
Comparative Analyses of Chloroplast Genomes Provide Comprehensive Insights into the Adaptive Evolution of Paphiopedilum (Orchidaceae)


 ,
, 
Abstract
1. Introduction
2. Methods
2.1. Sampling and Sequencing
2.2. Genome Assembly and Annotation
2.3. Genome Sequence Characteristic Analysis
2.4. Comparative Genomic Analysis
2.5. Repeat Sequence and SSR Element Analyses
2.6. Phylogenetic Analyses
2.7. Selective Pressure Estimation
3. Results
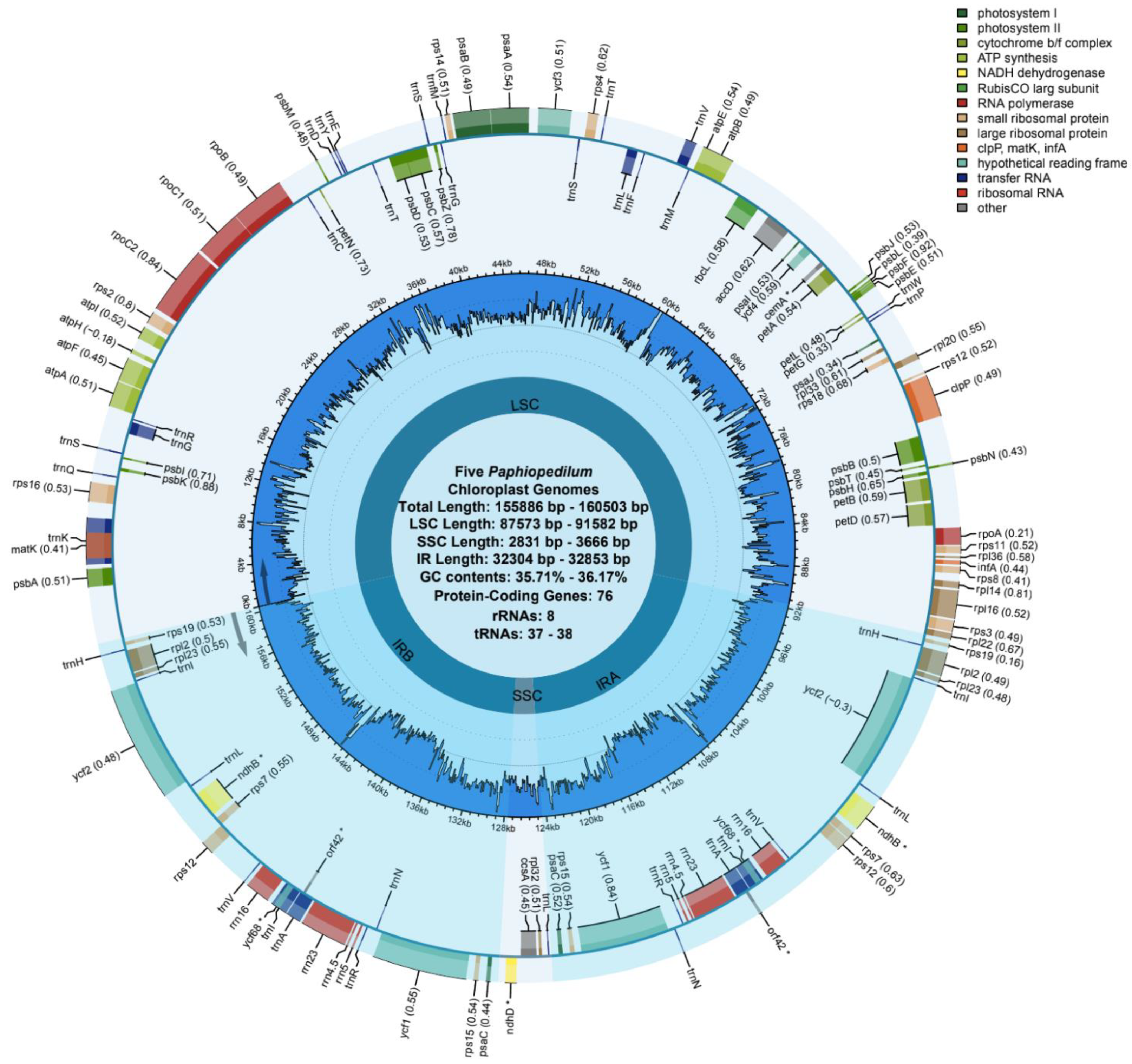
3.1. Length and Features of Cp Genome
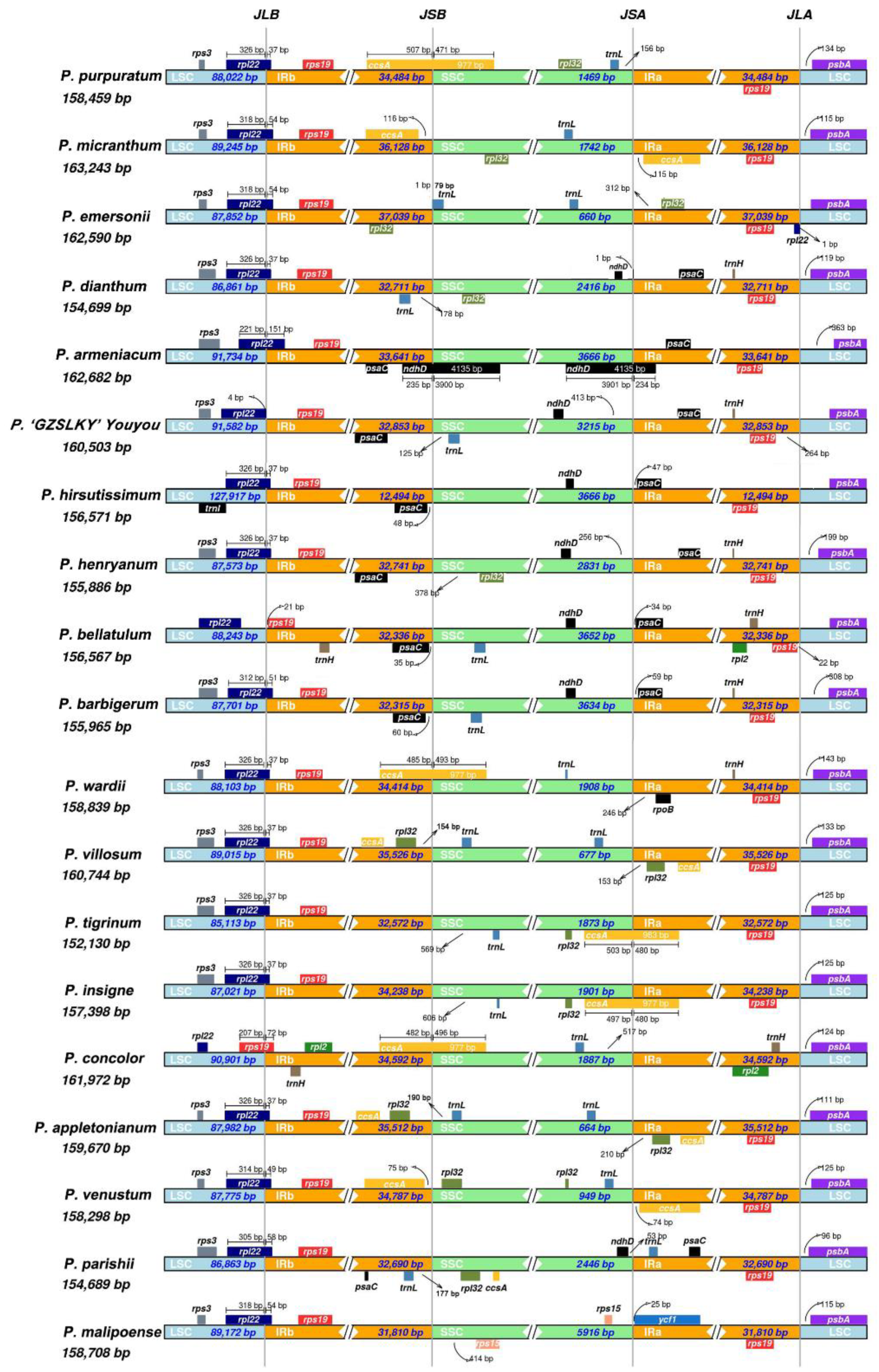
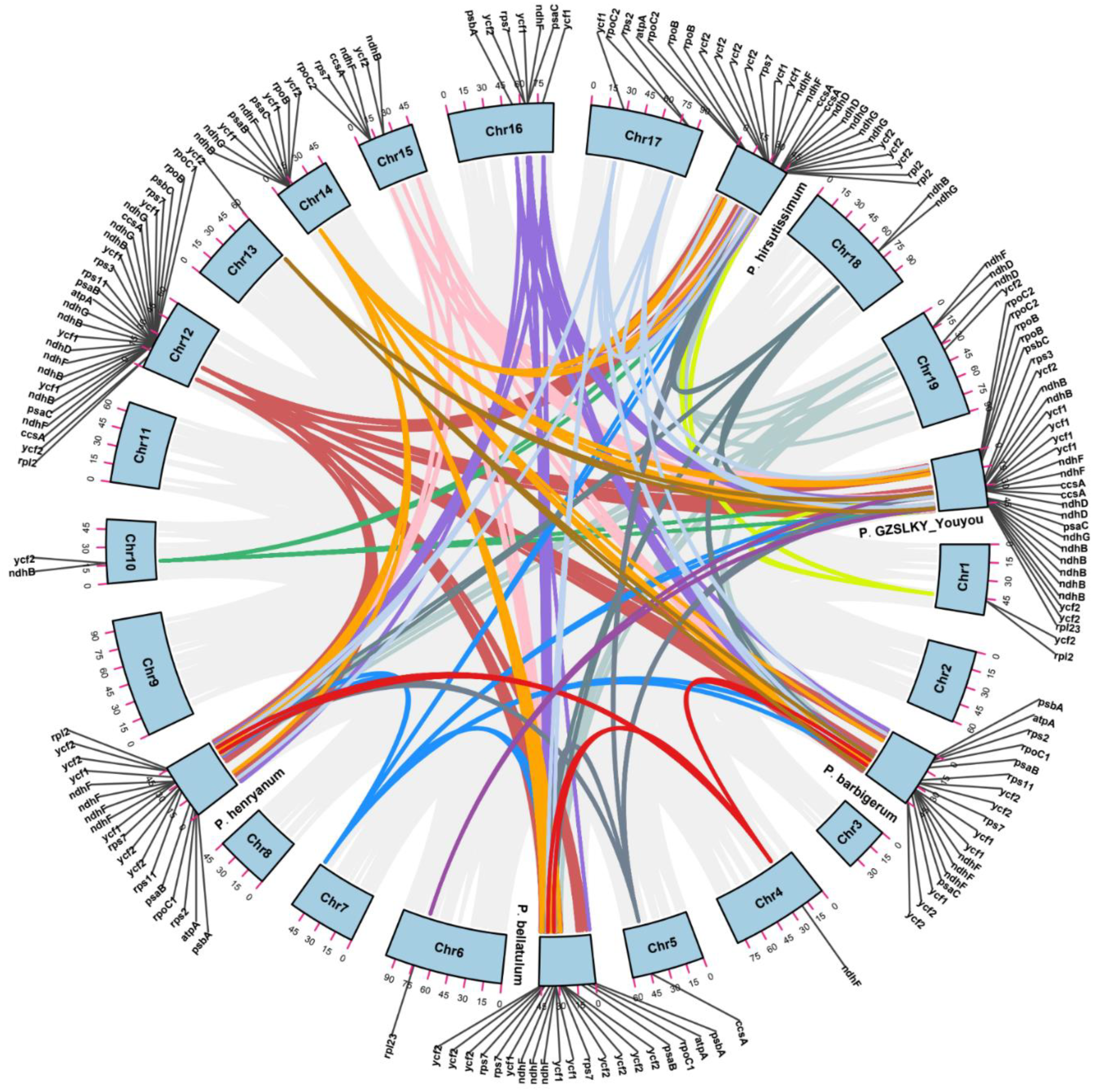
3.2. Comparative Cp Genomic Analysis
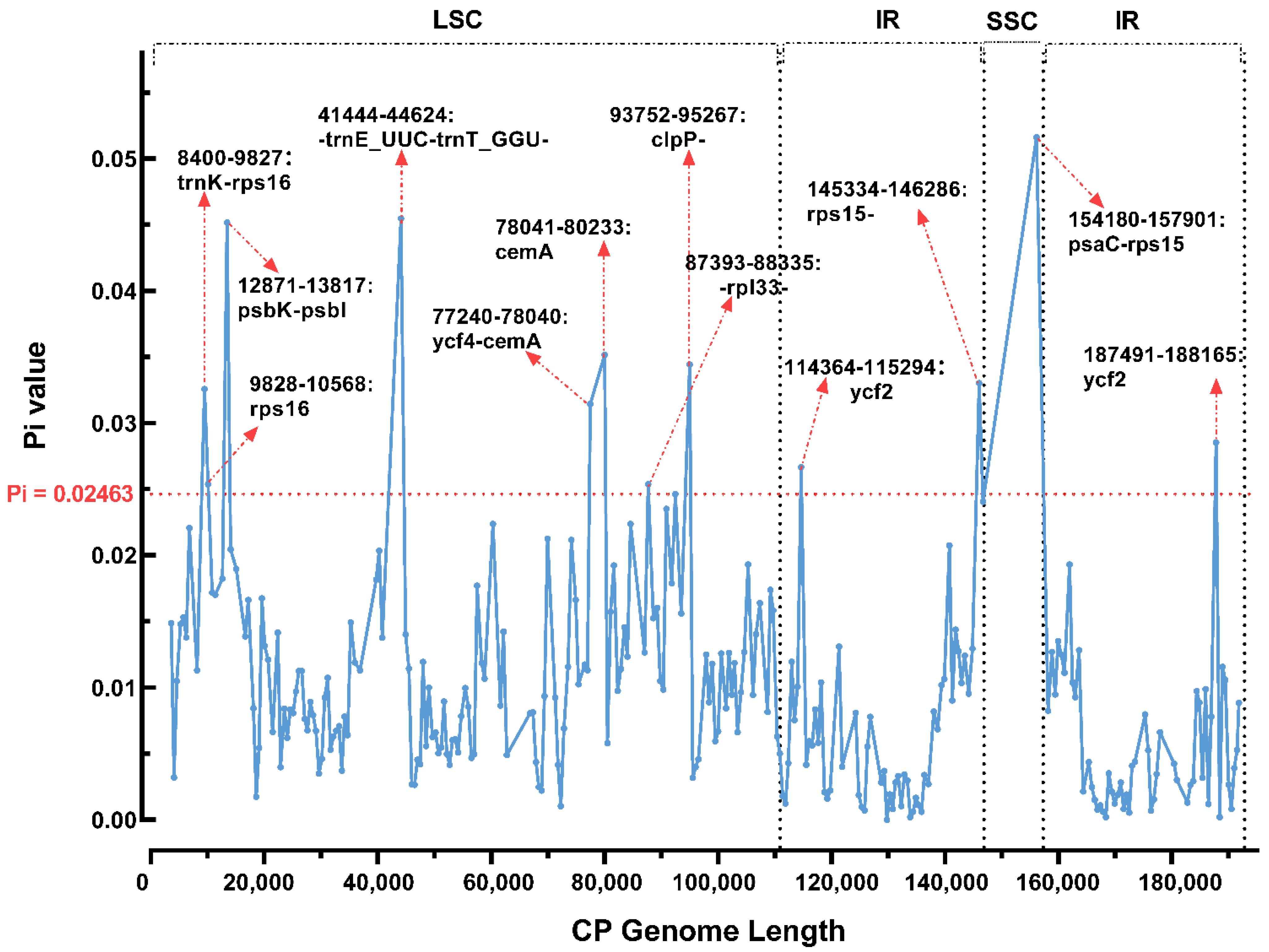
3.3. Repeat Structure and SSR Analysis
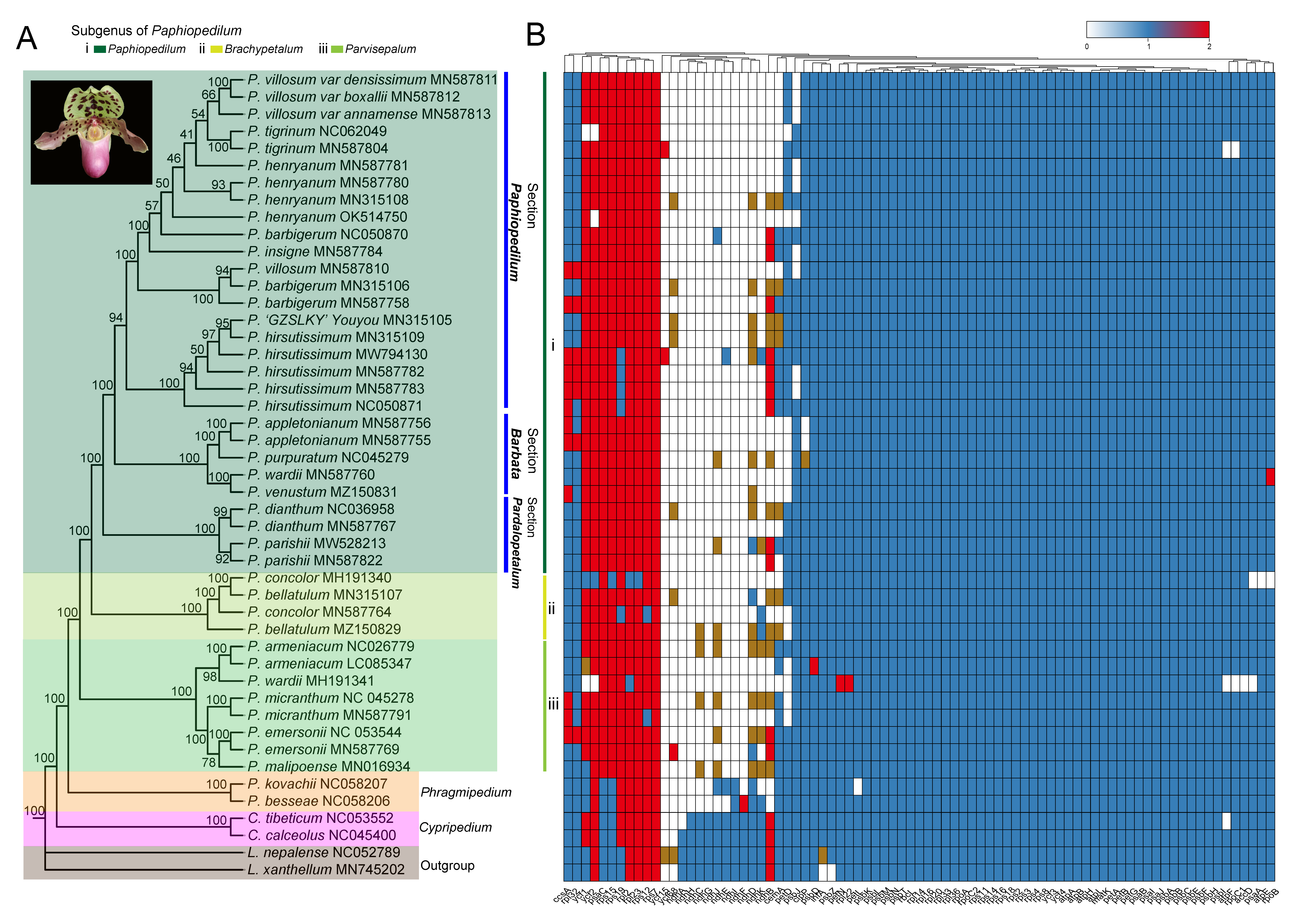
3.4. Phylogenomic Analysis
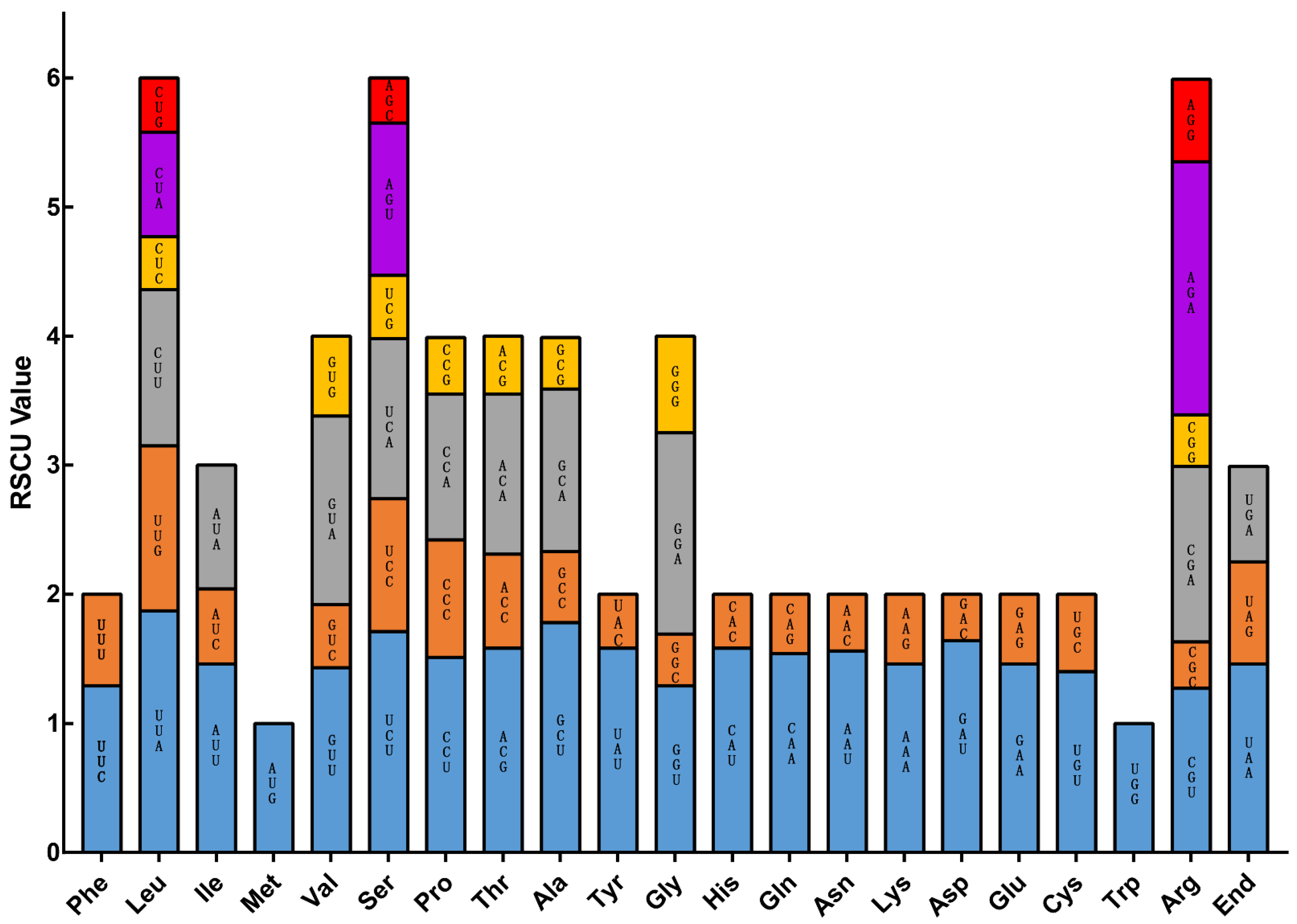
3.5. Selective Pressure Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, Z.; Liao, R.; Yang, T.; Dong, X.; Lan, D.; Qin, R.; Liu, H. Analysis of six chloroplast genomes provides insight into the evolution of Chrysosplenium (Saxifragaceae). BMC Genom. 2020, 21, 621. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.Z.; Liu, Y.L.; Zhang, D.; Li, W.; Gao, J.; Liu, Y.; Li, K.; Shi, C.; Zhao, Y.; Zhao, Y.J.; et al. Evolution of Oryza chloroplast genomes promoted adaptation to diverse ecological habitats. Commun. Biol. 2019, 2, 278. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zheng, Y. Dynamic evolution and phylogenomic analysis of the chloroplast genome in Schisandraceae. Sci. Rep. 2018, 8, 9285. [Google Scholar] [CrossRef] [PubMed]
- Munyao, J.N.; Dong, X.; Yang, J.X.; Mbandi, E.M.; Wanga, V.O.; Oulo, M.A.; Saina, J.K.; Musili, P.M.; Hu, G.W. Complete chloroplast genomes of Chlorophytum comosum and Chlorophytum gallabatense: Genome structures, comparative and phylogenetic analysis. Plants 2020, 9, 296. [Google Scholar] [CrossRef] [PubMed]
