [CrossRef]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, N.; et al. Tet-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef]
- Wang, X.L.; Song, S.H.; Wu, Y.S.; Li, Y.L.; Chen, T.T.; Huang, Z.Y.; Liu, S.; Dunwell, T.L.; Pfeifer, G.P.; Dunwell, J.M.; et al. Genome-Wide Mapping of 5-Hydroxymethylcytosine in Three Rice Cultivars Reveals Its Preferential Localization in Transcriptionally Silent Transposable Element Genes. J. Exp. Bot. 2015, 66, 6651–6663. [Google Scholar] [CrossRef]
- Terragni, J.; Bitinaite, J.; Zheng, Y.; Pradhan, S. Biochemical Characterization of Recombinant β-Glucosyltransferase and Analysis of Global 5-Hydroxymethylcytosine in Unique Genomes. Biochemistry 2012, 51, 1009–1019. [Google Scholar] [CrossRef]
- Yao, Q.; Song, C.X.; He, C.; Kumaran, D.; Dunn, J.J. Heterologous Expression and Purification of Arabidopsis thaliana VIM1 Protein: In Vitro Evidence for Its Inability to Recognize Hydroxymethylcytosine, a Rare Base in Arabidopsis DNA. Protein Expr. Purif. 2012, 83, 104–111. [Google Scholar] [CrossRef]
- Liu, S.; Dunwell, T.L.; Pfeifer, G.P.; Dunwell, J.M.; Ullah, I.; Wang, Y. Detection of Oxidation Products of 5-Methyl-2′-Deoxycytidine in Arabidopsis DNA. PLoS ONE 2013, 8, e84620. [Google Scholar] [CrossRef] [PubMed]
- Moricová, P.; Ondřej, V.; Navrátilová, B.; Luhová, L. Changes of DNA Methylation and Hydroxymethylation in Plant Protoplast Cultures. Acta Biochim. Pol. 2013, 60, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Shin, H.; Eichman, B.F.; Huh, J.H. Excision of 5-Hydroxymethylcytosine by DEMETER Family DNA Glycosylases. Biochem. Biophys. Res. Commun. 2014, 446, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Vanyushin, B.F.; Ashapkin, V.V. DNA Methylation in Higher Plants: Past, Present and Future. Biochim. Biophys. Acta 2011, 1809, 360–368. [Google Scholar] [CrossRef]
- Zhang, M.; Kimatu, J.N.; Xu, K.; Liu, B. DNA Cytosine Methylation in Plant Development. J. Genet. Genom. 2010, 37, 1–12. [Google Scholar] [CrossRef]
- Hirsch, S.; Baumberger, R.; Grossniklaus, U. Epigenetic Variation, Inheritance, and Selection in Plant Populations. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2012; Volume 77, pp. 97–104. [Google Scholar]
- Du, J.; Johnson, L.M.; Groth, M.; Feng, S.; Hale, C.J.; Li, S.; Vashisht, A.A.; Gallego-Bartolome, J.; Wohlschlegel, J.A.; Patel, D.J. Mechanism of DNA Methylation-Directed Histone Methylation by KRYPTONITE. Mol. Cell 2014, 55, 495–504. [Google Scholar] [CrossRef]
- Zemach, A.; Kim, M.Y.; Hsieh, P.-H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The Arabidopsis Nucleosome Remodeler DDM1 Allows DNA Methyltransferases to Access H1-Containing Heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef]
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-CG Methylation Patterns Shape the Epigenetic Landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014, 21, 64–72. [Google Scholar] [CrossRef]
- Ortega-Galisteo, A.P.; Morales-Ruiz, T.; Ariza, R.R.; Roldán-Arjona, T. Arabidopsis DEMETER-LIKE Proteins DML2 and DML3 Are Required for Appropriate Distribution of DNA Methylation Marks. Plant. Mol. Biol. 2008, 67, 671–681. [Google Scholar] [CrossRef]
- Teerawanichpan, P.; Krittanai, P.; Chauvatcharin, N.; Narangajavana, J. Purification and Characterization of Rice DNA Methyltransferase. Plant. Physiol. Biochem. 2009, 47, 671–680. [Google Scholar] [CrossRef]
- Pradhan, S.; Cummings, M.; Roberts, R.J.; Adams, R.L.P. Isolation, Characterization and Baculovirus-Mediated Expression of the CDNA Encoding Cytosine DNA Methyltransferase from Pisum Sativum. Nucleic Acids Res. 1998, 26, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Steward, N.; Kusano, T.; Sano, H. Expression of ZmMET1, a Gene Encoding a DNA Methyltransferase from Maize, Is Associated Not Only with DNA Replication in Actively Proliferating Cells, but Also with Altered DNA Methylation Status in Cold-Stressed Quiescent Cells. Nucleic Acids Res. 2000, 28, 3250–3259. [Google Scholar] [CrossRef] [PubMed]
- Lindroth, A.M.; Cao, X.; Jackson, J.P.; Zilberman, D.; McCallum, C.M.; Henikoff, S.; Jacobsen, S.E. Requirement of CHROMOMETHYLASE3 for Maintenance of CpXpG Methylation. Science 2001, 292, 2077–2080. [Google Scholar] [CrossRef] [PubMed]
- Bartee, L.; Malagnac, F.; Bender, J. Arabidopsis Cmt3 Chromomethylase Mutations Block Non-CG Methylation and Silencing of an Endogenous Gene. Genes Dev. 2001, 15, 1753–1758. [Google Scholar] [CrossRef]
- Fujimoto, R.; Sasaki, T.; Nishio, T. Characterization of DNA Methyltransferase Genes in Brassica Rapa. Genes Genet. Syst. 2006, 81, 235–242. [Google Scholar] [CrossRef]
- Cao, X.; Jacobsen, S.E. Locus-Specific Control of Asymmetric and CpNpG Methylation by the DRM and CMT3 Methyltransferase Genes. Proc. Natl. Acad. Sci. USA 2002, 99, 16491–16498. [Google Scholar] [CrossRef]
- Zhang, X.; Jacobsen, S.E. Genetic Analyses of DNA Methyltransferases in Arabidopsis thaliana. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2006. [Google Scholar]
- Pavlopoulou, A.; Kossida, S. Plant Cytosine-5 DNA Methyltransferases: Structure, Function, and Molecular Evolution. Genomics 2007, 90, 530–541. [Google Scholar] [CrossRef]
- Zhu, J.-K. Active DNA Demethylation Mediated by DNA Glycosylases. Annu. Rev. Genet. 2009, 43, 143. [Google Scholar] [CrossRef]
- Wu, S.C.; Zhang, Y. Active DNA Demethylation: Many Roads Lead to Rome. Nat. Rev. Mol. Cell Biol. 2010, 11, 607–620. [Google Scholar] [CrossRef]
- Bhutani, N.; Burns, D.M.; Blau, H.M. DNA Demethylation Dynamics. Cell 2011, 146, 866–872. [Google Scholar] [CrossRef]
- La, H.; Ding, B.; Mishra, G.P.; Zhou, B.; Yang, H.; del Rosario Bellizzi, M.; Chen, S.; Meyers, B.C.; Peng, Z.; Zhu, J.-K. A 5-Methylcytosine DNA Glycosylase/Lyase Demethylates the Retrotransposon Tos17 and Promotes Its Transposition in Rice. Proc. Natl. Acad. Sci. USA 2011, 108, 15498–15503. [Google Scholar] [CrossRef] [PubMed]
- Ono, A.; Yamaguchi, K.; Fukada-Tanaka, S.; Terada, R.; Mitsui, T.; Iida, S. A Null Mutation of ROS1a for DNA Demethylation in Rice Is Not Transmittable to Progeny. Plant. J. 2012, 71, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Nabel, C.S.; Manning, S.A.; Kohli, R.M. The Curious Chemical Biology of Cytosine: Deamination, Methylation, and Oxidation as Modulators of Genomic Potential. ACS Chem. Biol. 2011, 7, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Kohli, R.M.; Zhang, Y. TET Enzymes, TDG and the Dynamics of DNA Demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef]
- Gehring, M.; Huh, J.H.; Hsieh, T.-F.; Penterman, J.; Choi, Y.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. DEMETER DNA Glycosylase Establishes MEDEA Polycomb Gene Self-Imprinting by Allele-Specific Demethylation. Cell 2006, 124, 495–506. [Google Scholar] [CrossRef]
