
An Integrated Analysis of Transcriptome and miRNA Sequencing Provides Insights into the Dynamic Regulations during Flower Morphogenesis in Petunia

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction, Library Construction, and Illumina Sequencing
2.3. Illumina Sequencing and Data Analysis
2.4. Differential Gene Expression Analysis
2.5. Co-Expression Network Analysis
2.6. Small RNA Sequencing and microRNA Screening
2.7. Differentially Expressed miRNAs (DEM) Analysis
2.8. miRNA Target Prediction and Annotation
2.9. qRT-PCR Verification of Small RNA and Transcriptome Sequencing
3. Results
3.1. Global Transcriptomic Changes during Petunia Flower Development
3.2. Differential Expression of Transcription Factor Genes
3.3. GO Enrichment and KEGG Pathway Analysis of DEGs
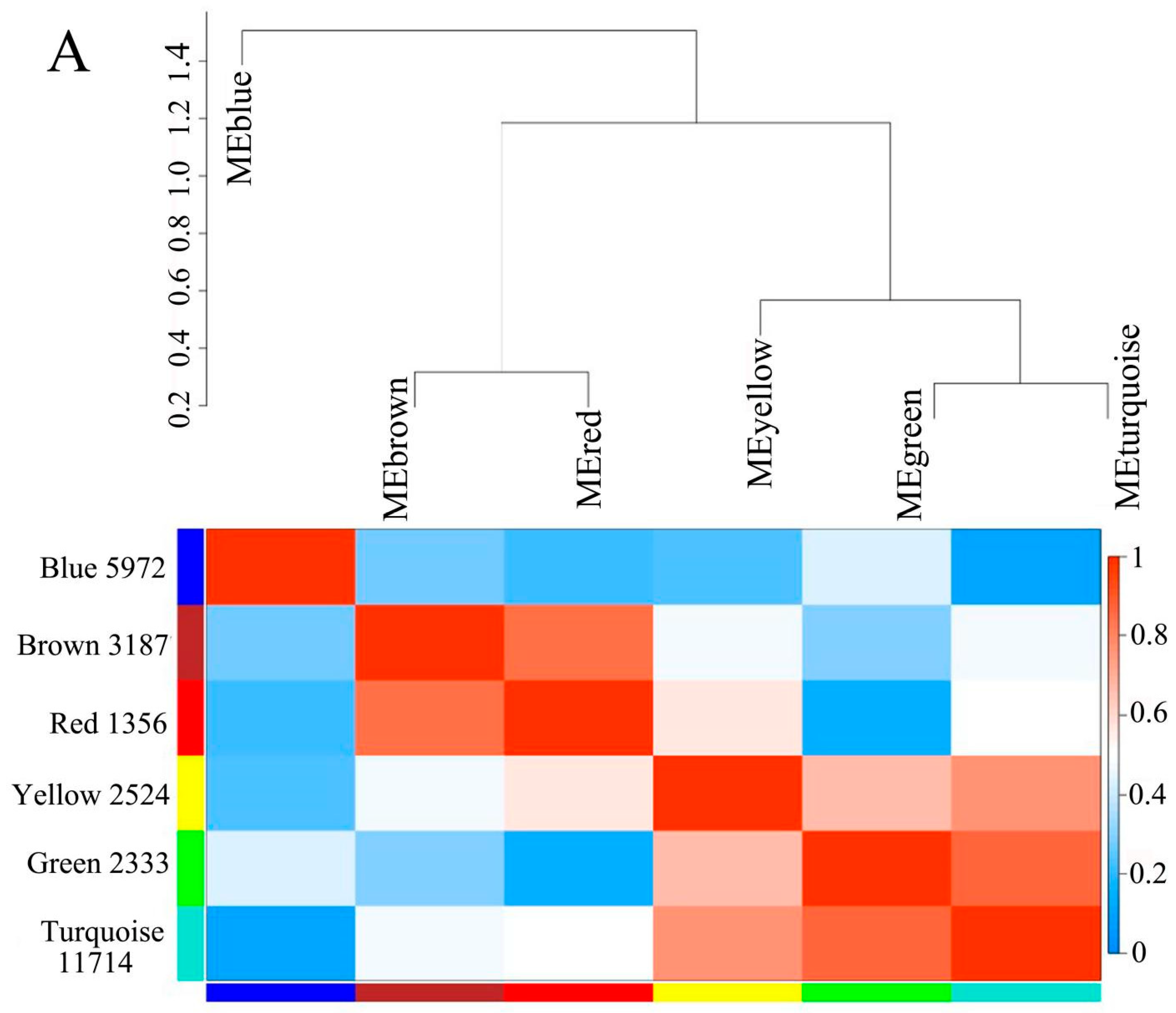
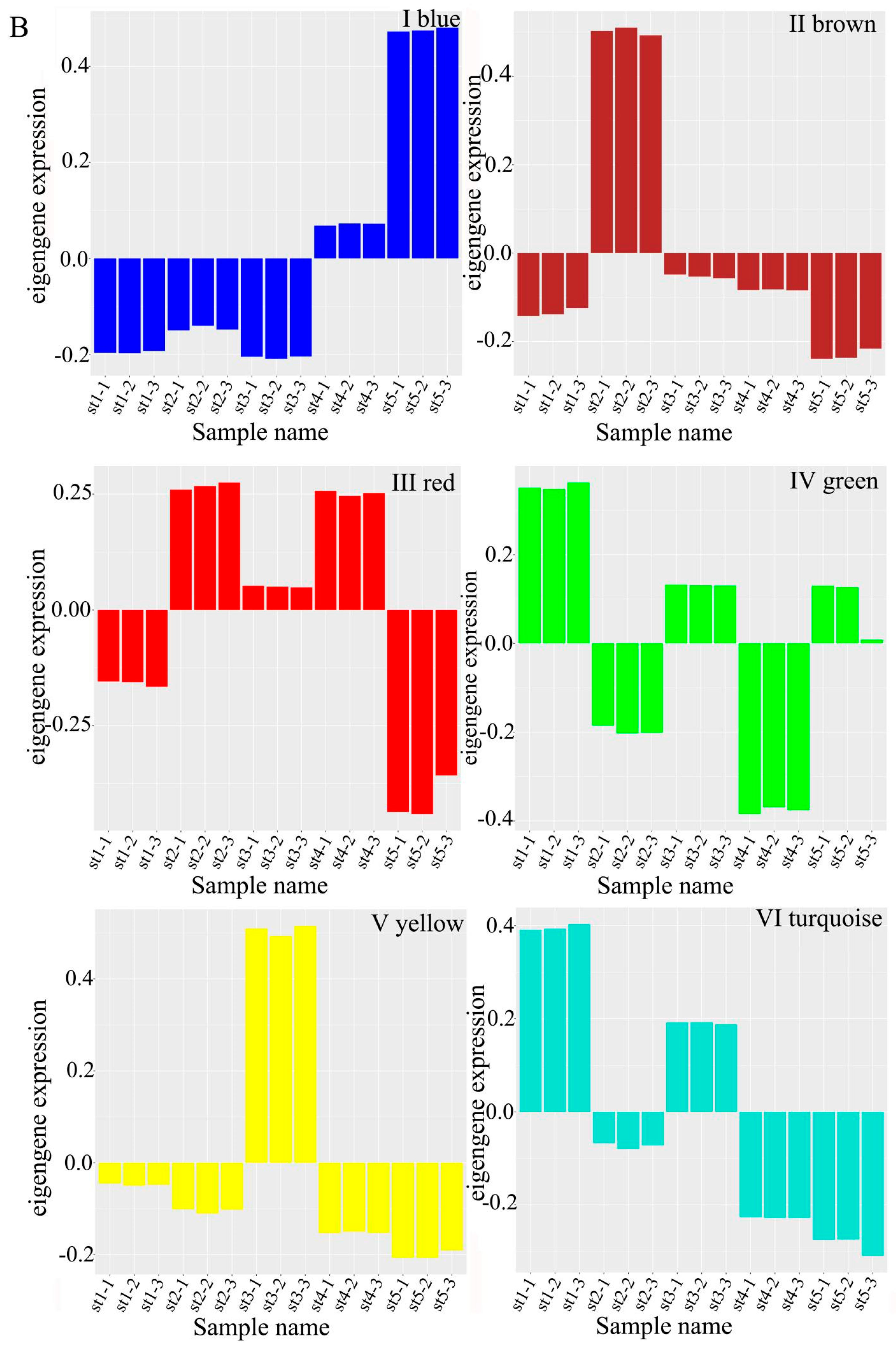
3.4. Gene Clustering Analysis and Co-Expression Network Construction
3.5. Analysis of sRNAs in Petunia
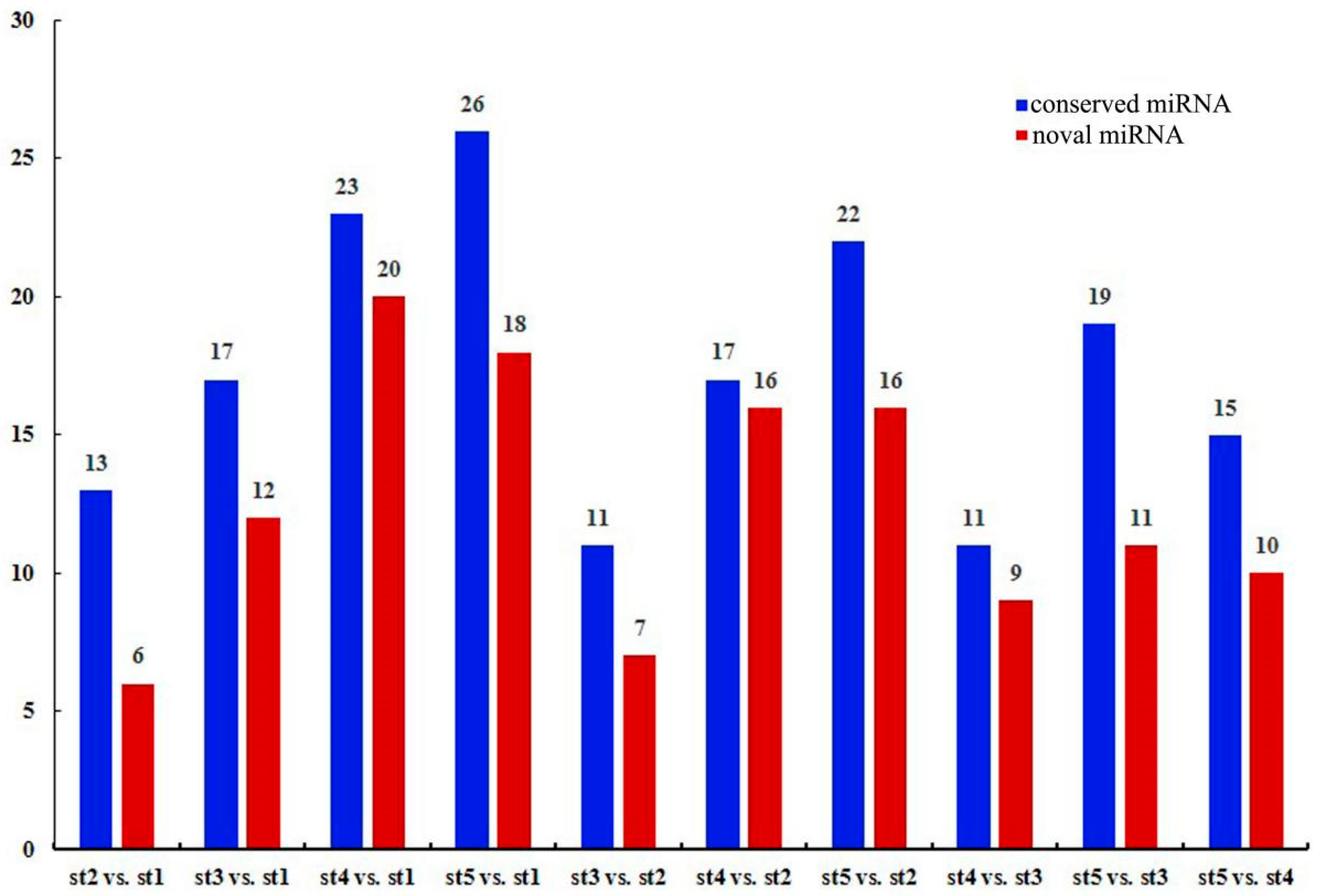
3.6. Differential Expression of miRNAs during Flower Development
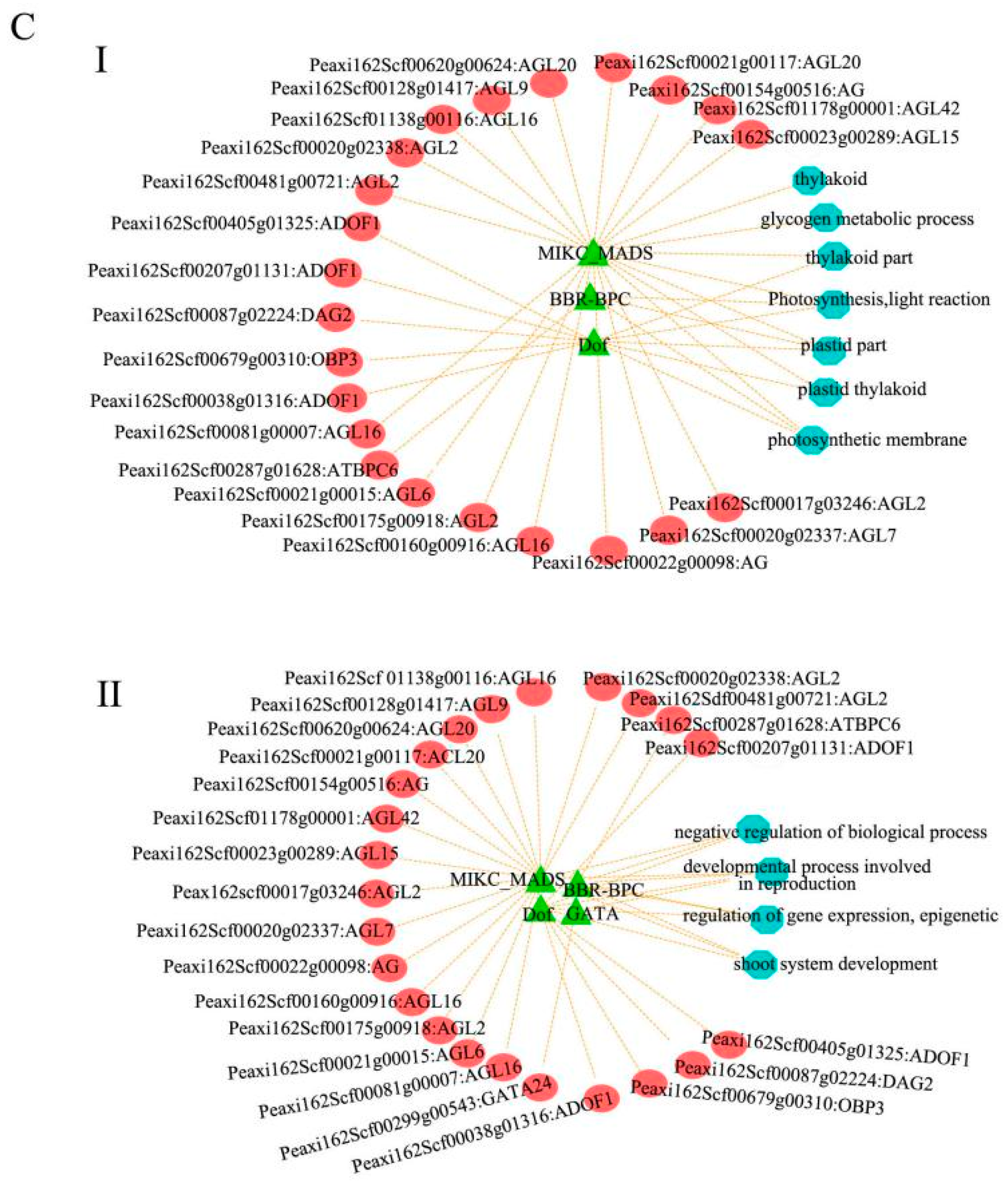
3.7. GO Enrichment Analysis and KEGG Analysis of Targets for DEMs
3.8. Correlation Analysis of miRNA–mRNA at Different Stages of Flower Development in Petunia
3.9. Validation of the Differentially Expressed Genes and miRNAs by Quantitative RT-PCR Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Shan, H.; Cheng, J.; Zhang, R.; Yao, X.; Kong, H. Developmental mechanisms involved in the diversification of flowers. Nat. Plants 2019, 5, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Krizek, B.A.; Fletcher, J.C. Molecular mechanisms of flower development: An armchair guide. Nat. Rev. Genet. 2005, 6, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Guo, Z.; Li, L. Evolutionary conservation of microRNA regulatory programs in plant flower development. Dev. Biol. 2013, 380, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.; Nasim, Z.; Susila, H.; Ahn, J.H. Evolution and functional diversification of FLOWERING LOCUS T/TERMINAL FLOWER 1 family genes in plants. Semin. Cell Dev. Biol. 2021, 109, 20–30. [Google Scholar] [CrossRef]
- Thomson, B.; Wellmer, F. Molecular regulation of flower development. Curr. Top. Dev. Biol. 2019, 131, 185–210. [Google Scholar] [CrossRef]
- Theißen, G.; Saedler, H. Floral quartets. Nature 2001, 409, 469–471. [Google Scholar] [CrossRef]
- Dubois, A.; Raymond, O.; Maene, M.; Baudino, S.; Langlade, N.B.; Boltz, V.; Vergne, P.; Bendahmane, M. Tinkering with the C-Function: A Molecular Frame for the Selection of Double Flowers in Cultivated Roses. PLoS ONE 2010, 5, e9288. [Google Scholar] [CrossRef]
- Kanno, A. Molecular Mechanism Regulating Floral Architecture in Monocotyledonous Ornamental Plants. Hortic. J. 2016, 85, 8–22. [Google Scholar] [CrossRef] [Green Version]
- Vroemen, C.W.; Mordhorst, A.P.; Albrecht, C.; Kwaaitaal, M.A.C.J.; De Vries, S.C. The CUP-SHAPED COTYLEDON3 Gene Is Required for Boundary and Shoot Meristem Formation in Arabidopsis. Plant Cell 2003, 15, 1563–1577. [Google Scholar] [CrossRef]
- Prunet, N.; Yang, W.; Das, P.; Meyerowitz, E.M.; Jack, T.P. SUPERMAN prevents class B gene expression and promotes stem cell termination in the fourth whorl of Arabidopsis thaliana flowers. Proc. Natl. Acad. Sci. USA 2017, 114, 7166–7171. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Irish, V.F. Temporal Control of Plant Organ Growth by TCP Transcription Factors. Curr. Biol. 2015, 25, 1765–1770. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; He, L.; Li, J.; Zhao, J.; Li, Z.; He, C. Regulatory change at Physalis Organ Size 1 correlates to natural variation in tomatillo reproductive organ size. Nat. Commun. 2014, 5, 4271. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Luo, Y.; Li, X.; Wang, L.; Xu, S.; Yang, J.; Weng, L.; Sato, S.; Tabata, S.; Ambrose, M.; et al. Genetic control of floral zygomorphy in pea (Pisum sativum L.). Proc. Natl. Acad. Sci. USA 2008, 105, 10414–10419. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, W.; Zhuang, L.; Zhu, Y.; Wang, F.; Chen, T.; Yang, J.; Ambrose, M.; Hu, Z.; Weller, J.L.; et al. Bigger organs and elephant ear-like Leaf1 control organ size and floral organ internal asymmetry in pea. J. Exp. Bot. 2019, 70, 179–191. [Google Scholar] [CrossRef]