- Niu, E.; Jiang, C.; Wang, W.; Zhang, Y.; Zhu, S. Chloroplast genome variation and evolutionary analysis of Olea europaea L. Genes 2020, 11, 879. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.L.; Jansen, R.K.; Chumley, T.W.; Kim, K.J. Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping Inversions. Mol. Biol. Evol. 2007, 24, 1161–1180. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Wu, J.; An, M.; Chen, H.; Zhao, X.; Jin, X.; Si, Q. Geographical distribution and relationship with environmental factors of Paphiopedilum Subgenus Brachypetalum Hallier (Orchidaceae) Taxa in southwest China. Diversity 2021, 13, 634. [Google Scholar] [CrossRef]
- Chen, Q.W.; Guo, Y.Q. Paphiopedilum plants in China: Scopes and review. Guangxi Agric. Sci. 2010, 41, 818–821. [Google Scholar]
- Sun, Y.; Zou, P.; Jiang, N.; Fang, Y.; Liu, G. Comparative analysis of the complete chloroplast genomes of nine Paphiopedilum species. Front. Genet. 2022, 12, 772415. [Google Scholar] [CrossRef]
- Ge, L.P.; Tang, L.; Luo, Y. The complete chloroplast genome of an endangered orchid Paphiopedilum spicerianum. Mitochondrial DNA B Res. 2020, 5, 3594–3595. [Google Scholar] [CrossRef]
- Vu, H.Y.; Tran, N.; Nguyen, T.D.; Vu, Q.L.; Bui, M.H.; Le, M.T.; Le, L. Complete chloroplast genome of Paphiopedilum delenatii and phylogenetic relationships among Orchidaceae. Plants 2020, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.Y.; Yang, J.X.; Bai, M.Z.; Zhang, G.Q.; Liu, Z.J. The chloroplast genome evolution of Venus slipper (Paphiopedilum): IR expansion, SSC contraction, and highly rearranged SSC regions. BMC Plant Biol. 2021, 21, 248. [Google Scholar] [CrossRef] [PubMed]
- Han, C.Y.; Ding, R.; Zong, X.Y.; Zhang, L.J.; Chen, X.H.; Qu, B. Structural characterization of Platanthera ussuriensis chloroplast genome and comparative analyses with other species of Orchidaceae. BMC Genom. 2022, 23, 84. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.C.; Liao, P.C.; Ko, Y.Z.; Chen, C.H.; Chiang, Y.C. Phylogeny and historical biogeography of Paphiopedilum Pfifitzer (Orchidaceae) based on nuclear and plastid DNA. Front. Plant Sci. 2022, 11, 126. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.B.; Jia, J.S.; Wang, C.L. Conservation strategy and potential advantages of the Chinese Paphiopedilum. Biodivers. Sci. 2003, 11, 491–498. [Google Scholar]