- Morales-Ruiz, T.; Ortega-Galisteo, A.P.; Ponferrada-Marín, M.I.; Martínez-Macías, M.I.; Ariza, R.R.; Roldán-Arjona, T. Demeter and Repressor of Silencing 1 Encode 5-Methylcytosine DNA Glycosylases. Proc. Natl. Acad. Sci. USA 2006, 103, 6853–6858. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, J.-K. Active DNA Demethylation in Plants and Animals. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2012; Volume 77, pp. 161–173. [Google Scholar]
- Zhang, P.; Li, X.; Wang, Y.; Guo, W.; Chachar, S.; Riaz, A.; Geng, Y.; Gu, X.; Yang, L. PRMT6 physically associates with nuclear factor Y to regulate photoperiodic flowering in Arabidopsis. aBiotech 2021, 2, 403–414. [Google Scholar] [CrossRef]
- Qian, W.; Miki, D.; Zhang, H.; Liu, Y.; Zhang, X.; Tang, K.; Kan, Y.; La, H.; Li, X.; Li, S. A Histone Acetyltransferase Regulates Active DNA Demethylation in Arabidopsis. Science 2012, 336, 1445–1448. [Google Scholar] [CrossRef]
- Yamamuro, C.; Miki, D.; Zheng, Z.; Ma, J.; Wang, J.; Yang, Z.; Dong, J.; Zhu, J.-K. Overproduction of Stomatal Lineage Cells in Arabidopsis Mutants Defective in Active DNA Demethylation. Nat. Commun. 2014, 5, 1–7. [Google Scholar] [CrossRef]
- Gong, Z.; Zhu, J.-K. Active DNA Demethylation by Oxidation and Repair. Cell Res. 2011, 21, 1649–1651. [Google Scholar] [CrossRef]
- Penterman, J.; Zilberman, D.; Huh, J.H.; Ballinger, T.; Henikoff, S.; Fischer, R.L. DNA Demethylation in the Arabidopsis Genome. Proc. Natl. Acad. Sci. USA 2007, 104, 6752–6757. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Morales-Ruiz, T.; Ariza, R.R.; Roldán-Arjona, T.; David, L.; Zhu, J.-K. ROS1, a Repressor of Transcriptional Gene Silencing in Arabidopsis, Encodes a DNA Glycosylase/Lyase. Cell 2002, 111, 803–814. [Google Scholar] [CrossRef]
- Agius, F.; Kapoor, A.; Zhu, J.-K. Role of the Arabidopsis DNA Glycosylase/Lyase ROS1 in Active DNA Demethylation. Proc. Natl. Acad. Sci. USA 2006, 103, 11796–11801. [Google Scholar] [CrossRef]
- Gehring, M.; Reik, W.; Henikoff, S. DNA Demethylation by DNA Repair. Trends Genet. 2009, 25, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Schoft, V.K.; Chumak, N.; Choi, Y.; Hannon, M.; Garcia-Aguilar, M.; Machlicova, A.; Slusarz, L.; Mosiolek, M.; Park, J.-S.; Park, G.T. Function of the DEMETER DNA Glycosylase in the Arabidopsis thaliana Male Gametophyte. Proc. Natl. Acad. Sci. USA 2011, 108, 8042–8047. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Lepère, G.; Jay, F.; Wang, J.; Bapaume, L.; Wang, Y.; Abraham, A.-L.; Penterman, J.; Fischer, R.L.; Voinnet, O. Dynamics and Biological Relevance of DNA Demethylation in Arabidopsis Antibacterial Defense. Proc. Natl. Acad. Sci. USA 2013, 110, 2389–2394. [Google Scholar] [CrossRef]
- Qian, W.; Miki, D.; Lei, M.; Zhu, X.; Zhang, H.; Liu, Y.; Li, Y.; Lang, Z.; Wang, J.; Tang, K. Regulation of Active DNA Demethylation by an α-Crystallin Domain Protein in Arabidopsis. Mol. Cell 2014, 55, 361–371. [Google Scholar] [CrossRef]
- Wang, C.; Dong, X.; Jin, D.; Zhao, Y.; Xie, S.; Li, X.; He, X.; Lang, Z.; Lai, J.; Zhu, J.-K. Methyl-CpG-Binding Domain Protein MBD7 Is Required for Active DNA Demethylation in Arabidopsis. Plant. Physiol. 2015, 167, 905–914. [Google Scholar] [CrossRef][Green Version]
- Lei, M.; Zhang, H.; Julian, R.; Tang, K.; Xie, S.; Zhu, J.-K. Regulatory Link between DNA Methylation and Active Demethylation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2015, 112, 3553–3557. [Google Scholar] [CrossRef]
- Williams, B.P.; Pignatta, D.; Henikoff, S.; Gehring, M. Methylation-Sensitive Expression of a DNA Demethylase Gene Serves as an Epigenetic Rheostat. PLoS Genet. 2015, 11, e1005142. [Google Scholar] [CrossRef]
- Zemach, A.; Kim, M.Y.; Silva, P.; Rodrigues, J.A.; Dotson, B.; Brooks, M.D.; Zilberman, D. Local DNA Hypomethylation Activates Genes in Rice Endosperm. Proc. Natl. Acad. Sci. USA 2010, 107, 18729–18734. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, G.; Qian, J. Transcription Factors as Readers and Effectors of DNA Methylation. Nat. Rev. Genet. 2016, 17, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Coelho, F.S.; Sangi, S.; Moraes, J.L.; da Silva Santos, W.; Gamosa, E.A.; Fernandes, K.V.S.; Grativol, C. Methyl-CpG Binding Proteins (MBD) Family Evolution and Conservation in Plants. Gene 2022, 824, 146404. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.J.; Scheibe, M.; Wongpalee, S.P.; Liu, W.; Cornett, E.M.; Vaughan, R.M.; Li, X.; Chen, W.; Xue, Y.; Zhong, Z. A DNA Methylation Reader Complex That Enhances Gene Transcription. Science 2018, 362, 1182–1186. [Google Scholar] [CrossRef]
- Wang, J.; Nan, N.; Li, N.; Liu, Y.; Wang, T.-J.; Hwang, I.; Liu, B.; Xu, Z.-Y. A DNA Methylation Reader–Chaperone Regulator–Transcription Factor Complex Activates OsHKT1; 5 Expression during Salinity Stress. Plant. Cell 2020, 32, 3535–3558. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qian, W.; Zhao, Y.; Wang, C.; Shen, J.; Zhu, J.-K.; Gong, Z. Antisilencing Role of the RNA-Directed DNA Methylation Pathway and a Histone Acetyltransferase in Arabidopsis. Proc. Natl. Acad. Sci. USA 2012, 109, 11425–11430. [Google Scholar] [CrossRef]
- Tang, K.; Lang, Z.; Zhang, H.; Zhu, J.K. The DNA Demethylase ROS1 Targets Genomic Regions with Distinct Chromatin Modifications. Nat. Plants 2016, 2, 1–10. [Google Scholar] [CrossRef]
- Mathieu, O.; Reinders, J.; Čaikovski, M.; Smathajitt, C.; Paszkowski, J. Transgenerational Stability of the Arabidopsis Epigenome Is Coordinated by CG Methylation. Cell 2007, 130, 851–862. [Google Scholar] [CrossRef]
- Hu, L.; Li, N.; Xu, C.; Zhong, S.; Lin, X.; Yang, J.; Zhou, T.; Yuliang, A.; Wu, Y.; Chen, Y.-R. Mutation of a Major CG Methylase in Rice Causes Genome-Wide Hypomethylation, Dysregulated Genome Expression, and Seedling Lethality. Proc. Natl. Acad. Sci. USA 2014, 111, 10642–10647. [Google Scholar] [CrossRef]
- Erhard, K.F.; Talbot, J.E.R.B.; Deans, N.C.; McClish, A.E.; Hollick, J.B. Nascent Transcription Affected by RNA Polymerase IV in Zea Mays. Genetics 2015, 199, 1107–1125. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, Y.; Luo, G.Z.; Wang, X.; Yue, Y.; Wang, X.; Zong, X.; Chen, K.; Yin, H.; Fu, Y.; et al. Abundant DNA 6mA Methylation during Early Embryogenesis of Zebrafish and Pig. Nat. Commun. 2016, 7, 13052. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.Z.; Wang, F.; Weng, X.; Chen, K.; Hao, Z.; Yu, M.; Deng, X.; Liu, J.; He, C. Characterization of Eukaryotic DNA N6-Methyladenine by a Highly Sensitive Restriction Enzyme-Assisted Sequencing. Nat. Commun. 2016, 7, 11301. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Blanco, M.A.; Gu, L.; Sendinc, E.; Liu, J.; Aristizábal-Corrales, D.; Hsu, C.H.; Aravind, L.; He, C.; Shi, Y. DNA Methylation on N6-Adenine in C. Elegans. Cell 2015, 161, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Luo, G.Z.; Chen, K.; Deng, X.; Yu, M.; Han, D.; Hao, Z.; Liu, J.; Lu, X.; Doré, L.C.; et al. N6-Methyldeoxyadenosine Marks Active Transcription Start Sites in Chlamydomonas. Cell 2015, 161, 879–892. [Google Scholar] [CrossRef]