- Koyama, T.; Ohme-Takagi, M.; Sato, F. Generation of serrated and wavy petals by inhibition of the activity of TCP transcription factors in Arabidopsis thaliana. Plant Signal. Behav. 2011, 6, 697–699. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Huang, J.; Parameswaran, S.; Ito, T.; Seubert, B.; Auer, M.; Rymaszewski, A.; Jia, G.; Owen, H.A.; Zhao, D. The SPOROCYTELESS/NOZZLE Gene Is Involved in Controlling Stamen Identity in Arabidopsis. Plant Physiol. 2009, 151, 1401–1411. [Google Scholar] [CrossRef] [Green Version]
- Ó’Maoiléidigh, D.S.; Wuest, S.E.; Rae, L.; Raganelli, A.; Ryan, P.T.; Kwaśniewska, K.; Das, P.; Lohan, A.J.; Loftus, B.; Graciet, E.; et al. Control of Reproductive Floral Organ Identity Specification in Arabidopsis by the C Function Regulator AGAMOUS. Plant Cell 2013, 25, 2482–2503. [Google Scholar] [CrossRef] [Green Version]
- Lohmann, J.; Hong, R.L.; Hobe, M.; Busch, M.A.; Parcy, F.; Simon, R.; Weigel, D. A Molecular Link between Stem Cell Regulation and Floral Patterning in Arabidopsis. Cell 2001, 105, 793–803. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Xu, Y.; Ng, K.-H.; Ito, T. A timing mechanism for stem cell maintenance and differentiation in the Arabidopsis floral meristem. Genes Dev. 2009, 23, 1791–1804. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Ito, T. Regulation of floral stem cell termination in Arabidopsis. Front. Plant Sci. 2015, 6, 17. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-W.; Czech, B.; Weigel, D. miR156-Regulated SPL Transcription Factors Define an Endogenous Flowering Pathway in Arabidopsis thaliana. Cell 2009, 138, 738–749. [Google Scholar] [CrossRef] [Green Version]
- Teotia, S.; Tang, G. To Bloom or Not to Bloom: Role of MicroRNAs in Plant Flowering. Mol. Plant 2015, 8, 359–377. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Li, J.; Liu, Y.; Zhang, Q.; Gao, Y.; Fang, K.; Cao, Q.; Qin, L.; Xing, Y. Roles of the GA-mediated SPL Gene Family and miR156 in the Floral Development of Chinese Chestnut (Castanea mollissima). Int. J. Mol. Sci. 2019, 20, 1577. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Ruan, H.; Yu, L.; Li, Q.; Song, Q.; Yang, S.; Gai, J. miR156b from Soybean CMS Line Modulates Floral Organ Development. J. Plant Biol. 2020, 63, 141–153. [Google Scholar] [CrossRef]
- Jung, J.-H.; Seo, Y.-H.; Seo, P.J.; Reyes, J.L.; Yun, J.; Chua, N.-H.; Park, C.-M. The GIGANTEA-Regulated MicroRNA172 Mediates Photoperiodic Flowering Independent of CONSTANS in Arabidopsis. Plant Cell 2007, 19, 2736–2748. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, J.; Yant, L.; Muerdter, F.; Küttner, F.; Schmid, M. Repression of Flowering by the miR172 Target SMZ. PLoS Biol. 2009, 7, e1000148. [Google Scholar] [CrossRef] [Green Version]
- Grigorova, B.; Mara, C.; Hollender, C.; Sijacic, P.; Chen, X.; Liu, Z. LEUNIG and SEUSS co-repressors regulate miR172 expression in Arabidopsis flowers. Development 2011, 138, 2451–2456. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Ping, X.; Cao, Y.; Jian, H.; Gao, Y.; Wang, J.; Tan, Y.; Xu, X.; Lu, K.; Li, J.; et al. Genome-wide exploration and characterization of miR172/euAP2 genes in Brassica napus L. for likely role in flower organ development. BMC Plant Biol. 2019, 19, 336. [Google Scholar] [CrossRef] [Green Version]