- Long, B.; Long, C.L. Amazing Paphiopedilum and its research status. Chin. J. Nat. 2006, 28, 341–344. [Google Scholar]
- Xu, Y.; Jia, R.; Zhou, Y.; Cheng, H.; Zhao, X.; Ge, H. Development and characterization of polymorphic EST-SSR markers for Paphiopedilum henryanum (Orchidaceae). Appl. Plant Sci. 2018, 6, e1152. [Google Scholar] [CrossRef]
- Zeng, S.J.; Chen, Z.L.; Wu, K.L.; Duan, J. Study on introduction and cultivation of Paphiopedilum distributed in China. Chin. Wild Plant Resour. 2010, 29, 53–58. [Google Scholar]
- Xiu, M.; Zhang, X.S.; Wei, X.L.; Hong, S.B.; Zheng, Y.H.; Zhang, K.B.; Chen, J.S.; Chen, C.; Lin, Y. Study on cultivation adaptability of introduced Paphiopedilum in Chaoshan area. Guangdong Agric. Sci. 2021, 48, 47–56. [Google Scholar]
- Tsiftsis, S.; Tsiripidis, I.; Karagiannakidou, V.; Alifragis, D. Niche analysis and conservation of the orchids of east Macedonia (NE Greece). Acta Oecol. 2008, 33, 27–35. [Google Scholar] [CrossRef]
- Chochai, A.; Leitch, I.J.; Ingrouille, M.J.; Fay, M.F. Molecular phylogenetics of Paphiopedilum (Cypripedioideae; Orchidaceae) based on nuclear ribosomal ITS and plastid sequences. Bot. J. Linn. Soc. 2012, 170, 176–196. [Google Scholar] [CrossRef][Green Version]
- Cox, A.V.; Pridgeon, A.M.; Albert, V.A.; Chase, M.W. Phylogenetics of the slipper orchids (Cypripedioideae, Orchidaceae): Nuclear rDNA ITS sequences. Plant Syst. Evol. 1997, 208, 197–223. [Google Scholar] [CrossRef]
- Guo, Y.Y.; Luo, Y.B.; Liu, Z.J.; Wang, X.Q. Reticulate evolution and sea-level fluctuations together drove species diversification of slipper orchids (Paphiopedilum) in South-East Asia. Mol. Ecol. 2015, 24, 2838–2855. [Google Scholar] [CrossRef] [PubMed]
- Smidt, E.D.C.; Páez, M.Z.; Vieira, L.D.N.; Viruel, J.; De Baura, V.A.; Balsanelli, E.; De Souza, E.M.; Chase, M.W. Characterization of sequence variability hotspots in Cranichideae plastomes (Orchidaceae, Orchidoideae). PLoS ONE 2020, 15, e0227991. [Google Scholar] [CrossRef]
- Yang, J.B.; Tang, M.; Li, H.T.; Zhang, Z.R.; Li, D.Z. Complete chloroplast genome of the genus Cymbidium: Lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 2013, 13, 84. [Google Scholar] [CrossRef]