- Zhang, G.; Huang, H.; Liu, D.; Cheng, Y.; Liu, X.; Zhang, W.; Yin, R.; Zhang, D.; Zhang, P.; Liu, J.; et al. N6-Methyladenine DNA Modification in Drosophila. Cell 2015, 161, 893–906. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, X.; Sheng, Y.; Liu, Y.; Gao, S. N6-Adenine DNA Methylation Is Associated with the Linker DNA of H2A.Z-Containing Well-Positioned Nucleosomes in Pol II-Transcribed Genes in Tetrahymena. Nucleic Acids Res. 2017, 45, 11594–11606. [Google Scholar] [CrossRef]
- Mondo, S.J.; Dannebaum, R.O.; Kuo, R.C.; Louie, K.B.; Bewick, A.J.; LaButti, K.; Haridas, S.; Kuo, A.; Salamov, A.; Ahrendt, S.R.; et al. Widespread Adenine N6-Methylation of Active Genes in Fungi. Nat. Genet. 2017, 49, 964–968. [Google Scholar] [CrossRef]
- Liang, Z.; Yu, G.; Liu, J.; Geng, Y.; Mao, J.; Wang, D.; Zhou, J.; Gu, X. The N6-Adenine Methylation in Yeast Genome Profiled by Single-Molecule Technology. J. Genet. Genom. 2018, 45, 223–225. [Google Scholar] [CrossRef]
- Wang, X.; Li, Z.; Zhang, Q.; Li, B.; Lu, C.; Li, W.; Cheng, T.; Xia, Q.; Zhao, P. DNA Methylation on N6-Adenine in Lepidopteran Bombyx Mori. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 815–825. [Google Scholar] [CrossRef]
- Wu, T.P.; Wang, T.; Seetin, M.G.; Lai, Y.; Zhu, S.; Lin, K.; Liu, Y.; Byrum, S.D.; Mackintosh, S.G.; Zhong, M.; et al. DNA Methylation on N-Adenine in Mammalian Embryonic Stem Cells. Nature 2016, 532, 329–333. [Google Scholar] [CrossRef]
- Yao, B.; Cheng, Y.; Wang, Z.; Li, Y.; Chen, L.; Huang, L.; Zhang, W.; Chen, D.; Wu, H.; Tang, B.; et al. DNA N6-Methyladenine Is Dynamically Regulated in the Mouse Brain Following Environmental Stress. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.-L.; Zhu, S.; He, M.-H.; Chen, Y.; Yu, G.-L.; Chen, D.; Xie, S.-Q.; Luo, F.; Liang, Z.; Gu, X.-F.; et al. N6-Methyladenine DNA Modification in Human Genome. Mol. Cell 2018, 71, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Shen, L.; Cui, X.; Bao, S.; Geng, Y.; Yu, G.; Liang, F.; Xie, S.; Lu, T.; Gu, X.; et al. DNA N-Adenine Methylation in Arabidopsis thaliana. Dev. Cell 2018, 45, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Shen, L.; Cui, X.; Bao, S.; Geng, Y.; Yu, G.; Liang, F.; Lu, T.; Gu, X.; Yu, H. DNA N6-Adenine Methylation in Arabidopsis thaliana. Dev. Cell 2017, 45, 406–416. [Google Scholar] [CrossRef]
- Zhou, C.; Wang, C.; Liu, H.; Zhou, Q.; Liu, Q.; Guo, Y.; Peng, T.; Song, J.; Zhang, J.; Chen, L.; et al. Identification and Analysis of Adenine N6-Methylation Sites in the Rice Genome. Nat. Plants 2018, 4, 554–563. [Google Scholar] [CrossRef]
- Zhang, Q.; Liang, Z.; Cui, X.; Ji, C.; Li, Y.; Zhang, P.; Liu, J.; Riaz, A.; Yao, P.; Liu, M.; et al. N(6)-Methyladenine DNA Methylation in Japonica and Indica Rice Genomes and Its Association with Gene Expression, Plant Development and Stress Responses. Mol. Plant. 2018, 11, 1492–1508. [Google Scholar] [CrossRef]
- Heyn, H.; Esteller, M. An Adenine Code for DNA: A Second Life for N6-Methyladenine. Cell 2015, 161, 710–713. [Google Scholar] [CrossRef]
- Luo, G.Z.; Blanco, M.A.; Greer, E.L.; He, C.; Shi, Y. DNA N6-Methyladenine: A New Epigenetic Mark in Eukaryotes? Nat. Rev. Mol. Cell Biol. 2015, 16, 705–710. [Google Scholar] [CrossRef]
- Fedoreyeva, L.I.; Vanyushin, B.F. N6-Adenine DNA-Methyltransferase in Wheat Seedlings. FEBS Lett. 2002, 514, 305–308. [Google Scholar] [CrossRef]
- Koziol, M.J.; Bradshaw, C.R.; Allen, G.E.; Costa, A.S.H.; Frezza, C.; Gurdon, J.B. Identification of Methylated Deoxyadenosines in Vertebrates Reveals Diversity in DNA Modifications. Nat. Struct. Mol. Biol. 2016, 23, 24–30. [Google Scholar] [CrossRef]
- Dhar, M.S.; Pethe, V.V.; Gupta, V.S.; Ranjekar, P.K. Predominance and Tissue Specificity of Adenine Methylation in Rice. Theor. Appl. Genet. 1990, 80, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Ashapkin, V.V.; Kutueva, L.I.; Vanyushin, B.F. The Gene for Domains Rearranged Methyltransferase (DRM2) in Arabidopsis thaliana Plants Is Methylated at Both Cytosine and Adenine Residues. FEBS Lett. 2002, 532, 367–372. [Google Scholar] [CrossRef]
- Schübeler, D. Function and Information Content of DNA Methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.A.; Zilberman, D. Evolution and Function of Genomic Imprinting in Plants. Genes Dev. 2015, 29, 2517–2531. [Google Scholar] [CrossRef] [PubMed]
- Vanyushin, B.F. JSM Genetics & Genomics Enzymatic DNA Methylation: What It Is Needed for in the Cell. JSM Genet. Genom. 2016, 3, 1010–1014. [Google Scholar]
- Feng, S.; Jacobsen, S.E.; Reik, W. Epigenetic Reprogramming in Plant and Animal Development. Science 2010, 330, 622–627. [Google Scholar] [CrossRef]
- Charlet, J.; Duymich, C.E.; Lay, F.D.; Mundbjerg, K.; Dalsgaard Sørensen, K.; Liang, G.; Jones, P.A. Bivalent Regions of Cytosine Methylation and H3K27 Acetylation Suggest an Active Role for DNA Methylation at Enhancers. Mol. Cell 2016, 62, 422–431. [Google Scholar] [CrossRef]
- Lang, Z.; Wang, Y.; Tang, K.; Tang, D.; Datsenka, T.; Cheng, J.; Zhang, Y.; Handa, A.K.; Zhu, J.-K. Critical Roles of DNA Demethylation in the Activation of Ripening-Induced Genes and Inhibition of Ripening-Repressed Genes in Tomato Fruit. Proc. Natl. Acad. Sci. USA 2017, 114, E4511–E4519. [Google Scholar] [CrossRef]
- Domcke, S.; Bardet, A.F.; Adrian Ginno, P.; Hartl, D.; Burger, L.; Schübeler, D. Competition between DNA Methylation and Transcription Factors Determines Binding of NRF1. Nature 2015, 528, 575–579. [Google Scholar] [CrossRef]
- Liang, Z.; Riaz, A.; Chachar, S.; Ding, Y.; Du, H.; Gu, X. Epigenetic modifications of mRNA and DNA methylation in plants. Mol. Plant 2020, 13, 14–30. [Google Scholar] [CrossRef]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun Bisulphite Sequencing of the Arabidopsis Genome Reveals DNA Methylation Patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. Reduced DNA Methylation in Arabidopsis thaliana Results in Abnormal Plant Development. Proc. Natl. Acad. Sci. USA 1996, 93, 8449–8454. [Google Scholar] [CrossRef]
- Xiao, W.; Gehring, M.; Choi, Y.; Margossian, L.; Pu, H.; Harada, J.J.; Goldberg, R.B.; Pennell, R.I.; Fischer, R.L. Imprinting of the MEA Polycomb Gene Is Controlled by Antagonism between MET1 Methyltransferase and DME Glycosylase. Dev. Cell 2003, 5, 891–901. [Google Scholar] [CrossRef]
- Xiao, W.; Custard, K.D.; Brown, R.C.; Lemmon, B.E.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. DNA Methylation Is Critical for Arabidopsis Embryogenesis and Seed Viability. Plant. Cell 2006, 18, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Rea, M.; Zheng, W.; Chen, M.; Braud, C.; Bhangu, D.; Rognan, T.N.; Xiao, W. Histone H1 Affects Gene Imprinting and DNA Methylation in Arabidopsis. Plant. J. 2012, 71, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, S.E.; Sakai, H.; Finnegan, E.J.; Cao, X.; Meyerowitz, E.M. Ectopic Hypermethylation of Flower-Specific Genes in Arabidopsis. Curr. Biol. 2000, 10, 179–186. [Google Scholar] [CrossRef]