- Chung, M.-Y.; Nath, U.K.; Vrebalov, J.; Gapper, N.; Lee, J.M.; Lee, D.-J.; Kil Kim, C.; Giovannoni, J. Ectopic expression of miRNA172 in tomato (Solanum lycopersicum) reveals novel function in fruit development through regulation of an AP2 transcription factor. BMC Plant Biol. 2020, 20, 283. [Google Scholar] [CrossRef]
- Baker, C.C.; Sieber, P.; Wellmer, F.; Meyerowitz, E.M. The early extra petals1 Mutant Uncovers a Role for MicroRNA miR164c in Regulating Petal Number in Arabidopsis. Curr. Biol. 2005, 15, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; López-Giráldez, F.; Townsend, J.; Irish, V.F. RBE controls microRNA164 expression to effect floral organogenesis. Development 2012, 139, 2161–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achard, P.; Herr, A.; Baulcombe, D.; Harberd, N.P. Modulation of floral development by a gibberellin-regulated microRNA. Development 2004, 131, 3357–3365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, R.; Palatnik, J.; Riester, M.; Schommer, C.; Schmid, M.; Weigel, D. Specific Effects of MicroRNAs on the Plant Transcriptome. Dev. Cell 2005, 8, 517–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Li, C.; Ding, G.; Jin, Y. Evolution of MIR159/319 microRNA genes and their post-transcriptional regulatory link to siRNA pathways. BMC Evol. Biol. 2011, 11, 122. [Google Scholar] [CrossRef] [Green Version]
- Nag, A.; King, S.; Jack, T. miR319a targeting of TCP4 is critical for petal growth and development in Arabidopsis. Proc. Natl. Acad. Sci. USA 2009, 106, 22534–22539. [Google Scholar] [CrossRef] [Green Version]
- Sarvepalli, K.; Nath, U. Hyper-activation of the TCP4 transcription factor in Arabidopsis thaliana accelerates multiple aspects of plant maturation. Plant J. 2011, 67, 595–607. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Jung, J.-H.; Reyes, J.L.; Kim, Y.-S.; Kim, S.-Y.; Chung, K.-S.; Kim, J.A.; Lee, M.; Lee, Y.; Kim, V.N.; et al. microRNA-directed cleavage of ATHB15 mRNA regulates vascular development in Arabidopsis inflorescence stems. Plant J. 2005, 42, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Ji, L.; Liu, X.; Yan, J.; Wang, W.; Yumul, R.E.; Kim, Y.J.; Dinh, T.T.; Liu, J.; Cui, X.; Zheng, B.; et al. ARGONAUTE10 and ARGONAUTE1 Regulate the Termination of Floral Stem Cells through Two MicroRNAs in Arabidopsis. PLoS Genet. 2011, 7, e1001358. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Wu, S.; Van Houten, J.; Wang, Y.; Ding, B.; Fei, Z.; Clarke, T.H.; Reed, J.W.; van der Knaap, E. Down-regulation of AUXIN RESPONSE FACTORS 6 and 8 by microRNA 167 leads to floral development defects and female sterility in tomato. J. Exp. Bot. 2014, 65, 2507–2520. [Google Scholar] [CrossRef] [Green Version]
- Mallory, A.C.; Bartel, D.P.; Bartel, B. MicroRNA-Directed Regulation of Arabidopsis AUXIN RESPONSE FACTOR17 Is Essential for Proper Development and Modulates Expression of Early Auxin Response Genes. Plant Cell 2005, 17, 1360–1375. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Huang, J.; Wang, Y.; Khanna, K.; Xie, Z.; Owen, H.A.; Zhao, D. The role of floral organs in carpels, an Arabidopsis loss-of-function mutation in MicroRNA160a, in organogenesis and the mechanism regulating its expression. Plant J. 2010, 62, 416–428. [Google Scholar] [CrossRef]
- Gerats, T.; Vandenbussche, M. A model system for comparative research: Petunia. Trends Plant Sci. 2005, 10, 251–256. [Google Scholar] [CrossRef]
- Cabral, A.R.; Beaudoin, G.; Choquette, M.; Lehmann, B.; Polônia, J.C. Supergene leaching and formation of platinum in alluvium: Evidence from Serro, Minas Gerais, Brazil. Miner. Pet. 2007, 90, 141–150. [Google Scholar] [CrossRef]