- Dong, W.L.; Wang, R.N.; Zhang, N.Y.; Fan, W.B.; Fang, M.F.; Li, Z.H. Molecular evolution of chloroplast genomes of Orchid species: Insights into phylogenetic relationship and adaptive evolution. Int. J. Mol. Sci. 2018, 19, 716. [Google Scholar] [CrossRef]
- Roma, L.; Cozzolino, S.; Schlu¨ter, P.M.; Scopece, G.; Cafasso, D. The complete plastid genomes of Ophrysiricolor and O. sphegodes (Orchidaceae) and comparative analyses with other orchids. PLoS ONE 2018, 13, e0204174. [Google Scholar] [CrossRef]
- Hu, C.; Jiang, K.; Zeng, X.; Huang, W. The complete chloroplast genome sequence of a critically endangered orchid Paphiopedilum gratrixianum (Orchidaceae). Mitochondrial DNA B Res. 2022, 7, 609–610. [Google Scholar] [CrossRef]
- Kao, H.; Zhao, Y.; Yang, M.; Sun, Y.; Cheng, J. The complete chloroplast genome sequences of an endangered orchid species Paphiopedilum parishii (Orchidaceae). Mitochondrial DNA B Res. 2021, 6, 2521–2522. [Google Scholar] [CrossRef]
- Górniak, M.; Szlachetko, D.L.; Olędrzyńska, N.; Naczk, A.M.; Mieszkowska, A.; Boss, L.; Ziętara, M.S. Species phylogeny versus gene trees: A case study of an incongruent data matrix based on Paphiopedilum Pfitz. (Orchidaceae). Int. J. Mol. Sci. 2021, 22, 11393. [Google Scholar] [CrossRef]
- Cox, A.V.; Abdelnour, G.J.; Bennett, M.D.; Leitch, I.J. Genome size and karyotype evolution in the slipper orchids (Cypripedioideae: Orchidaceae). Am. J. Bot. 1998, 85, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.F.; Yang, Z.J.; Wang, B.Q.; Lv, F.B.; Zhang, X. Karyotypes of 12 Species of Paphiopedilum subgenus Paphiopedilum. J. Trop. Subtrop. Bot. 2011, 19, 152–158. [Google Scholar]
- Yang, Z.J. Study on Cytology and the Relationship in Plants of Paphiopedilum (Cypripedioideae: Orchidaceae). Master’s Thesis, Northwest A&F University, Yangling, China, 2006. [Google Scholar]