- Kim, M.; Ohr, H.; Lee, J.W.; Hyun, Y.; Fischer, R.L.; Choi, Y. Temporal and Spatial Downregulation of Arabidopsis MET1 Activity Results in Global DNA Hypomethylation and Developmental Defects. Mol. Cells 2014, 17, 280. [Google Scholar] [CrossRef]
- Choi, Y.; Gehring, M.; Johnson, L.; Hannon, M.; Harada, J.J.; Goldberg, R.B.; Jacobsen, S.E.; Fischer, R.L. DEMETER, a DNA Glycosylase Domain Protein, Is Required for Endosperm Gene Imprinting and Seed Viability in Arabidopsis. Cell 2002, 110, 33–42. [Google Scholar] [CrossRef]
- Kawakatsu, T.; Nery, J.R.; Castanon, R.; Ecker, J.R. Dynamic DNA Methylation Reconfiguration during Seed Development and Germination. Genome Biol. 2017, 18, 1–12. [Google Scholar] [CrossRef]
- Candaele, J.; Demuynck, K.; Mosoti, D.; Beemster, G.T.S.; Inze, D.; Nelissen, H. Differential Methylation during Maize Leaf Growth Targets Developmentally Regulated Genes. Plant Physiol. 2014, 164, 1350–1364. [Google Scholar] [CrossRef]
- Shen, H.; He, H.; Li, J.; Chen, W.; Wang, X.; Guo, L.; Peng, Z.; He, G.; Zhong, S.; Qi, Y. Genome-Wide Analysis of DNA Methylation and Gene Expression Changes in Two Arabidopsis Ecotypes and Their Reciprocal Hybrids. Plant Cell 2012, 24, 875–892. [Google Scholar] [CrossRef]
- Msogoya, T.J.; Grout, B.W.; Roberts, A. Reduction in Genome Size and DNA Methylation Alters Plant and Fruit Development in Tissue Culture Induced Off-Type Banana (Musa spp.). J. Anim. Plant. Sci. 2011, 3, 1450–1456. [Google Scholar]
- Zhong, S.; Fei, Z.; Chen, Y.-R.; Zheng, Y.; Huang, M.; Vrebalov, J.; McQuinn, R.; Gapper, N.; Liu, B.; Xiang, J. Single-Base Resolution Methylomes of Tomato Fruit Development Reveal Epigenome Modifications Associated with Ripening. Nat. Biotechnol. 2013, 31, 154–159. [Google Scholar] [CrossRef]
- Liu, R.; How-Kit, A.; Stammitti, L.; Teyssier, E.; Rolin, D.; Mortain-Bertrand, A.; Halle, S.; Liu, M.; Kong, J.; Wu, C. A DEMETER-like DNA Demethylase Governs Tomato Fruit Ripening. Proc. Natl. Acad. Sci. USA 2015, 112, 10804–10809. [Google Scholar] [CrossRef]
- Xu, J.; Xu, H.; Xu, Q.; Deng, X. Characterization of DNA Methylation Variations During Fruit Development and Ripening of Sweet Orange. Plant. Mol. Biol. Rep. 2015, 33, 1–11. [Google Scholar] [CrossRef]
- Baulcombe, D.C.; Dean, C. Epigenetic Regulation in Plant Responses to the Environment. Cold Spring Harb. Perspect. Biol. 2014, 6, a019471. [Google Scholar] [CrossRef] [PubMed]
- Probst, A.V.; Scheid, O.M. Stress-Induced Structural Changes in Plant Chromatin. Curr. Opin. Plant Biol. 2015, 27, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Brown, R.C.; Lemmon, B.E.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. Regulation of Seed Size by Hypomethylation of Maternal and Paternal Genomes. Plant Physiol. 2006, 142, 1160–1168. [Google Scholar] [CrossRef]
- Furner, I.J.; Matzke, M. Methylation and Demethylation of the Arabidopsis Genome. Curr. Opin. Plant Biol. 2011, 14, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P. DNA Methylation Systems and Targets in Plants. FEBS Lett. 2011, 585, 2008–2015. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.-Q.; Zhang, Y.; Zhou, S.-R.; Hu, W.-Y.; Wu, X.-T.; Ye, Y.-J.; Xiao, Y.-P.; Li, X.; Xue, H.-W. Global Analysis Reveals the Crucial Roles of DNA Methylation during Rice Seed Development. Plant Physiol. 2015, 168, 1417–1432. [Google Scholar] [CrossRef] [PubMed]
- An, Y.C.; Goettel, W.; Han, Q.; Bartels, A.; Liu, Z.; Xiao, W. Dynamic Changes of Genome-Wide DNA Methylation during Soybean Seed Development. Sci. Rep. 2017, 7, 12263. [Google Scholar] [CrossRef] [PubMed]
- Kou, H.P.; Li, Y.; Song, X.X.; Ou, X.F.; Xing, S.C.; Ma, J.; von Wettstein, D.; Liu, B. Heritable Alteration in DNA Methylation Induced by Nitrogen-Deficiency Stress Accompanies Enhanced Tolerance by Progenies to the Stress in Rice (Oryza sativa L.). J. Plant. Physiol. 2011, 168, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xin, C.; Cai, J.; Zhou, Q.; Dai, T.; Cao, W.; Jiang, D. Heat Priming Induces Trans-Generational Tolerance to High Temperature Stress in Wheat. Front. Plant Sci. 2016, 7, 501. [Google Scholar] [CrossRef]
- Kathiria, P.; Sidler, C.; Golubov, A.; Kalischuk, M.; Kawchuk, L.M.; Kovalchuk, I. Tobacco Mosaic Virus Infection Results in an Increase in Recombination Frequency and Resistance to Viral, Bacterial, and Fungal Pathogens in the Progeny of Infected Tobacco Plants. Plant Physiol. 2010, 153, 1859–1870. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, K.J.F.; Jansen, J.J.; van Dijk, P.J.; Biere, A. Stress-induced DNA Methylation Changes and Their Heritability in Asexual Dandelions. New Phytol. 2010, 185, 1108–1118. [Google Scholar] [CrossRef]
- Johnsen, Ø.; Dæhlen, O.G.; Østreng, G.; Skrøppa, T. Daylength and Temperature during Seed Production Interactively Affect Adaptive Performance of Picea Abies Progenies. New Phytol. 2005, 168, 589–596. [Google Scholar] [CrossRef]
- Johannes, F.; Porcher, E.; Teixeira, F.K.; Saliba-Colombani, V.; Simon, M.; Agier, N.; Bulski, A.; Albuisson, J.; Heredia, F.; Audigier, P. Assessing the Impact of Transgenerational Epigenetic Variation on Complex Traits. PLoS Genet. 2009, 5, e1000530. [Google Scholar] [CrossRef]
- Schmid, M.W.; Heichinger, C.; Coman Schmid, D.; Guthörl, D.; Gagliardini, V.; Bruggmann, R.; Aluri, S.; Aquino, C.; Schmid, B.; Turnbull, L.A. Contribution of Epigenetic Variation to Adaptation in Arabidopsis. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Herman, J.J.; Sultan, S.E. DNA Methylation Mediates Genetic Variation for Adaptive Transgenerational Plasticity. Proc. R. Soc. B Biol. Sci. 2016, 283, 20160988. [Google Scholar] [CrossRef]
- Mercé, C.; Bayer, P.E.; Tay Fernandez, C.; Batley, J.; Edwards, D. Induced Methylation in Plants as a Crop Improvement Tool: Progress and Perspectives. Agronomy 2020, 10, 1484. [Google Scholar] [CrossRef]
- Molinier, J.; Ries, G.; Zipfel, C.; Hohn, B. Transgeneration Memory of Stress in Plants. Nature 2006, 442, 1046–1049. [Google Scholar] [CrossRef] [PubMed]
- Lukens, L.N.; Zhan, S. The Plant Genome’s Methylation Status and Response to Stress: Implications for Plant Improvement. Curr. Opin. Plant Biol. 2007, 10, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Bossdorf, O.; Richards, C.L.; Pigliucci, M. Epigenetics for Ecologists. Ecol. Lett. 2008, 11, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Fieldes, M.A.; Schaeffer, S.M.; Krech, M.J.; Brown, J.C.L. DNA Hypomethylation in 5-Azacytidine-Induced Early-Flowering Lines of Flax. Theor. Appl. Genet. 2005, 111, 136–149. [Google Scholar] [CrossRef]
- Fieldes, M.A. Heritable Effects of 5-Azacytidine Treatments on the Growth and Development of Flax (Linum usitatissimum) Genotrophs and Genotypes. Genome 1994, 37, 1–11. [Google Scholar] [CrossRef]