- Faraco, M.; Spelt, C.; Bliek, M.; Verweij, W.; Hoshino, A.; Espen, L.; Prinsi, B.; Jaarsma, R.; Tarhan, E.; de Boer, A.H.; et al. Hyperacidification of Vacuoles by the Combined Action of Two Different P-ATPases in the Tonoplast Determines Flower Color. Cell Rep. 2014, 6, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Cna’Ani, A.; Spitzer-Rimon, B.; Ravid, J.; Farhi, M.; Masci, T.; Aravena-Calvo, J.; Ovadis, M.; Vainstein, A. Two showy traits, scent emission and pigmentation, are finely coregulated by the MYB transcription factor PH 4 in petunia flowers. New Phytol. 2015, 208, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Colquhoun, T.A.; Verdonk, J.; Schimmel, B.; Tieman, D.M.; Underwood, B.A.; Clark, D.G. Petunia floral volatile benzenoid/phenylpropanoid genes are regulated in a similar manner. Phytochemistry 2010, 71, 158–167. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bombarely, A.; Moser, M.; Amrad, A.; Bao, M.; Bapaume, L.; Barry, C.; Bliek, M.; Boersma, M.R.; Borghi, L.; Bruggmann, R.; et al. Insight into the evolution of the Solanaceae from the parental genomes of Petunia hybrida. Nat. Plants 2016, 2, 16074. [Google Scholar] [CrossRef] [Green Version]
- Tedder, P.; Zubko, E.; Westhead, D.R.; Meyer, P. Small RNA analysis in Petunia hybrida identifies unusual tissue-specific expression patterns of conserved miRNAs and of a 24mer RNA. RNA 2009, 15, 1012–1020. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, J.N.; Chand, S.K.; Joshi, R.K. Multiple microRNAs Regulate the Floral Development and Sex Differentiation in the Dioecious Cucurbit Coccinia grandis (L.) Voigt. Plant Mol. Biol. Rep. 2019, 37, 111–128. [Google Scholar] [CrossRef]
- Chen, D.; Yan, W.; Fu, L.-Y.; Kaufmann, K. Architecture of gene regulatory networks controlling flower development in Arabidopsis thaliana. Nat. Commun. 2018, 9, 4534. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.K.; Garg, R.; Jain, M. A global view of transcriptome dynamics during flower development in chickpea by deep sequencing. Plant Biotechnol. J. 2013, 11, 691–701. [Google Scholar] [CrossRef]
- Liu, K.; Feng, S.; Pan, Y.; Zhong, J.; Chen, Y.; Yuan, C.; Li, H. Transcriptome Analysis and Identification of Genes Associated with Floral Transition and Flower Development in Sugar Apple (Annona squamosa L.). Front. Plant Sci. 2016, 7, 1695. [Google Scholar] [CrossRef] [Green Version]
- Roberts, W.R.; Roalson, E.H. Comparative transcriptome analyses of flower development in four species of Achimenes (Gesneriaceae). BMC Genom. 2017, 18, 240. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Li, Z.; Jin, W.; Fang, Y.; Yang, Q.; Xiang, J. Transcriptome analysis and identification of genes associated with flower development in Rhododendron pulchrum Sweet (Ericaceae). Gene 2018, 679, 108–118. [Google Scholar] [CrossRef]
- Zhao, T.; Specht, C.D.; Dong, Z.; Ye, Y.; Liu, H.; Liao, J. Transcriptome analyses provide insights into development of the Zingiber zerumbet flower, revealing potential genes related to floral organ formation and patterning. Plant Growth Regul. 2020, 90, 331–345. [Google Scholar] [CrossRef]
- Brun, O.; Lebon, G.; Duchêne, E.; Magné, C.; Clément, C. Flower abscission and inflorescence carbohydrates in sensitive and non-sensitive cultivars of grapevine. Sex. Plant Reprod. 2004, 17, 71–79. [Google Scholar] [CrossRef]
- Lebon, G.; Rondeau, M.; Sanchez, L.; Barka, E.A.; Vaillant-Gaveau, N. Modulation of the Activity of Enzymes Involved in Carbohydrate Metabolism during Flower Development of Grapevine (Vitis vinifera L.). Open J. Plant Sci. 2016, 1, 010–017. [Google Scholar] [CrossRef] [Green Version]
- Lemoine, R.; La Camera, S.; Atanassova, R.; Dédaldéchamp, F.; Allario, T.; Pourtau, N.; Bonnemain, J.L.; Laloi, M.; Coutos-Thévenot, P.; Maurousset, L.; et al. Source-to-sink transport of sugar and regulation by environmental factors. Front. Plant Sci. 2013, 4, 272. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, R. Carbohydrate Availability in Relation to Fruitlet Abscission in Citrus. Ann. Bot. 2001, 87, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Rolland, F.; Baena-Gonzalez, E.; Sheen, J. Sugar sensing and signaling in plants: Conserved and Novel Mechanisms. Annu. Rev. Plant Biol. 2006, 57, 675–709. [Google Scholar] [CrossRef] [Green Version]