- Kim, H.T.; Kim, J.S.; Moore, M.J.; Neubig, K.M.; Williams, N.H.; Whitten, W.M.; Kim, J.H. Seven new complete plastome sequences reveal rampant independent loss of the ndh gene family across Orchids and associated instability of the inverted repeat/small single-copy region boundaries. PLoS ONE 2015, 10, e0142215. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phyto Chem. Bull. 1987, 19, 11–15. [Google Scholar]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Giannoulatou, E.; Park, S.H.; Humphreys, D.T.; Ho, J.W.K. Verification and validation of bioinformatics software without a gold standard: A case study of BWA and Bowtie. BMC Bioinform. 2014, 15, S15. [Google Scholar] [CrossRef]
- Chen, X.D.; Peng, D.H.; Lan, S.R.; Chen, J.; Fu, W.Q. The complete chloroplast genome sequence of Paphiopedilum purpuratum (Orchidaceae). Mitochondrial DNA B 2019, 4, 3910–3911. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Liu, C.; Shi, L.; Zhu, Y.; Chen, H.; Zhang, J.; Lin, X.; Guan, X. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genom. 2012, 13, 715. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Zheng, S.; Poczai, P.; Hyvönen, J.; Tang, J.; Amiryousefi, A. Chloroplot: An online program for the versatile plotting of organelle genomes. Front. Genet. 2020, 11, 1123. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A cross-platform and ultrafast Toolkit for FASTA/Q file manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative Toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating Maximum-Likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Ivica, L.; Peer, B. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A Toolkit incorporating Gamma-Series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef]
- Cribb, P.J.; Kell, S.P.; Dixon, K.W.; Barrett, R.L. Orchid conservation: A global perspective. In Orchid Conservation; Natural History Publications: Kota Kinabalu, Malaysia, 2003. [Google Scholar]
- Li, Z.Y.; Wu, Y.L.; Peng, K. Micro-morphological characters of leaf epidermis of ten species in genus Paphiopedilum. Bull. Bot. Res. 2014, 34, 723–729. [Google Scholar]
- Shi, J.Z.; Chen, H.; An, M.; Zhang, Y.; Ye, C.; Wu, J. Analyses on Distribution Characteristics and Protection Effect of Wild Paphiopedilum in Guizhou Province. Guihaia 2021, 1–10. Available online: https://kns.cnki.net/kcms/detail/45.1134.Q.20210324.1433.012.html (accessed on 26 March 2021).