- Boyko, A.; Kathiria, P.; Zemp, F.J.; Yao, Y.; Pogribny, I.; Kovalchuk, I. Transgenerational Changes in the Genome Stability and Methylation in Pathogen-Infected Plants: (Virus-Induced Plant Genome Instability). Nucleic Acids Res. 2007, 35, 1714–1725. [Google Scholar] [CrossRef]
- Akimoto, K.; Katakami, H.; Kim, H.-J.; Ogawa, E.; Sano, C.M.; Wada, Y.; Sano, H. Epigenetic Inheritance in Rice Plants. Ann. Bot. 2007, 100, 205–217. [Google Scholar] [CrossRef]
- Tani, E.; Polidoros, A.N.; Nianiou-Obeidat, I.; Tsaftaris, A.S. DNA Methylation Patterns Are Differently Affected by Planting Density in Maize Inbreds and Their Hybrids. Maydica 2005, 50, 19. [Google Scholar]
- Ashikawa, I. Surveying CpG Methylation at 5′-CCGG in the Genomes of Rice Cultivars. Plant. Mol. Biol. 2001, 45, 31–39. [Google Scholar] [CrossRef]
- Cervera, M.-T.; Ruiz-García, L.; Martinez-Zapater, J. Analysis of DNA Methylation in Arabidopsis thaliana Based on Methylation-Sensitive AFLP Markers. Mol. Genet. Genom. 2002, 268, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Salmon, A.; Clotault, J.; Jenczewski, E.; Chable, V.; Manzanares-Dauleux, M.J. Brassica Oleracea Displays a High Level of DNA Methylation Polymorphism. Plant Sci. 2008, 174, 61–70. [Google Scholar] [CrossRef]
- Keyte, A.L.; Percifield, R.; Liu, B.; Wendel, J.F. Infraspecific DNA Methylation Polymorphism in Cotton (Gossypium hirsutum L.). J. Hered. 2006, 97, 444–450. [Google Scholar] [CrossRef]
- Cubas, P.; Vincent, C.; Coen, E. An Epigenetic Mutation Responsible for Natural Variation in Floral Symmetry. Nature 1999, 401, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Kalisz, S.; Purugganan, M.D. Epialleles via DNA Methylation: Consequences for Plant Evolution. Trends Ecol. Evol. 2004, 19, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Preuss, D. Strand-Biased DNA Methylation Associated with Centromeric Regions in Arabidopsis. Proc. Natl. Acad. Sci. USA 2003, 100, 11133–11138. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.Z.; Xu, C.G.; Maroof, S.; Zhang, Q. Patterns of Cytosine Methylation in an Elite Rice Hybrid and Its Parental Lines, Detected by a Methylation-Sensitive Amplification Polymorphism Technique. Mol. Gen. Genet. 1999, 261, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chai, Y.; Liu, B. Epigenetic Inheritance and Variation of DNA Methylation Level and Pattern in Maize Intra-Specific Hybrids. Plant. Sci. 2007, 172, 930–938. [Google Scholar] [CrossRef]
- Xu, R.; Li, H.; Qin, R.; Wang, L.; Li, L.; Wei, P.; Yang, J. Gene Targeting Using the Agrobacterium Tumefaciens-Mediated CRISPR-Cas System in Rice. Rice 2014, 7, 1–4. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, S.; Wang, W.; Xiong, X.; Meng, F.; Cui, X. Efficient Targeted Mutagenesis in Potato by the CRISPR/Cas9 System. Plant Cell Rep. 2015, 34, 1473–1476. [Google Scholar] [CrossRef]
- Jia, H.; Wang, N. Targeted Genome Editing of Sweet Orange Using Cas9/SgRNA. PLoS ONE 2014, 9, e93806. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Chen, L.; Liu, X.; Sun, S.; Wu, C.; Jiang, B.; Han, T.; Hou, W. CRISPR/Cas9-Mediated Genome Editing in Soybean Hairy Roots. PLoS ONE 2015, 10, e0136064. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.; Nekrasov, V.; Lippman, Z.B.; van Eck, J. Efficient Gene Editing in Tomato in the First Generation Using the Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-Associated9 System. Plant Physiol. 2014, 166, 1292–1297. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Zhang, K.; Chen, K.; Gao, C. Targeted Mutagenesis in Zea Mays Using TALENs and the CRISPR/Cas System. J. Genet. Genom. 2014, 41, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lu, X.; Shu, N.; Wang, S.; Wang, J.; Wang, D.; Guo, L.; Ye, W. Targeted Mutagenesis in Cotton (Gossypium hirsutum L.) Using the CRISPR/Cas9 System. Sci. Rep. 2017, 7, 1–7. [Google Scholar] [CrossRef]
- Mlambo, T.; Nitsch, S.; Hildenbeutel, M.; Romito, M.; Müller, M.; Bossen, C.; Diederichs, S.; Cornu, T.I.; Cathomen, T.; Mussolino, C. Designer Epigenome Modifiers Enable Robust and Sustained Gene Silencing in Clinically Relevant Human Cells. Nucleic Acids Res. 2018, 46, 4456–4468. [Google Scholar] [CrossRef] [PubMed]
- Springer, N.M. Epigenetics and Crop Improvement. Trends Genet. 2013, 29, 241–247. [Google Scholar] [CrossRef]
- Hegarty, M.J.; Barker, G.L.; Wilson, I.D.; Abbott, R.J.; Edwards, K.J.; Hiscock, S.J. Transcriptome Shock after Interspecific Hybridization in Senecio Is Ameliorated by Genome Duplication. Curr. Biol. 2006, 16, 1652–1659. [Google Scholar] [CrossRef]
- Buggs, R.J.A.; Zhang, L.; Miles, N.; Tate, J.A.; Gao, L.; Wei, W.; Schnable, P.S.; Barbazuk, W.B.; Soltis, P.S.; Soltis, D.E. Transcriptomic Shock Generates Evolutionary Novelty in a Newly Formed, Natural Allopolyploid Plant. Curr. Biol. 2011, 21, 551–556. [Google Scholar] [CrossRef]
- Chen, K.; Wang, Y.; Zhang, R.; Zhang, H.; Gao, C. CRISPR/Cas Genome Editing and Precision Plant Breeding in Agriculture. Annu. Rev. Plant. Biol. 2019, 70, 667–697. [Google Scholar] [CrossRef]
- Metje-Sprink, J.; Menz, J.; Modrzejewski, D.; Sprink, T. DNA-Free Genome Editing: Past, Present and Future. Front. Plant Sci. 2019, 9, 1957. [Google Scholar] [CrossRef]
- Agapito-Tenfen, S.Z.; Okoli, A.S.; Bernstein, M.J.; Wikmark, O.-G.; Myhr, A.I. Revisiting Risk Governance of GM Plants: The Need to Consider New and Emerging Gene-Editing Techniques. Front. Plant Sci. 2018, 9, 1874. [Google Scholar] [CrossRef] [PubMed]
- Davison, J.; Ammann, K. New GMO Regulations for Old: Determining a New Future for EU Crop Biotechnology. GM Crop. Food 2017, 8, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Gayacharan; Joel, A.J. Epigenetic Responses to Drought Stress in Rice (Oryza sativa L.). Physiol. Mol. Biol. Plants 2013, 19, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Mutum, R.D.; Balyan, S.C.; Kansal, S.; Agarwal, P.; Kumar, S.; Kumar, M.; Raghuvanshi, S. Evolution of Variety-specific Regulatory Schema for Expression of Osa-miR408 in Indica Rice Varieties under Drought Stress. FEBS J. 2013, 280, 1717–1730. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-S.; Pan, Y.-J.; Zhao, X.-Q.; Dwivedi, D.; Zhu, L.-H.; Ali, J.; Fu, B.-Y.; Li, Z.-K. Drought-Induced Site-Specific DNA Methylation and Its Association with Drought Tolerance in Rice (Oryza sativa L.). J. Exp. Bot. 2011, 62, 1951–1960. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, X.; Pan, Y.; Zhu, L.; Fu, B.; Li, Z. DNA Methylation Changes Detected by Methylation-Sensitive Amplified Polymorphism in Two Contrasting Rice Genotypes under Salt Stress. J. Genet. Genom. 2011, 38, 419–424. [Google Scholar] [CrossRef]
- Forestan, C.; Farinati, S.; Rouster, J.; Lassagne, H.; Lauria, M.; Dal Ferro, N.; Varotto, S. Control of Maize Vegetative and Reproductive Development, Fertility, and RRNAs Silencing by HISTONE DEACETYLASE 108. Genetics 2018, 208, 1443–1466. [Google Scholar] [CrossRef]