- Neta-Sharir, I.; Shoseyov, O.; Weiss, D. Sugars enhance the expression of gibberellin-induced genes in developing petunia flowers. Physiol. Plant. 2000, 109, 196–202. [Google Scholar] [CrossRef]
- Shin, D.H.; Choi, M.-G.; Lee, H.K.; Cho, M.; Choi, S.-B.; Choi, G.; Park, Y.-I. Calcium dependent sucrose uptake links sugar signaling to anthocyanin biosynthesis in Arabidopsis. Biochem. Biophys. Res. Commun. 2013, 430, 634–639. [Google Scholar] [CrossRef]
- Paul, M.; Pellny, T.; Goddijn, O. Enhancing photosynthesis with sugar signals. Trends Plant Sci. 2001, 6, 197–200. [Google Scholar] [CrossRef]
- Sawicki, M.; Jacquens, L.; Baillieul, F.; Clément, C.; Jacquard, C.; Vaillant-Gaveau, N. Distinct regulation in inflorescence carbohydrate metabolism according to grapevine cultivars during floral development. Physiol. Plant. 2015, 154, 447–467. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, M.; Courteaux, B.; Rabenoelina, F.; Baillieul, F.; Clement, C.; Barka, E.; Jacquard, C.; Vaillant-Gaveau, N. Leaf vs. inflorescence: Differences in photosynthetic activity of grapevine. Photosynthetics 2017, 55, 58–68. [Google Scholar] [CrossRef]
- Liu, J.; Osbourn, A.; Ma, P. MYB Transcription Factors as Regulators of Phenylpropanoid Metabolism in Plants. Mol. Plant 2015, 8, 689–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Q.J.; Wang, T.; Guo, Q.S.; Yang, F.; Chen, J.M.; Zhang, W.Y. Combined Metabolomic and Transcriptomic Analysis Reveals Redirection of the Phenylpropanoid Metabolic Flux in Different Colored Medicinal Chrysanthemum morifolium. Ind. Crops Prod. 2021, 164, 113343. [Google Scholar] [CrossRef]
- Xie, D.-Y.; Sharma, S.B.; Paiva, N.L.; Ferreira, D.; Dixon, R.A. Role of Anthocyanidin Reductase, Encoded by BANYULS in Plant Flavonoid Biosynthesis. Science 2003, 299, 396–399. [Google Scholar] [CrossRef] [Green Version]
- Feng, G.; Huang, S.; Liu, Y.; Xiao, F.; Liu, J.; Zhang, Z.; Chen, Q.; Mao, Y.; Cao, X.; Wang, Y.; et al. The transcriptome analyses of Tagetes erecta provides novel insights into secondary metabolite biosynthesis during flower development. Gene 2018, 660, 18–27. [Google Scholar] [CrossRef]
- Patra, B.; Schluttenhofer, C.; Wu, Y.; Pattanaik, S.; Yuan, L. Transcriptional regulation of secondary metabolite biosynthesis in plants. Biochim. Biophys. Acta 2013, 1829, 1236–1247. [Google Scholar] [CrossRef]
- Zhang, B.; Chopra, D.; Schrader, A.; Hülskamp, M. Evolutionary comparison of competitive protein-complex formation of MYB, bHLH, and WDR proteins in plants. J. Exp. Bot. 2019, 70, 3197–3209. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.-J.; Geng, Z.; Wang, Y.; Wang, Y.; Liu, S.; Chen, C.; Song, A.; Jiang, J.; Chen, S.; Chen, F. A novel transcription factor CmMYB012 inhibits flavone and anthocyanin biosynthesis in response to high temperatures in chrysanthemum. Hortic. Res. 2021, 8, 1–16. [Google Scholar] [CrossRef]
- Matsubara, K.; Shimamura, K.; Kodama, H.; Kokubun, H.; Watanabe, H.; Basualdo, I.L.; Ando, T. Green corolla segments in a wild Petunia species caused by a mutation in FBP2, a SEPALLATA-like MADS box gene. Planta 2008, 228, 401–409. [Google Scholar] [CrossRef]
- Pan, Z.; Chen, Y.-Y.; Du, J.; Chen, Y.; Chung, M.; Tsai, W.; Wang, C.-N.; Chen, H. Flower development of Phalaenopsis orchid involves functionally divergent SEPALLATA-like genes. New Phytol. 2014, 202, 1024–1042. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Wang, T.; McGie, T.; Voogd, C.; Allan, A.C.; Hellens, R.P.; Varkonyi-Gasic, E. Overexpression of the kiwifruit SVP3 gene affects reproductive development and suppresses anthocyanin biosynthesis in petals, but has no effect on vegetative growth, dormancy, or flowering time. J. Exp. Bot. 2014, 65, 4985–4995. [Google Scholar] [CrossRef] [Green Version]