- Jiang, K.; Miao, L.Y.; Wang, Z.W.; Ni, Z.Y.; Hu, C.; Zeng, X.H.; Huang, W.C. Chloroplast genome analysis of two medicinal Coelogyne spp. (Orchidaceae) shed light on the genetic information, comparative genomics, and species identification. Plants 2020, 9, 1332. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.Y.; Yang, J.X.; Li, H.K.; Zhao, H.S. Chloroplast genomes of two species of Cypripedium: Expanded genome size and proliferation of AT-Biased repeat sequences. Front. Plant Sci. 2021, 12, 609729. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Tian, J.; Yang, J.; Dong, X.; Zhong, Z.; Mwachala, G.; Zhang, C.; Hu, G.; Wang, Q. Comparative and phylogenetic analyses of six Kenya Polystachya (Orchidaceae) species based on the complete chloroplast genome sequences. BMC Plant Biol. 2022, 22, 177. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Li, D.; Feng, X.; Yue, L.; Zhao, X. The Complete Chloroplast Genome Sequence of Clivia gardenii. Mol. Plant Breed. 2021, 1–8. Available online: https://kns.cnki.net/kcms/detail/46.1068.S.20210722.1708.032.html (accessed on 23 July 2021).
- Li, J.; Yang, Q.; Liu, Z.L. The complete chloroplast genome sequence of Liparis japonica (Orchidaceae). Mitochondrial DNA B Res. 2019, 4, 2405–2406. [Google Scholar] [CrossRef]
- Feng, H.; Cao, T.; Xu, H.; Liu, Y.; Han, Y.; Sun, W.; Liu, Y. Characterization of the complete chloroplast genome of Bletilla striata (Orchidaceae: Bletilla), the herb in China. Mitochondrial DNA B Res. 2019, 4, 3542–3543. [Google Scholar] [CrossRef]
- Charlesworth, B. Genetic recombination: Patterns in the genome. Curr. Biol. 1994, 4, 182–184. [Google Scholar] [CrossRef]
- Foerstner, K.U.; von Mering, C.; Hooper, S.D.; Bork, P. Environments shape the nucleotide composition of genomes. EMBO Rep. 2005, 6, 1208–1213. [Google Scholar] [CrossRef]
- Jia, Q.; Wu, H.; Zhou, X.; Gao, J.; Zhao, W.; Aziz, J.; Wei, J.; Hou, L.; Wu, S.; Zhang, Y.; et al. A “GC-rich” method for mammalian gene expression: A dominant role of non-coding DNA GC content in regulation of mammalian gene expression. Sci. China Life Sci. 2010, 53, 94–100. [Google Scholar] [CrossRef]
- Zhu, T.T.; Zhang, L.; Chen, W.S.; Yin, J.; Li, Q. Analysis of chloroplast genomes in 1342 plants. Genom. Appl. Biol. 2017, 36, 4323–4333. [Google Scholar]
- Wu, C.S.; Chaw, S.M. Highly rearranged and size-variable chloroplast genomes in conifers II clade (cupressophytes): Evolution towards shorter intergenic spacers. Plant Biotechnol. J. 2014, 12, 344–353. [Google Scholar] [CrossRef]
- Wu, C.S.; Wang, Y.N.; Hsu, C.Y.; Lin, C.P.; Chaw, S.M. Loss of different inverted repeat copies from the chloroplast genomes of pinaceae and cupressophytes and influence of heterotachy on the evaluation of gymnosperm phylogeny. Genome Biol. Evol. 2011, 3, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schneeweiss, G.M.; dePamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef]
- Zhang, X.M.; Li, D.Z.; Gao, L.M. Phylogeographical study on Taxus wallichiana var. mairei (Lemée & Léveillé) LK Fu & Nan Li. Acta Bot. Boreali-Occident. Sin. 2012, 32, 1983–1989. [Google Scholar]
- Ruhlman, T.A.; Jansen, R.K. The plastid genomes of flowering plants. In Chloroplast Biotechnology; Springer: Berlin/Heidelberg, Germany, 2014; pp. 3–38. [Google Scholar]
- Ruhlman, T.A.; Jansen, R.K. Chapter eight—Aberration or analogy? The atypical plastomes of geraniaceae. In Advances in Botanical Research; Chaw, S.M., Jansen, R.K., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 85, pp. 223–262. [Google Scholar]
- Dugas, D.V.; Hernandez, D.; Koenen, E.J.M.; Schwarz, E.; Straub, S.; Hughes, C.E.; Jansen, R.K.; Nageswara-Rao, M.; Staats, M.; Trujillo, J.T.; et al. Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions and accelerated rate of evolution in clpP. Sci. Rep. 2015, 5, 16958. [Google Scholar] [CrossRef] [PubMed]
- Blazier, J.C.; Jansen, R.K.; Mower, J.P.; Govindu, M.; Zhang, J.; Weng, M.L.; Ruhlman, T.A. Variable presence of the inverted repeat and plastome stability in Erodium. Ann. Bot. 2016, 117, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Rabah, S.O.; Shrestha, B.; Hajrah, N.H.; Sabir, M.J.; Alharby, H.F.; Sabir, M.J.; Alhebshi, A.M.; Sabir, J.S.M.; Gilbert, L.E.; Ruhlman, T.A.; et al. Passiflora plastome sequencing reveals widespread genomic rearrangements. J. Syst. Evol. 2019, 57, 1–14. [Google Scholar] [CrossRef]
- Weng, M.L.; Ruhlman, T.A.; Jansen, R.K. Expansion of inverted repeat does not decrease substitution rates in Pelargonium plastid genomes. New Phytol. 2017, 214, 842–851. [Google Scholar] [CrossRef]
- Niu, Z.; Xue, Q.; Zhu, S.; Sun, J.; Liu, W.; Ding, X. The complete plastome sequences of four orchid species: Insights into the evolution of the Orchidaceae and the utility of plastomic mutational hotspots. Front. Plant Sci. 2017, 8, 715. [Google Scholar] [CrossRef]
- Pan, I.C.; Liao, D.C.; Wu, F.H.; Daniell, H.; Singh, N.D.; Chang, C.; Shih, M.C.; Chan, M.T.; Lin, C.S. Complete chloroplast genome sequence of an orchid model plant candidate: Erycina pusilla apply in tropical Oncidium breeding. PLoS ONE 2012, 7, e34738. [Google Scholar] [CrossRef]
- Lin, C.S.; Chen, J.J.W.; Huang, Y.T.; Chan, M.T.; Daniell, H.; Chang, W.J.; Hsu, C.T.; Liao, D.C.; Wu, F.H.; Lin, S.Y.; et al. The location and translocation of ndh genes of chloroplast origin in the Orchidaceae family. Sci. Rep. 2015, 5, 9040. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Chen, J.J.W.; Chiu, C.C.; Hsiao, H.C.W.; Yang, C.J.; Jin, X.H.; Leebens-Mack, J.; de Pamphilis, C.W.; Huang, Y.T.; Yang, L.H.; et al. Concomitant loss of NDH complex-related genes within chloroplast and nuclear genomes in some orchids. Plant J. 2017, 90, 994–1006. [Google Scholar] [CrossRef] [PubMed]