- Xu, J.; Wang, Q.; Freeling, M.; Zhang, X.; Xu, Y.; Mao, Y.; Tang, X.; Wu, F.; Lan, H.; Cao, M. Natural Antisense Transcripts Are Significantly Involved in Regulation of Drought Stress in Maize. Nucleic Acids Res. 2017, 45, 5126–5141. [Google Scholar] [CrossRef]
- Sosa-Valencia, G.; Palomar, M.; Covarrubias, A.A.; Reyes, J.L. The Legume MiR1514a Modulates a NAC Transcription Factor Transcript to Trigger PhasiRNA Formation in Response to Drought. J. Exp. Bot. 2017, 68, 2013–2026. [Google Scholar] [CrossRef]
- Kulcheski, F.R.; Oliveira, L.F.; Molina, L.G.; Almerão, M.P.; Rodrigues, F.A.; Marcolino, J.; Abdelnoor, R.V. Identification of Novel Soybean MicroRNAs Involved in Abiotic and Biotic Stresses. BMC Genom. 2011, 12, 307. [Google Scholar] [CrossRef] [PubMed]
- Surdonja, K.; Eggert, K.; Hajirezaei, M.-R.; Harshavardhan, V.T.; Seiler, C.; von Wirén, N.; Sreenivasulu, N.; Kuhlmann, M. Increase of DNA Methylation at the HvCKX2. 1 Promoter by Terminal Drought Stress in Barley. Epigenomes 2017, 1, 9. [Google Scholar] [CrossRef]
- Temel, A.; Janack, B.; Humbeck, K. Drought Stress-Related Physiological Changes and Histone Modifications in Barley Primary Leaves at HSP17 Gene. Agronomy 2017, 7, 43. [Google Scholar] [CrossRef]
- Kantar, M.; Unver, T.; Budak, H. Regulation of Barley MiRNAs upon Dehydration Stress Correlated with Target Gene Expression. Funct. Integr. Genom. 2010, 10, 493–507. [Google Scholar] [CrossRef]
- Benoit, M.; Drost, H.-G.; Catoni, M.; Gouil, Q.; Lopez-Gomollon, S.; Baulcombe, D.; Paszkowski, J. Environmental and Epigenetic Regulation of Rider Retrotransposons in Tomato. PLoS Genet. 2019, 15, e1008370. [Google Scholar] [CrossRef]
- González, R.M.; Ricardi, M.M.; Iusem, N.D. Epigenetic Marks in an Adaptive Water Stress-Responsive Gene in Tomato Roots under Normal and Drought Conditions. Epigenetics 2013, 8, 864–872. [Google Scholar] [CrossRef]
- González, R.M.; Ricardi, M.M.; Iusem, N.D. Atypical Epigenetic Mark in an Atypical Location: Cytosine Methylation at Asymmetric (CNN) Sites within the Body of a Non-Repetitive Tomato Gene. BMC Plant Biol. 2011, 11, 1–11. [Google Scholar] [CrossRef]
- Labra, M.; Ghiani, A.; Citterio, S.; Sgorbati, S.; Sala, F.; Vannini, C.; Ruffini-Castiglione, M.; Bracale, M. Analysis of Cytosine Methylation Pattern in Response to Water Deficit in Pea Root Tips. Plant Biol. 2002, 4, 694–699. [Google Scholar] [CrossRef]
- Shui, X.-R.; Chen, Z.-W.; Li, J.-X. MicroRNA Prediction and Its Function in Regulating Drought-Related Genes in Cowpea. Plant Sci. 2013, 210, 25–35. [Google Scholar] [CrossRef]
- De la Rosa, C.; Covarrubias, A.A.; Reyes, J.L. A Dicistronic Precursor Encoding MiR398 and the Legume-specific MiR2119 Coregulates CSD1 and ADH1 MRNAs in Response to Water Deficit. Plant Cell Environ. 2019, 42, 133–144. [Google Scholar] [CrossRef]
- Abid, G.; Mingeot, D.; Muhovski, Y.; Mergeai, G.; Aouida, M.; Abdelkarim, S.; Aroua, I.; el Ayed, M.; M’hamdi, M.; Sassi, K. Analysis of DNA Methylation Patterns Associated with Drought Stress Response in Faba Bean (Vicia faba L.) Using Methylation-Sensitive Amplification Polymorphism (MSAP). Environ. Exp. Bot. 2017, 142, 34–44. [Google Scholar] [CrossRef]
- Arshad, M.; Gruber, M.; Hannoufa, A. Transcriptome Analysis of MicroRNA156 Overexpression Alfalfa Roots under Drought Stress. Sci. Rep. 2018, 8, 9363. [Google Scholar] [CrossRef] [PubMed]
- Hajyzadeh, M.; Turktas, M.; Khawar, K.M.; Unver, T. MiR408 Overexpression Causes Increased Drought Tolerance in Chickpea. Gene 2015, 555, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Khandal, H.; Parween, S.; Roy, R.; Meena, M.K.; Chattopadhyay, D. MicroRNA Profiling Provides Insights into Post-Transcriptional Regulation of Gene Expression in Chickpea Root Apex under Salinity and Water Deficiency. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Kumar, S.; Beena, A.S.; Awana, M.; Singh, A. Physiological, Biochemical, Epigenetic and Molecular Analyses of Wheat (Triticum aestivum) Genotypes with Contrasting Salt Tolerance. Front. Plant Sci. 2017, 8, 1151. [Google Scholar] [CrossRef]
- Zhong, L.; Xu, Y.; Wang, J. DNA-Methylation Changes Induced by Salt Stress in Wheat Triticum Aestivum. Afr. J. Biotechnol. 2009, 8, 6201–6207. [Google Scholar]
- Zhu, N.; Cheng, S.; Liu, X.; Du, H.; Dai, M.; Zhou, D.-X.; Yang, W.; Zhao, Y. The R2R3-Type MYB Gene OsMYB91 Has a Function in Coordinating Plant Growth and Salt Stress Tolerance in Rice. Plant. Sci. 2015, 236, 146–156. [Google Scholar] [CrossRef]
- Karan, R.; DeLeon, T.; Biradar, H.; Subudhi, P.K. Salt Stress Induced Variation in DNA Methylation Pattern and Its Influence on Gene Expression in Contrasting Rice Genotypes. PLoS ONE 2012, 7, e40203. [Google Scholar] [CrossRef]
- Ferreira, L.J.; Donoghue, M.T.A.; Barros, P.; Saibo, N.J.; Santos, A.P.; Oliveira, M.M. Uncovering Differentially Methylated Regions (DMRs) in a Salt-Tolerant Rice Variety under Stress: One Step towards New Regulatory Regions for Enhanced Salt Tolerance. Epigenomes 2019, 3, 4. [Google Scholar] [CrossRef]
- Marconi, G.; Pace, R.; Traini, A.; Raggi, L.; Lutts, S.; Chiusano, M.; Guiducci, M.; Falcinelli, M.; Benincasa, P.; Albertini, E. Use of MSAP Markers to Analyse the Effects of Salt Stress on DNA Methylation in Rapeseed (Brassica napus Var. Oleifera). PLoS ONE 2013, 8, e75597. [Google Scholar] [CrossRef]
- Sako, K.; Kim, J.-M.; Matsui, A.; Nakamura, K.; Tanaka, M.; Kobayashi, M.; Saito, K.; Nishino, N.; Kusano, M.; Taji, T.; et al. Ky-2, a Histone Deacetylase Inhibitor, Enhances High-Salinity Stress Tolerance in Arabidopsis thaliana. Plant Cell Physiol. 2016, 57, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Zhao, L.; Zheng, X.; Gautam, M.; Yue, M.; Hou, J.; Chen, Z.; Wang, P.; Li, L. Dynamic Changes in Histone Modification Are Associated with Upregulation of Hsf and RRNA Genes during Heat Stress in Maize Seedlings. Protoplasma 2019, 256, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Ku, L.; Chen, Y.; Wang, W. Comparative Proteomic Analysis of Leaves between Photoperiod-Sensitive and Photoperiod-Insensitive Maize Inbred Seedlings under Long Day Treatments. Acta Physiol. Plant. 2015, 37, 1–7. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.; Sun, Q.; Yao, Y. Characterization of Small RNAs Derived from TRNAs, RRNAs and SnoRNAs and Their Response to Heat Stress in Wheat Seedlings. PLoS ONE 2016, 11, e0150933. [Google Scholar] [CrossRef]
- Hossain, M.S.; Kawakatsu, T.; Kim, K.D.; Zhang, N.; Nguyen, C.T.; Khan, S.M.; Batek, J.M.; Joshi, T.; Schmutz, J.; Grimwood, J. Divergent Cytosine DNA Methylation Patterns in Single-cell, Soybean Root Hairs. New Phytol. 2017, 214, 808–819. [Google Scholar] [CrossRef]