- Jaakola, L.; Poole, M.; Jones, M.O.; Kämäräinen-Karppinen, T.; Koskimäki, J.J.; Hohtola, A.; Häggman, H.; Fraser, P.D.; Manning, K.; King, G.J.; et al. A SQUAMOSA MADS Box Gene Involved in the Regulation of Anthocyanin Accumulation in Bilberry Fruits. Plant Physiol. 2010, 153, 1619–1629. [Google Scholar] [CrossRef] [Green Version]
- Mara, C.D.; Irish, V.F. Two GATA Transcription Factors Are Downstream Effectors of Floral Homeotic Gene Action in Arabidopsis. Plant Physiol. 2008, 147, 707–718. [Google Scholar] [CrossRef] [Green Version]
- Lymperopoulos, P.; Msanne, J.; Rabara, R. Phytochrome and Phytohormones: Working in Tandem for Plant Growth and Development. Front. Plant Sci. 2018, 9, 1037. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-C.; Wang, N.; Xu, H.-F.; Jiang, S.-H.; Fang, H.-C.; Su, M.-Y.; Zhang, Z.-Y.; Zhang, T.-L.; Chen, X.-S. Auxin regulates anthocyanin biosynthesis through the Aux/IAA-ARF signaling pathway in apple. Hortic. Res. 2018, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.Q.; Xing, W.; Luo, P.; Zhang, F.J.; Jin, X.L.; Zhang, M.H. Comparative transcriptome analysis of Rosa chinensis ‘Slater’s crimson China’ provides insights into the crucial factors and signaling pathways in heat stress response. Plant Physiol. Biochem. 2019, 142, 312–331. [Google Scholar] [CrossRef]
- He, C.; Liu, X.; Da Silva, J.A.T.; Liu, N.; Zhang, M.; Duan, J. Transcriptome sequencing and metabolite profiling analyses provide comprehensive insight into molecular mechanisms of flower development in Dendrobium officinale (Orchidaceae). Plant Mol. Biol. 2020, 104, 529–548. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, K.; Zeng, S.; Silva, J.A.T.D.; Zhao, X.; Tian, C.-E.; Xia, H.; Duan, J. Transcriptome analysis of Cymbidium sinense and its application to the identification of genes associated with floral development. BMC Genom. 2013, 14, 279. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Zhang, Y.; Zhen, X.; Guo, D.; Guo, C.; Shu, Y. Transcriptome sequencing and expression profiling of genes involved in daylily (Hemerocallis citrina Borani) flower development. Biotechnol. Biotechnol. Equip. 2020, 34, 542–548. [Google Scholar] [CrossRef]
- Sunkar, R.; Zhu, J.-K. Novel and Stress-Regulated MicroRNAs and Other Small RNAs from Arabidopsis. Plant Cell 2004, 16, 2001–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Zhang, Y.-C.; Wang, C.-Y.; Luo, Y.-C.; Huang, Q.-J.; Chen, S.-Y.; Zhou, H.; Qu, L.-H.; Chen, Y.-Q. Expression analysis of phytohormone-regulated microRNAs in rice, implying their regulation roles in plant hormone signaling. FEBS Lett. 2009, 583, 723–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.H.; Han, S.J.; Yoon, E.K.; Lee, W.S. ‘Evidence of an auxin signal pathway, microRNA167-ARF8-GH3, and its response to exogenous auxin in cultured rice cells’. Nucleic Acids Res. 2006, 34, 1892–1899. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chen, Y.-Q. Insights into the mechanism of plant development: Interactions of miRNAs pathway with phytohormone response. Biochem. Biophys. Res. Commun. 2009, 384, 1–5. [Google Scholar] [CrossRef]
- Chen, Z.-H.; Bao, M.-L.; Sun, Y.-Z.; Yang, Y.-J.; Xu, X.-H.; Wang, J.-H.; Han, N.; Bian, H.-W.; Zhu, M.-Y. Regulation of auxin response by miR393-targeted transport inhibitor response protein 1 is involved in normal development in Arabidopsis. Plant Mol. Biol. 2011, 77, 619–629. [Google Scholar] [CrossRef]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.-W.; Weigel, D.; Poethig, R.S. The Sequential Action of miR156 and miR172 Regulates Developmental Timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef] [Green Version]