- Bock, R.; Timmis, J.N. Reconstructing evolution: Gene transfer from plastids to the nucleus. BioEssays 2008, 30, 556–566. [Google Scholar] [CrossRef]
- Martin, W.; Herrmann, R.G. Gene transfer from organelles to the nucleus: How much, what happens, and why?1. Plant Physiol. 1998, 118, 9–17. [Google Scholar] [CrossRef]
- Liu, Q.; Dou, S.; Ji, Z.; Xue, Q. Synonymous codon usage and gene function are strongly related in Oryza sativa. Biosystems 2005, 80, 123–131. [Google Scholar] [CrossRef]
- Srivastava, D.; Shanker, A. Identification of simple sequence repeats in chloroplast genomes of Magnoliids through bioinformatics approach. Interdiscip. Sci. 2016, 8, 327–336. [Google Scholar] [CrossRef]
- Bierne, N.; Eyre-Walker, A. The problem of counting sites in the estimation of the synonymous and nonsynonymous substitution rates: Implications for the correlation between the synonymous substitution rate and codon usage bias. Genetics 2003, 165, 1587–1597. [Google Scholar] [CrossRef]
- Liang, H.; Zhang, Y.; Deng, J.; Gao, G.; Ding, C.; Zhang, L.; Yang, R. The complete chloroplast genome sequences of 14 Curcuma species: Insights into genome evolution and phylogenetic relationships within Zingiberales. Front. Genet. 2020, 11, 802. [Google Scholar] [CrossRef]
- Xu, C.; Ben, A.; Cai, X. Analysis of synonymous codon usage in chloroplast genome of Phalaenopsis aphrodite subsp. formosana. Mol. Plant Breed. 2010, 8, 945–950. [Google Scholar]
- Kuroda, H.; Maliga, P. Sequences downstream of the translation initiation codon are important determinants of translation efficiency in chloroplasts. Plant Physiol. 2001, 125, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.W.; Yang, J.S.; Kim, I.; Yang, J.; Min, B.E.; Kim, S.; Jung, G.Y. Predictive design of mRNA translation initiation region to control prokaryotic translation efficiency. Metab. Eng. 2013, 15, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Hoch, B.; Maier, R.M.; Appel, K.; Igloi, G.L.; Kössel, H. Editing of a chloroplast mRNA by creation of an initiation codon. Nature 1991, 353, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.S.; Li, P.; Qiu, Y.X. The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: Comparative genomic and phylogenetic analyses. Front. Plant Sci. 2016, 7, 2054. [Google Scholar] [CrossRef] [PubMed]
- Khakhlova, O.; Bock, R. Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 2006, 46, 85–94. [Google Scholar] [CrossRef]
- Raven, J.A.; Beardall, J.; Larkum, A.W.D.; Sánchez-Baracaldo, P. Interactions of photosynthesis with genome size and function. Philos. Trans. R. Soc. B 2013, 368, 20120264. [Google Scholar] [CrossRef]
- Rohde, K. Latitudinal gradients in species diversity: The search for the primary cause. Oikos 1992, 65, 514–527. [Google Scholar] [CrossRef]
- Yang, Z.; Wong, W.S.W.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef]
- Hilu, K.W.; Borsch, T.; Müller, K.; Soltis, D.E.; Soltis, P.S.; Savolainen, V.; Chase, M.W.; Powell, M.P.; Alice, L.A.; Evans, R.; et al. Angiosperm phylogeny based on matK sequence information. Am. J. Bot. 2003, 90, 1758–1776. [Google Scholar] [CrossRef]
- Huang, J.L.; Sun, G.L.; Zhang, D.M. Molecular evolution and phylogeny of the angiosperm ycf2 gene. J. Syst. Evol. 2010, 48, 240–248. [Google Scholar] [CrossRef]
- Zhong, Q.; Yang, S.; Sun, X.; Wang, L.; Li, Y. The complete chloroplast genome of the Jerusalem artichoke (Helianthus tuberosus L.) and an adaptive evolutionary analysis of the ycf2 gene. Peer J. 2019, 7, e7596. [Google Scholar] [CrossRef] [PubMed]
- Kohzuma, K.; Sato, Y.; Ito, H.; Okuzaki, A.; Watanabe, M.; Kobayashi, H.; Nakano, M.; Yamatani, H.; Masuda, Y.; Nagashima, Y.; et al. The non-mendelian green cotyledon gene in soybean encodes a small subunit of photosystem II. Plant Physiol. 2017, 173, 2138–2147. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, Z.; He, J.; Cheng, J.; Xie, L. The complete chloroplast genome sequences of a highly endangered orchid species Paphiopedilum barbigerum (Orchidaceae). Mitochondrial DNA B Res. 2019, 2, 2928–2929. [Google Scholar] [CrossRef]
- Hou, N.; Wang, G.; Zhu, Y.; Wang, L.; Xu, J. The complete chloroplast genome of the rare and endangered herb Paphiopedilum dianthum (Asparagales: Orchidaceae). Conserv. Genet. Resour. 2018, 10, 709–712. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, M.; He, J.; Cheng, J.; Xie, L. Complete chloroplast genome sequences of an important horticultural orchid: Paphiopedilum hirsutissimum (Orchidaceae). Mitochondrial DNA B Res. 2019, 2, 2950–2951. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | P. barbigerum | P. bellatulum | P. henryanum | P. hirsutissimum | P. ‘GZSLKY’ Youyou | |
|---|---|---|---|---|---|---|
| Accession No. | MN315106 | MN315107 | MN315108 | MN315109 | MN315105 | |
| Total Length | 155,965 | 156,567 | 155,886 | 156,571 | 160,503 | |
| GC (%) | 35.72 | 35.71 | 35.89 | 36.17 | 36.15 | |
| LSC | Length (bp) | 87,701 | 88,243 | 87,573 | 87,990 | 91,582 |
| Length (%) | 56.23 | 56.36 | 56.18 | 56.20 | 57.06 | |
| GC (%) | 33.17 | 33.26 | 33.36 | 33.68 | 33.83 | |
| SSC | Length (bp) | 3646 | 3652 | 2831 | 3666 | 3215 |
| Length (%) | 2.34 | 2.33 | 1.82 | 2.34 | 2.00 | |
| GC (%) | 28.47 | 28.83 | 28.86 | 29.51 | 29.80 | |
| IRa | Length (bp) | 32,304 | 32,336 | 32,741 | 32,467 | 32,853 |
| Length (%) | 20.71 | 20.65 | 21.00 | 20.74 | 20.47 | |