- Gao, G.; Li, J.; Li, H.; Li, F.; Xu, K.; Yan, G.; Chen, B.; Qiao, J.; Wu, X. Comparison of the Heat Stress Induced Variations in DNA Methylation between Heat-Tolerant and Heat-Sensitive Rapeseed Seedlings. Breed. Sci. 2014, 64, 125–133. [Google Scholar] [CrossRef]
- Hu, X.; Wu, X.; Li, C.; Lu, M.; Liu, T.; Wang, Y.; Wang, W. Abscisic Acid Refines the Synthesis of Chloroplast Proteins in Maize (Zea Mays) in Response to Drought and Light. PLoS ONE 2012, 7, e49500. [Google Scholar] [CrossRef]
- Hu, X.; Lu, M.; Li, C.; Liu, T.; Wang, W.; Wu, J.; Tai, F.; Li, X.; Zhang, J. Differential Expression of Proteins in Maize Roots in Response to Abscisic Acid and Drought. Acta Physiol. Plant. 2011, 33, 2437–2446. [Google Scholar] [CrossRef]
- Steward, N.; Ito, M.; Yamaguchi, Y.; Koizumi, N.; Sano, H. Periodic DNA Methylation in Maize Nucleosomes and Demethylation by Environmental Stress. J. Biol. Chem. 2002, 277, 37741–37746. [Google Scholar] [CrossRef]
- Zhang, B.; Tieman, D.M.; Jiao, C.; Xu, Y.; Chen, K.; Fei, Z.; Giovannoni, J.J.; Klee, H.J. Chilling-Induced Tomato Flavor Loss Is Associated with Altered Volatile Synthesis and Transient Changes in DNA Methylation. Proc. Natl. Acad. Sci. USA 2016, 113, 12580–12585. [Google Scholar] [CrossRef]
- Habu, Y.; Kakutani, T.; Paszkowski, J. Epigenetic Developmental Mechanisms in Plants: Molecules and Targets of Plant Epigenetic Regulation. Curr. Opin. Genet. Dev. 2001, 11, 215–220. [Google Scholar] [CrossRef]
- Kakutani, T. Epi-Alleles in Plants: Inheritance of Epigenetic Information over Generations. Plant Cell Physiol. 2002, 43, 1106–1111. [Google Scholar] [CrossRef] [PubMed]
- Messeguer, R.; Ganal, M.W.; Steffens, J.C.; Tanksley, S.D. Characterization of the Level, Target Sites and Inheritance of Cytosine Methylation in Tomato Nuclear DNA. Plant Mol. Biol. 1991, 16, 753–770. [Google Scholar] [CrossRef] [PubMed]
- Fieldes, M.A.; Amyot, L.M. Epigenetic Control of Early Flowering in Flax Lines Induced by 5-Azacytidine Applied to Germinating Seed. J. Hered. 1999, 90, 199–206. [Google Scholar] [CrossRef][Green Version]
- Bird, A. DNA Methylation Patterns and Epigenetic Memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Ehrlich, M.; Lacey, M. DNA Hypomethylation and Hemimethylation in Cancer. Epigenetic Alter. Oncog. 2013, 754, 31–56. [Google Scholar]
- Gehring, M.; Henikoff, S. DNA Methylation Dynamics in Plant Genomes. Biochim. Biophys. Acta 2007, 1769, 276–286. [Google Scholar] [CrossRef]
- Aina, R.; Sgorbati, S.; Santagostino, A.; Labra, M.; Ghiani, A.; Citterio, S. Specific Hypomethylation of DNA Is Induced by Heavy Metals in White Clover and Industrial Hemp. Physiol. Plant. 2004, 121, 472–480. [Google Scholar] [CrossRef]
- Galaud, J.; Gaspar, T.; Boyer, N. Inhibition of Internode Growth Due to Mechanical Stress in Bryonia Dioica: Relationship between Changes in DNA Methylation and Ethylene Metabolism. Physiol. Plant. 1993, 87, 25–30. [Google Scholar] [CrossRef]
- Hashida, S.; Kitamura, K.; Mikami, T.; Kishima, Y. Temperature Shift Coordinately Changes the Activity and the Methylation State of Transposon Tam3 in Antirrhinum Majus. Plant Physiol. 2003, 132, 1207–1216. [Google Scholar] [CrossRef]
- Peng, H.; Zhang, J. Plant Genomic DNA Methylation in Response to Stresses: Potential Applications and Challenges in Plant Breeding. Prog. Nat. Sci. 2009, 19, 1037–1045. [Google Scholar] [CrossRef]
- Erdmann, R.M.; Picard, C.L. RNA-Directed DNA Methylation. PLoS Genet. 2020, 16, e1009034. [Google Scholar] [CrossRef] [PubMed]
- Bond, D.M.; Baulcombe, D.C. Epigenetic Transitions Leading to Heritable, RNA-Mediated de Novo Silencing in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2015, 112, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, A.; Inaba, J.; Shimura, H.; Otagaki, S.; Tsukahara, S.; Matsuzawa, A.; Kim, B.M.; Goto, K.; Masuta, C. Virus-mediated Efficient Induction of Epigenetic Modifications of Endogenous Genes with Phenotypic Changes in Plants. Plant J. 2011, 65, 156–168. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, Y.; He, H.; He, G.; Deng, X.W. From Hybrid Genomes to Heterotic Trait Output: Challenges and Opportunities. Curr. Opin. Plant Biol. 2022, 66, 102193. [Google Scholar] [CrossRef]
- Chen, Z.J. Genomic and Epigenetic Insights into the Molecular Bases of Heterosis. Nat. Rev. Genet. 2013, 14, 471–482. [Google Scholar] [CrossRef]
- He, G.; He, H.; Deng, X.W. Epigenetic Variations in Plant Hybrids and Their Potential Roles in Heterosis. J. Genet. Genom. 2013, 40, 205–210. [Google Scholar] [CrossRef]
- Chodavarapu, R.K.; Feng, S.; Ding, B.; Simon, S.A.; Lopez, D.; Jia, Y.; Wang, G.-L.; Meyers, B.C.; Jacobsen, S.E.; Pellegrini, M. Transcriptome and Methylome Interactions in Rice Hybrids. Proc. Natl. Acad. Sci. USA 2012, 109, 12040–12045. [Google Scholar] [CrossRef]
- Wang, X.; Elling, A.A.; Li, X.; Li, N.; Peng, Z.; He, G.; Sun, H.; Qi, Y.; Liu, X.S.; Deng, X.W. Genome-Wide and Organ-Specific Landscapes of Epigenetic Modifications and Their Relationships to MRNA and Small RNA Transcriptomes in Maize. Plant Cell 2009, 21, 1053–1069. [Google Scholar] [CrossRef]
- Sinha, P.; Singh, V.K.; Saxena, R.K.; Kale, S.M.; Li, Y.; Garg, V.; Meifang, T.; Khan, A.W.; Kim, K.D.; Chitikineni, A. Genome-wide Analysis of Epigenetic and Transcriptional Changes Associated with Heterosis in Pigeonpea. Plant Biotechnol. J. 2020, 18, 1697–1710. [Google Scholar] [CrossRef]
- Borges, F.; Donoghue, M.T.A.; LeBlanc, C.; Wear, E.E.; Tanurdžić, M.; Berube, B.; Brooks, A.; Thompson, W.F.; Hanley-Bowdoin, L.; Martienssen, R.A. Loss of Small-RNA-Directed DNA Methylation in the Plant Cell Cycle Promotes Germline Reprogramming and Somaclonal Variation. Curr. Biol. 2021, 31, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Xing, F.; Jia, Q.; Zhang, Q.; Hu, T.; Wu, B.; Shao, L.; Zhao, Y.; Zhang, Q.; Zhou, D.-X. Parental Variation in CHG Methylation Is Associated with Allelic-Specific Expression in Elite Hybrid Rice. Plant Physiol. 2021, 186, 1025–1041. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Xing, M.; Zhao, Z.; Gu, Y.; Xiao, Y.; Liu, Q.; Xue, H. DNA Methylation Modification in Heterosis Initiation through Analyzing Rice Hybrid Contemporary Seeds. Crop. J. 2021, 9, 1179–1190. [Google Scholar] [CrossRef]
- Lv, Z.; Zhang, W.; Wu, Y.; Huang, S.; Zhou, Y.; Zhang, A.; Deng, X.; Xu, C.; Xu, Z.; Gong, L. Extensive Allele-level Remodeling of Histone Methylation Modification in Reciprocal F1 Hybrids of Rice Subspecies. Plant J. 2019, 97, 571–586. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Li, Z.H.; Jiang, L.L.; Yu, X.M.; Ngezahayo, F.; Liu, B. Grain-Yield Heterosis in Zea mays L. Shows Positive Correlation with Parental Difference in CHG Methylation. Crop Sci. 2010, 50, 2338–2346. [Google Scholar] [CrossRef]
- Jahnke, S.; Sarholz, B.; Thiemann, A.; Kühr, V.; Gutiérrez-Marcos, J.F.; Geiger, H.H.; Piepho, H.-P.; Scholten, S. Heterosis in Early Seed Development: A Comparative Study of F1 Embryo and Endosperm Tissues 6 Days after Fertilization. Theor. Appl. Genet. 2010, 120, 389–400. [Google Scholar] [CrossRef]