- Yant, L.; Mathieu, J.; Dinh, T.T.; Ott, F.; Lanz, C.; Wollmann, H.; Chen, X.; Schmid, M. Orchestration of the floral transition and floral development in Arabidopsis by the bifunctional transcription factor APETALA2. Plant Cell 2010, 22, 2156–2170. [Google Scholar] [CrossRef] [Green Version]
- Galvão, V.C.; Horrer, D.; Küttner, F.; Schmid, M. Spatial control of flowering by DELLA proteins in Arabidopsis thaliana. Development 2012, 139, 4072–4082. [Google Scholar] [CrossRef] [Green Version]
- Yanai, O.; Shani, E.; Russ, D.; Ori, N. Gibberellin partly mediates LANCEOLATE activity in tomato. Plant J. 2011, 68, 571–582. [Google Scholar] [CrossRef]
- Silva, G.F.F.; Silva, E.M.; Correa, J.P.O.; Vicente, M.H.; Jiang, N.; Notini, M.M.; Junior, A.C.; De Jesus, F.A.; Frederico, A.; Castilho, P.; et al. Tomato floral induction and flower develop-ment are orchestrated by the interplay between gibberellin and two unrelated microRNA-controlled modules. New Phytol. 2019, 221, 1328–1344. [Google Scholar] [CrossRef] [Green Version]
- Bari, R.; Pant, B.D.; Stitt, M.; Golm, S.P. PHO2, MicroRNA399, and PHR1 Define a Phosphate-Signaling Pathway in Plants. Plant Physiol. 2006, 141, 988–999. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Fang, Y.-N.; Wu, X.-M.; Qing, M.; Li, C.-C.; Xie, K.-D.; Deng, X.-X.; Guo, W.-W. The miR399-CsUBC24 Module Regulates Reproductive Development and Male Fertility in Citrus. Plant Physiol. 2020, 183, 1681–1695. [Google Scholar] [CrossRef]
- Sun, Y.; Xiong, X.; Wang, Q.; Zhu, L.; Wang, L.; He, Y.; Zeng, H. Integrated Analysis of Small RNA, Transcriptome, and Degradome Sequencing Reveals the MiR156, MiR5488 and MiR399 Are Involved in the Regulation of Male Sterility in PTGMS Rice. Int. J. Mol. Sci. 2021, 22, 2260. [Google Scholar] [CrossRef]
- Chen, Q.; Jing, D.; Wang, S.; Xu, F.; Bao, C.; Luo, M.; Guo, Q. The Putative Role of the NAC Transcription Factor EjNACL47 in Cell Enlargement of Loquat (Eriobotrya japonica Lindl.). Horticulturae 2021, 7, 323. [Google Scholar] [CrossRef]
- Jian, W.; Zheng, Y.; Yu, T.; Cao, H.; Chen, Y.; Cui, Q.; Xu, C.; Li, Z. SlNAC6, A NAC transcription factor, is involved in drought stress response and reproductive process in tomato. J. Plant Physiol. 2021, 264, 153483. [Google Scholar] [CrossRef]
- Allen, P.D.; Gansle, K.A. Global Implications of the Terrorist Attacks of September 11, 2001. New Glob. Dev. 2003, 19, 3–4. [Google Scholar] [CrossRef]
- Mallory, A.C.; Dugas, D.; Bartel, D.P.; Bartel, B. MicroRNA Regulation of NAC-Domain Targets Is Required for Proper Formation and Separation of Adjacent Embryonic, Vegetative, and Floral Organs. Curr. Biol. 2004, 14, 1035–1046. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.; Wei, W.; Li, Y.; Kan, L.; Wang, F.; Zhang, X.; Li, F.; Liu, Z.; Kang, C. Conserved and novel roles of miR164- CUC 2 regulatory module in specifying leaf and floral organ morphology in strawberry. New Phytol. 2019, 224, 480–492. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison Group | Number of Up-Regulated Genes | Number of Down-Regulated Genes |
|---|---|---|
| st2_vs_st1 | 4784 | 3521 |
| st3_vs_st1 | 2271 | 734 |
| st4_vs_st1 | 5782 | 4688 |
| st5_vs_st1 | 6354 | 6320 |
| st3_vs_st2 | 2474 | 3416 |
| st4_vs_st2 | 2473 | 2253 |
| st5_vs_st2 | 4935 | 5183 |
| st4_vs_st3 | 4642 | 3632 |
| st5_vs_st3 | 5775 | 6223 |
| st5_vs_st4 | 2716 | 3296 |
| miRNA | Target_Gene | Annotation | Score |
|---|---|---|---|
| Pa-miRn2/t0000048_x74694 | Peaxi162Scf00038g02039 | T-complex protein 1 subunit eta | 3 |
| Pa-miRn2/t0000048_x74694 | Peaxi162Scf00275g00008 | T-complex protein 1 subunit eta | 3 |
| Pa-miRn5/t0000087_x39090 | Peaxi162Scf00026g02810 | UDP-galactose transporter 3 (UGT3) | 3 |