| GC (%) | 39.58 | 39.42 | 39.56 | 39.91 | 39.70 | |
| IRb | Length (bp) | 32,314 | 32,336 | 32,741 | 32,448 | 32,853 |
| Length (%) | 20.72 | 20.65 | 21.00 | 20.72 | 20.47 | |
| GC (%) | 39.58 | 39.42 | 39.56 | 39.91 | 39.70 | |
| gene counts | total | 122 | 122 | 121 | 121 | 122 |
| protein-coding genes | 76 | 76 | 76 | 76 | 76 | |
| tRNA | 38 | 38 | 37 | 37 | 38 | |
| rRNA | 8 | 8 | 8 | 8 | 8 | |
| Categories | Group of Genes | Genes Contents |
|---|---|---|
| Genes for Self-replication | tRNA genes | trnR-UCU, trnF-GAA, trnV-UAC+, trnP-UGG, trnD-GUC, trnE-UUC, trnY-GUA, trnM-CAU, trnA-UGC+, trnfM-CAU, trnV-GAC, trnW-CCA, trnK-UUU+, trnN-GUU, trnS-GGA, trnI-CAU, trnL-UAA+, trnC-GCA, trnT-UGU, trnS-GCU, trnI-GAU+, trnR-ACG, trnH-GUG, trnS-UGA, trnG-GCC, trnG-UCC+, trnQ-UUG, trnT-GGU, trnL-CAA, trnL-UAG |
| rRNA genes | rrn16S, rrn4.5S, rrn23S, rrn5S | |
| Small subunit of ribosome | rps16+, rps2, rps14, rps4, rps18, rps12++, rps11, rps8, rps3, rps19, rps7, rps15, rps12 | |
| Large subunit of ribosome | rpl33, rpl20, rpl36, rpl14, rpl16+, rpl22, rpl2+, rpl23, rpl32 | |
| DNA dependent RNA polymerase | rpoC2, rpoC1+, rpoB, rpoA | |
| photosynthesis | Subunits of NADH-dehydrogenase | ndhB *, ndhD * |
| Subunits of photosystem I | psaB, psaA, psaI, psaJ, psaC | |
| Subunits of photosystem II | psbA, psbK, psbI, psbM, psbD, psbC, psbZ, psbJ, psbL, psbF, psbE, psbB, psbT, psbN, psbH | |
| Subunits of cytochrome b/f complex | petN, petA, petL, petG, petD+, petD, petB+ | |
| Subunits of ATP synthase | atpA, atpF+, atpH, atpI, atpE, atpB | |
| Large subunit of rubisco | rbcL | |
| Others | Maturase | matK |
| Protease | clpP++ | |
| Envelope membrane protein | cemA * | |
| Subunit of acetyl-CoA-carboxylase | accD | |
| C-type cytochrome synthesis gene | ccsA | |
| Translational initiation factor 1 | infA | |
| unknown function | tRNA genes | ycf3++, ycf4, ycf1, ycf2, orf42 *, ycf68 * |
| Microsatellite Sequence | Interspersed Repeat Sequence | Tandem Repeats | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mono- | Di- | Tri- | Tetra- | Penta- | Hexa- | Total | Forward | Reverse | Complement | Palindromic | Total | 1–30 | 31–60 | 61–90 | >90 | Total | |
| P. barbigerum | 70 | 24 | 18 | 17 | 6 | 13 | 148 | 279 | 97 | 56 | 260 | 692 | 156 | 34 | 10 | 8 | 208 |
| P. bellatulum | 60 | 25 | 16 | 17 | 11 | 10 | 139 | 130 | 68 | 60 | 118 | 376 | 153 | 18 | 6 | 10 | 187 |
| P. henryanum | 66 | 24 | 15 | 16 | 7 | 7 | 135 | 172 | 57 | 31 | 157 | 417 | 103 | 22 | 10 | 12 | 147 |
| P. hirsutissimum | 61 | 17 | 13 | 23 | 5 | 4 | 123 | 217 | 176 | 44 | 176 | 613 | 114 | 20 | 4 | 12 | 150 |
| P. ‘GZSLKY’ Youyou | 57 | 16 | 16 | 25 | 9 | 6 | 129 | 188 | 68 | 16 | 148 | 420 | 122 | 18 | 4 | 14 | 158 |
| P. appletonianum | 58 | 27 | 13 | 18 | 16 | 6 | 138 | 195 | 187 | 150 | 192 | 724 | 179 | 22 | 6 | 2 | 209 |
| P. armeniacum | 64 | 45 | 15 | 21 | 5 | 5 | 155 | 260 | 225 | 180 | 244 | 909 | 185 | 26 | 7 | 4 | 222 |
| P. concolor | 68 | 24 | 17 | 19 | 12 | 7 | 147 | 142 | 127 | 93 | 120 | 482 | 177 | 16 | 4 | 0 | 197 |
| P. dianthum | 49 | 26 | 16 | 18 | 3 | 5 | 117 | 90 | 68 | 38 | 79 | 275 | 138 | 14 | 4 | 0 | 156 |
| P. emersonii | 68 | 45 | 19 | 25 | 6 | 8 | 171 | 246 | 217 | 134 | 182 | 779 | 170 | 25 | 5 | 1 | 201 |
| P. insigne | 65 | 22 | 25 | 18 | 5 | 7 | 142 | 173 | 56 | 44 | 153 | 426 | 130 | 21 | 3 | 2 | 156 |
| P. malipoense | 53 | 34 | 14 | 23 | 3 | 6 | 133 | 155 | 142 | 88 | 132 | 517 | 132 | 15 | 1 | 0 | 148 |
| P. tigrinum | 71 | 26 | 25 | 17 | 6 | 9 | 154 | 230 | 81 | 42 | 193 | 546 | 149 | 20 | 1 | 1 | 171 |
| P. micranthum | 54 | 37 | 23 | 21 | 7 | 6 | 148 | 52 | 89 | 51 | 86 | 278 | 163 | 20 | 2 | 2 | 187 |
| P. parishii | 54 | 26 | 16 | 18 | 2 | 7 | 123 | 116 | 116 | 56 | 102 | 390 | 135 | 15 | 7 | 1 | 158 |
| P. purpuratum | 56 | 24 | 20 | 17 | 13 | 5 | 135 | 137 | 109 | 77 | 122 | 445 | 213 | 31 | 5 | 0 | 249 |
| P. venustum | 66 | 25 | 20 | 24 | 16 | 5 | 156 | 186 | 181 | 90 | 122 | 579 | 178 | 20 | 3 | 1 | 202 |
| P. villosum | 74 | 33 | 25 | 20 | 10 | 12 | 174 | 289 | 146 | 103 | 258 | 796 | 185 | 27 | 7 | 4 | 223 |
| P. wardii | 63 | 30 | 15 | 25 | 9 | 5 | 147 | 200 | 170 | 127 | 186 | 683 | 212 | 30 | 4 | 3 | 249 |
| Total | 1177 | 530 | 341 | 382 | 151 | 133 | 2714 | 3457 | 2380 | 1480 | 3030 | 10,347 | 2994 | 414 | 93 | 77 | 3578 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Ye, H.; Zhang, N.; Ma, J.; Wang, J.; Hu, G.; Li, M.; Zhao, P. Comparative Analyses of Chloroplast Genomes Provide Comprehensive Insights into the Adaptive Evolution of Paphiopedilum (Orchidaceae). Horticulturae 2022, 8, 391. https://doi.org/10.3390/horticulturae8050391
Liu H, Ye H, Zhang N, Ma J, Wang J, Hu G, Li M, Zhao P. Comparative Analyses of Chloroplast Genomes Provide Comprehensive Insights into the Adaptive Evolution of Paphiopedilum (Orchidaceae). Horticulturae. 2022; 8(5):391. https://doi.org/10.3390/horticulturae8050391
Chicago/Turabian StyleLiu, Hengzhao, Hang Ye, Naiyu Zhang, Jiayu Ma, Jiangtao Wang, Guojia Hu, Mengdi Li, and Peng Zhao. 2022. "Comparative Analyses of Chloroplast Genomes Provide Comprehensive Insights into the Adaptive Evolution of Paphiopedilum (Orchidaceae)" Horticulturae 8, no. 5: 391. https://doi.org/10.3390/horticulturae8050391
APA StyleLiu, H., Ye, H., Zhang, N., Ma, J., Wang, J., Hu, G., Li, M., & Zhao, P. (2022). Comparative Analyses of Chloroplast Genomes Provide Comprehensive Insights into the Adaptive Evolution of Paphiopedilum (Orchidaceae). Horticulturae, 8(5), 391. https://doi.org/10.3390/horticulturae8050391