- Lin, J.-Y.; Le, B.H.; Chen, M.; Henry, K.F.; Hur, J.; Hsieh, T.-F.; Chen, P.-Y.; Pelletier, J.M.; Pellegrini, M.; Fischer, R.L. Similarity between Soybean and Arabidopsis Seed Methylomes and Loss of Non-CG Methylation Does Not Affect Seed Development. Proc. Natl. Acad. Sci. USA 2017, 114, E9730–E9739. [Google Scholar] [CrossRef]
- Cao, S.; Wang, L.; Han, T.; Ye, W.; Liu, Y.; Sun, Y.; Moose, S.P.; Song, Q.; Chen, Z.J. Small RNAs Mediate Transgenerational Inheritance of Genome-Wide Trans-Acting Epialleles in Maize. Genome Biol. 2022, 23, 1–25. [Google Scholar] [CrossRef]
- Tao, Z.; Shen, L.; Gu, X.; Wang, Y.; Yu, H.; He, Y. Embryonic Epigenetic Reprogramming by a Pioneer Transcription Factor in Plants. Nature 2017, 551, 124–128. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, Y.; Ren, X.; Yao, D.; Song, Y.; Fan, S.; Li, X.; Zhang, Z.; Yang, S.; Zhang, J. Heterosis and Differential DNA Methylation in Soybean Hybrids and Their Parental Lines. Plants 2022, 11, 1136. [Google Scholar] [CrossRef]
- Wada, Y.; Miyamoto, K.; Kusano, T.; Sano, H. Association between Up-Regulation of Stress-Responsive Genes and Hypomethylation of Genomic DNA in Tobacco Plants. Mol. Genet. Genom. 2004, 271, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Tatra, G.S.; Miranda, J.; Chinnappa, C.C.; Reid, D.M. Effect of Light Quality and 5-azacytidine on Genomic Methylation and Stem Elongation in Two Ecotypes of Stellaria Longipes. Physiol. Plant. 2000, 109, 313–321. [Google Scholar] [CrossRef]
- Fieldes, M.A.; Harvey, C.G. Differences in Developmental Programming and Node Number at Flowering in the 5-Azacytidine-Induced Early Flowering Flax Lines and Their Controls. Int. J. Plant. Sci. 2004, 165, 695–706. [Google Scholar] [CrossRef]
- Peng, H.; Zhang, J.; Wu, X. The Ploidy Effects in Plant Gene Expression: Progress, Problems and Prospects. Sci. China Ser. C Life Sci. 2008, 51, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-Wide Mapping of in Vivo Protein-DNA Interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef]
- Zilberman, D.; Henikoff, S. Genome-Wide Analysis of DNA Methylation Patterns. Development 2007, 134, 3959–3965. [Google Scholar] [CrossRef]
- Dyachenko, O.V.; Zakharchenko, N.S.; Shevchuk, T.V.; Bohnert, H.J.; Cushman, J.C.; Buryanov, Y.I. Effect of Hypermethylation of CCWGG Sequences in DNA of Mesembryanthemum Crystallinum Plants on Their Adaptation to Salt Stress. Biochemistry 2006, 71, 461–465. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, P.; Wang, Y.; Hu, G.; Guo, W.; Gu, X.; Pu, L. Plant synthetic epigenomic engineering for crop improvement. Sci. China Life Sci. 2022. Available online: https://engine.scichina.com/doi/10.1007/s11427-021-2131-6 (accessed on 10 June 2022).


{kind=link}
| Modification | Substrate Specificity | Putative Functions | Example Proteins/Domains | ||
|---|---|---|---|---|---|
| Human | Arabidopsis | Rice | |||
| 5mC | Cytosine | Repression | |||
| Writer | DNMT1 | MET1 | |||
| DNMT3 | CMT3 | ||||
| CMT2 | |||||
| DRM2 | |||||
| Eraser | TET1 | ROS1 | |||
| TET2 | DME | ||||
| TDG | DML2 | ||||
| DML3 | |||||
| Reader | MECP2 | SUVH2 | SUVH7 | ||
| MBD | SUVH9 | ||||
| 6mA | Deoxyadenosine | Activation | |||
| Writer | N6AMT1 | AtN6AMT1 | OsN6AMT1 | ||
| Eraser | ALKBH1 | AtALKBH1 | OsALKBH1 | ||
| Abiotic Stress | Crop | Epigenetic Mechanism(s) | Reference |
|---|---|---|---|
| Drought | Rice | Hypomethylation | [208] |
| Up-regulation of miR408 expression | [209] | ||
| Site-specific DNA methylation | [210,211] | ||
| Maize | Modifications of H3K4me3 and H3K9ac dynamics | [212] | |
| Enrichment in H3K36me3, H3K9ac, and H3K4me3 | [213] | ||
| Soybean | miR1514a modulation of a NAC transcription factor transcript | [214] | |
| Up-regulation of isomiRNAs | [215] | ||
| Barley | Hc-siRNA-mediated hyper-methylation at CYTOKININ-OXIDASE 2.1 promoter | [216] | |
| Increase in H3 and loss in H3K9me2 | [217] | ||
| Accumulation of miR408 transcripts | [218] | ||
| Tomato | RNA-dependent DNA methylation | [219] | |
| Increased Asr1 and Asr2 expression due to demethylation of putative regulatory and transcribed regions | [220,221] | ||
| Pea | Hypermethylation of cytosine residues | [221] | |
| Cowpea | Increase of P5CS transcripts and very low expression of vun-miR5021 and vun-miR156b-3p | [222] | |
| Bean | Dicistronic arrangement of miR398a and miR2119 | [223] | |
| Faba bean | Increased DNA demethylation | [224] | |
| Alfalfa | Overexpression of miR156 | [225] | |
| Chickpea | Accumulation of miR408 transcripts | [226] | |
| Accumulation of miRNAs at root apex | [227] | ||
| Salinity | Wheat | Hypermethylation of cytosines at HKT genes | [228] |
| 5-mC depletion | [229] | ||
| Rice | Demethylation at promoter region of OsMYB91 gene and rapid histone modifications at OsMYB9 locus | [230] | |
| DNA methylation | [231,232] | ||
| Rapeseed | Increased DNA demethylation | [233] | |
| Chickpea | Accumulation of miRNAs at root apex | [227] | |
| Arabidopsis | Increased acetylation of histone H4 at AtSOS1 due to inhibition of de-acetylase | [234] | |
| Heat | Maize | H3K4me2 and H3K9ac alterations | [235] |
| Increased histone acetylation and decreased H3K9me3 | [236] | ||
| Wheat | Increased histone demethylation of the various genes | [237] | |
| Soyabean | Hypomethylation of cytosine | [238] | |
| Rapeseed | Increased DNA demethylation | [239] | |
| Cold | Maize | Enrichment in H3K9ac and decrease in DNA methylation and H3K9me2 | [240] |
| Reduction in histone acetylation in euchromatin-associated gene regions | [241] | ||
| DNA demethylation | [242] | ||
| Tomato | Increased DNA methylation | [243] | |
| Arabidopsis | Non-CG hypermethylation under cold and low light stress | [244] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaikh, A.A.; Chachar, S.; Chachar, M.; Ahmed, N.; Guan, C.; Zhang, P. Recent Advances in DNA Methylation and Their Potential Breeding Applications in Plants. Horticulturae 2022, 8, 562. https://doi.org/10.3390/horticulturae8070562
Shaikh AA, Chachar S, Chachar M, Ahmed N, Guan C, Zhang P. Recent Advances in DNA Methylation and Their Potential Breeding Applications in Plants. Horticulturae. 2022; 8(7):562. https://doi.org/10.3390/horticulturae8070562
Chicago/Turabian StyleShaikh, Aamir Ali, Sadaruddin Chachar, Muzafaruddin Chachar, Nazir Ahmed, Changfei Guan, and Pingxian Zhang. 2022. "Recent Advances in DNA Methylation and Their Potential Breeding Applications in Plants" Horticulturae 8, no. 7: 562. https://doi.org/10.3390/horticulturae8070562
APA StyleShaikh, A. A., Chachar, S., Chachar, M., Ahmed, N., Guan, C., & Zhang, P. (2022). Recent Advances in DNA Methylation and Their Potential Breeding Applications in Plants. Horticulturae, 8(7), 562. https://doi.org/10.3390/horticulturae8070562