| Pa-miRn5/t0000087_x39090 | Peaxi162Scf00284g00524 | S-adenosyl-L-methionine-dependent methyltransferases superfamily protein | 3 |
| Pa-miRn8/t0000114_x29467 | Peaxi162Scf00213g00939 | Sequence-specific DNA binding transcription factors (Trihelix) | 3 |
| Pa-miRn8/t0000114_x29467 | Peaxi162Scf01021g00126 | Proline-rich spliceosome-associated (PSP) family protein | 2.8 |
| Pa-miRn14/t0000356_x10375 | Peaxi162Scf00055g00142 | Exocyst subunit exo70 family protein B1 | 3 |
| Pa-miRn14/t0000356_x10375 | Peaxi162Scf00488g00924 | RING/U-box superfamily protein | 2.5 |
| Pa-miRn14/t0000356_x10375 | Peaxi162Scf01205g00173 | RNA-binding protein | 3 |
| Pa-miRn15/t0000510_x7859 | Peaxi162Scf00304g01121 | Oxidoreductase, zinc-binding dehydrogenase family protein | 3 |
| Pa-miRn18/t0001319_x3709 | Peaxi162Scf00684g00120 | YEATS family protein | 2.8 |
| Pa-miRn22/t0014836_x652 | Peaxi162Scf00906g00224 | Unknown protein | 3 |
| Pa-miR2/nta-miR477a | Peaxi162Scf01026g00268 | Double-stranded DNA binding | 3 |
| Pa-miR3/nta-miR477b | Peaxi162Scf00295g00014 | DNA-directed RNA polymerases I, II, and III subunit RPABC1 | 3 |
| Pa-miR3/nta-miR477b | Peaxi162Scf00853g00311 | Flotillin-like protein 1 | 2.8 |
| Pa-miR37/stu-miR398b-3p | Peaxi162Scf00498g00025 | 60S ribosomal protein L31 | 3 |
| Pa-miR38/stu-miR399f-3p | Peaxi162Scf01012g00019 | Nucleotide sugar transporter family protein | 3 |
| Pa-miR39/stu-miR399i-3p | Peaxi162Scf00016g02216 | Protein of unknown function (DUF579) | 3 |
| Pa-miR39/stu-miR399i-3p | Peaxi162Scf00095g00913 | Mitogen-activated protein kinase 16 | 3 |
| Pa-miR39/stu-miR399i-3p | Peaxi162Scf00589g00322 | NAC domain containing protein 82 (NAC) | 3 |
| Pa-miR40/stu-miR399o-3p | Peaxi162Scf00767g00521 | Beta-galactosidase | 2.8 |
| Pa-miR43/stu-miR530 | Peaxi162Scf00819g00414 | 4,5-DOPA dioxygenase extradiol | 3 |
| Pa-miR44/stu-miR6024-5p | Peaxi162Scf00042g01612 | Late blight resistance protein, putative | 3 |
| Pa-miR15/sly-miR390b-5p | Peaxi162Scf00102g01878 | Serine/threonine-protein kinase 25 | 3 |
| Pa-miR15/sly-miR390b-5p | Peaxi162Scf00173g00046 | Diphosphomevalonate decarboxylase | 3 |
| Pa-miR15/sly-miR390b-5p | Peaxi162Scf00286g00028 | Leucine-rich receptor-like protein kinase family protein | 2.8 |
| Pa-miR15/sly-miR390b-5p | Peaxi162Scf00620g00014 | Leucine-rich receptor-like protein kinase family protein | 3 |
| Pa-miR15/sly-miR390b-5p | Peaxi162Scf00732g10028 | Leucine-rich receptor-like protein kinase family protein | 2.5 |
| Pa-miR19/sly-miR396b | Peaxi162Scf00016g03253 | Pyruvate kinase family protein | 3 |
| Pa-miR19/sly-miR396b | Peaxi162Scf00074g00526 | Unknown protein | 2.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Q.; Jin, X.; Liu, C.; Wen, Y. An Integrated Analysis of Transcriptome and miRNA Sequencing Provides Insights into the Dynamic Regulations during Flower Morphogenesis in Petunia. Horticulturae 2022, 8, 284. https://doi.org/10.3390/horticulturae8040284
Yu Q, Jin X, Liu C, Wen Y. An Integrated Analysis of Transcriptome and miRNA Sequencing Provides Insights into the Dynamic Regulations during Flower Morphogenesis in Petunia. Horticulturae. 2022; 8(4):284. https://doi.org/10.3390/horticulturae8040284
Chicago/Turabian StyleYu, Qiuxiu, Xiaoling Jin, Caixian Liu, and Yafeng Wen. 2022. "An Integrated Analysis of Transcriptome and miRNA Sequencing Provides Insights into the Dynamic Regulations during Flower Morphogenesis in Petunia" Horticulturae 8, no. 4: 284. https://doi.org/10.3390/horticulturae8040284
APA StyleYu, Q., Jin, X., Liu, C., & Wen, Y. (2022). An Integrated Analysis of Transcriptome and miRNA Sequencing Provides Insights into the Dynamic Regulations during Flower Morphogenesis in Petunia. Horticulturae, 8(4), 284. https://doi.org/10.3390/horticulturae8040284