
New Insight into the Phylogeny and Taxonomy of Cultivated and Related Species of Crataegus in China, Based on Complete Chloroplast Genome Sequencing

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and DNA Extraction
2.2. Chloroplast Genome Sequencing, Assembly, Annotation, and Visualization
2.3. Analysis of Microsatellites and Repeat Sequences
2.4. Variation Hotspots Detection and Sequence Divergence Analysis
2.5. Comparative Genome Analysis
2.6. Phylogenomic Analysis
3. Results
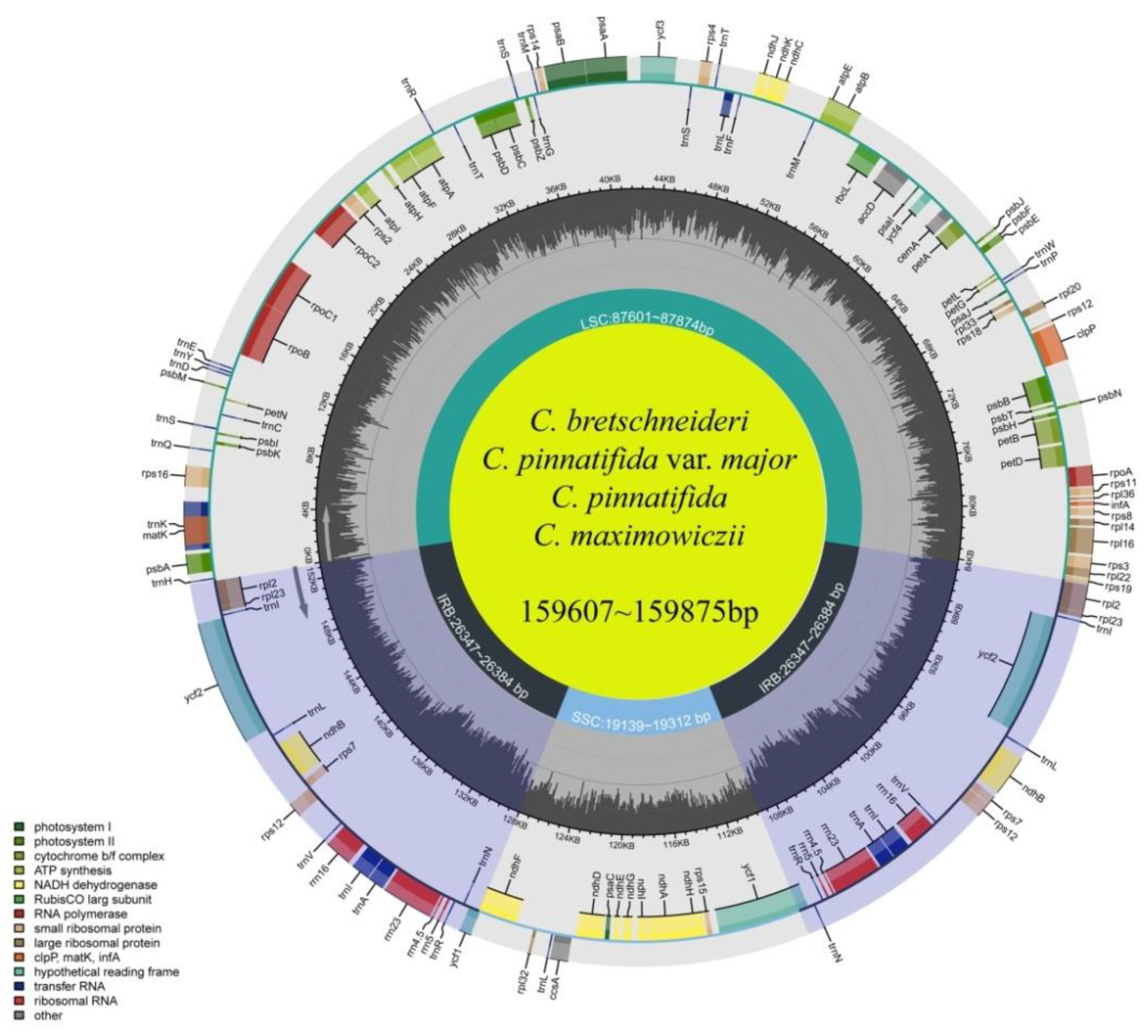
3.1. Genome Organization and Features
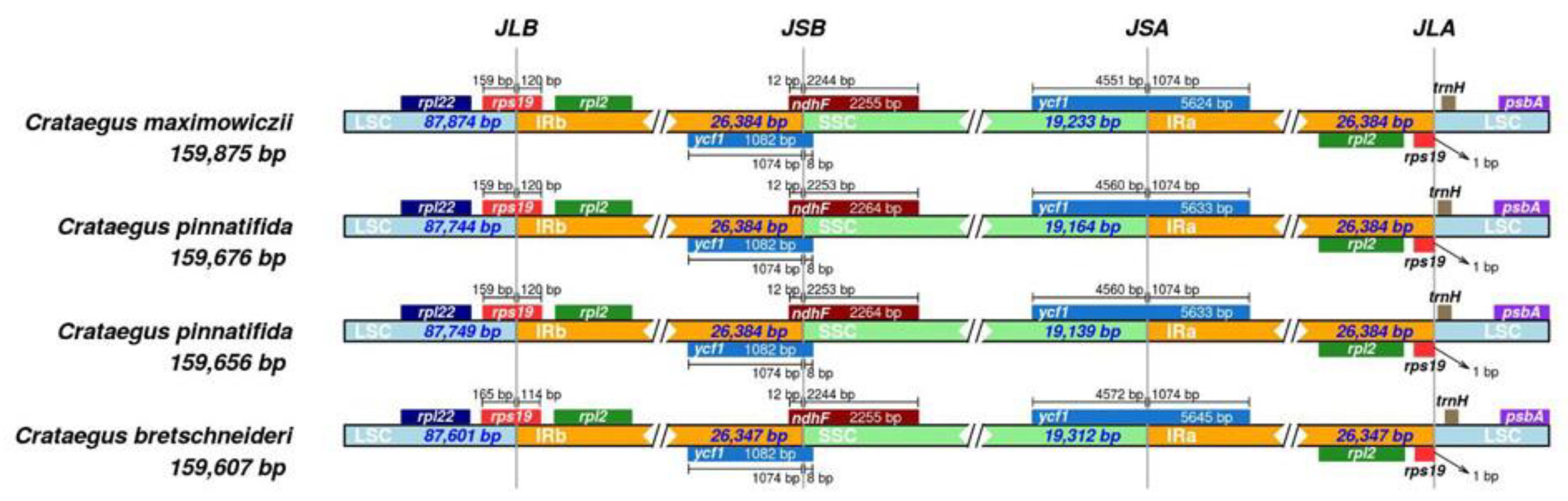
3.2. IR Expansion and Shrinkage
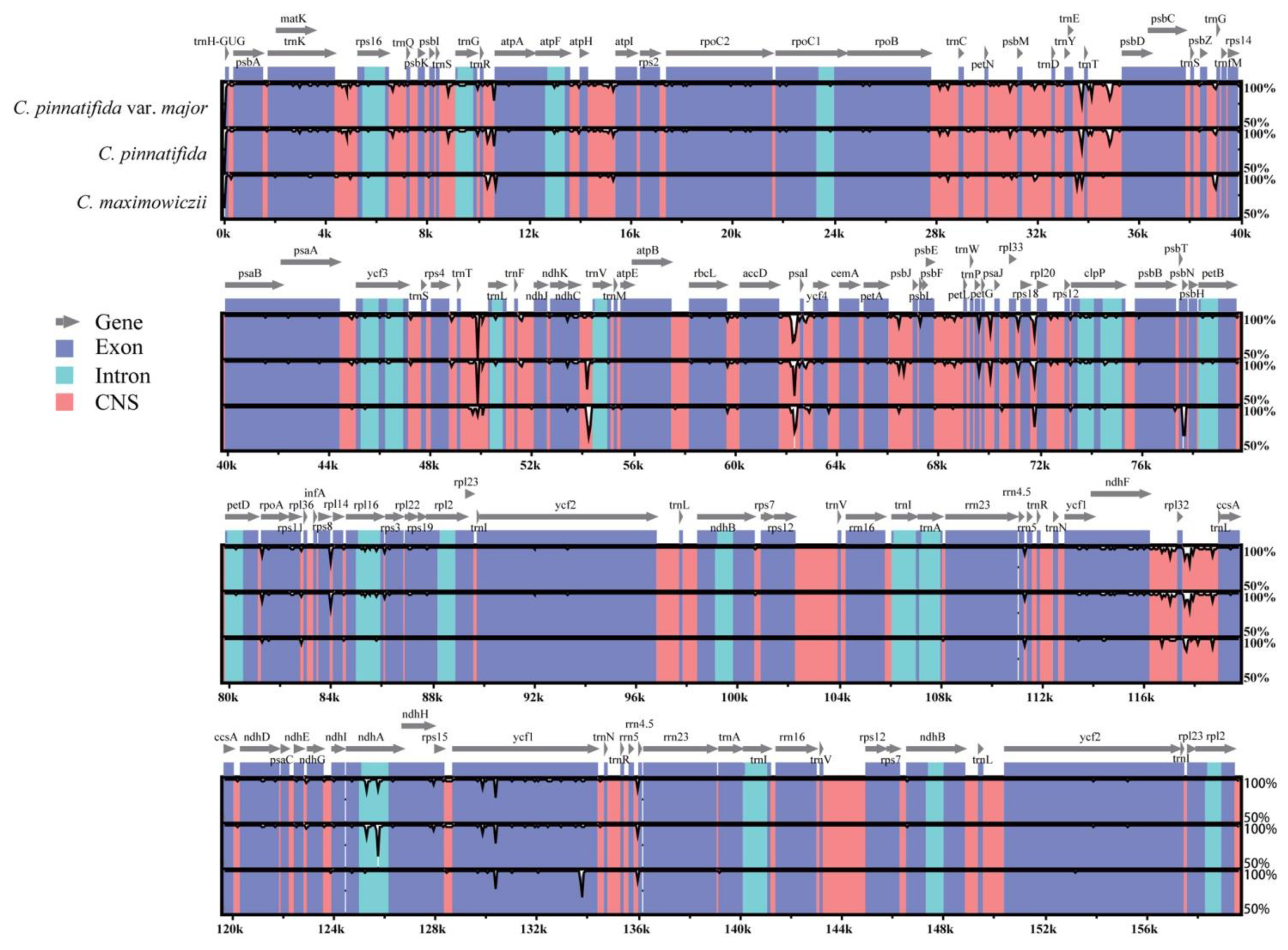
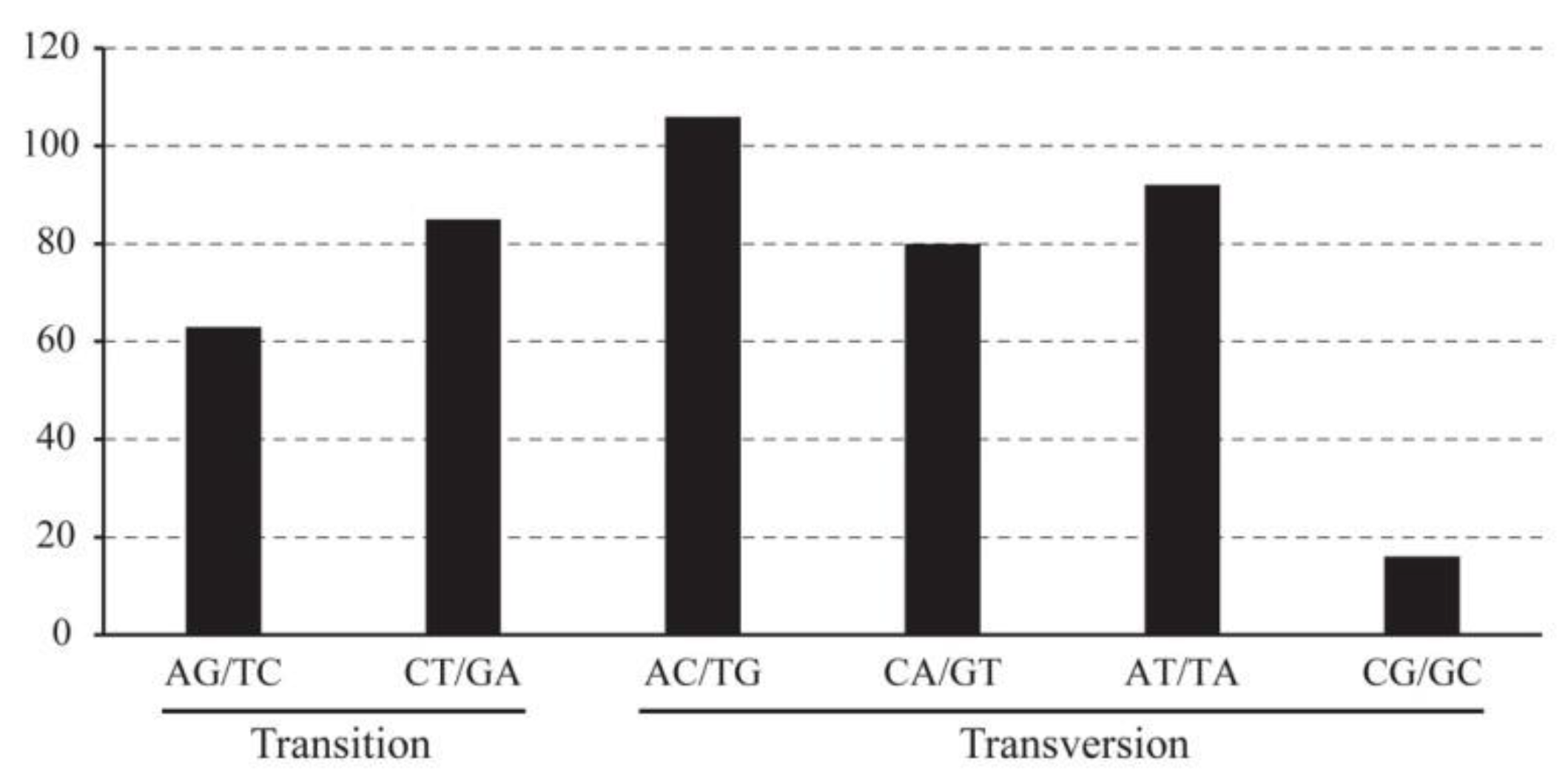
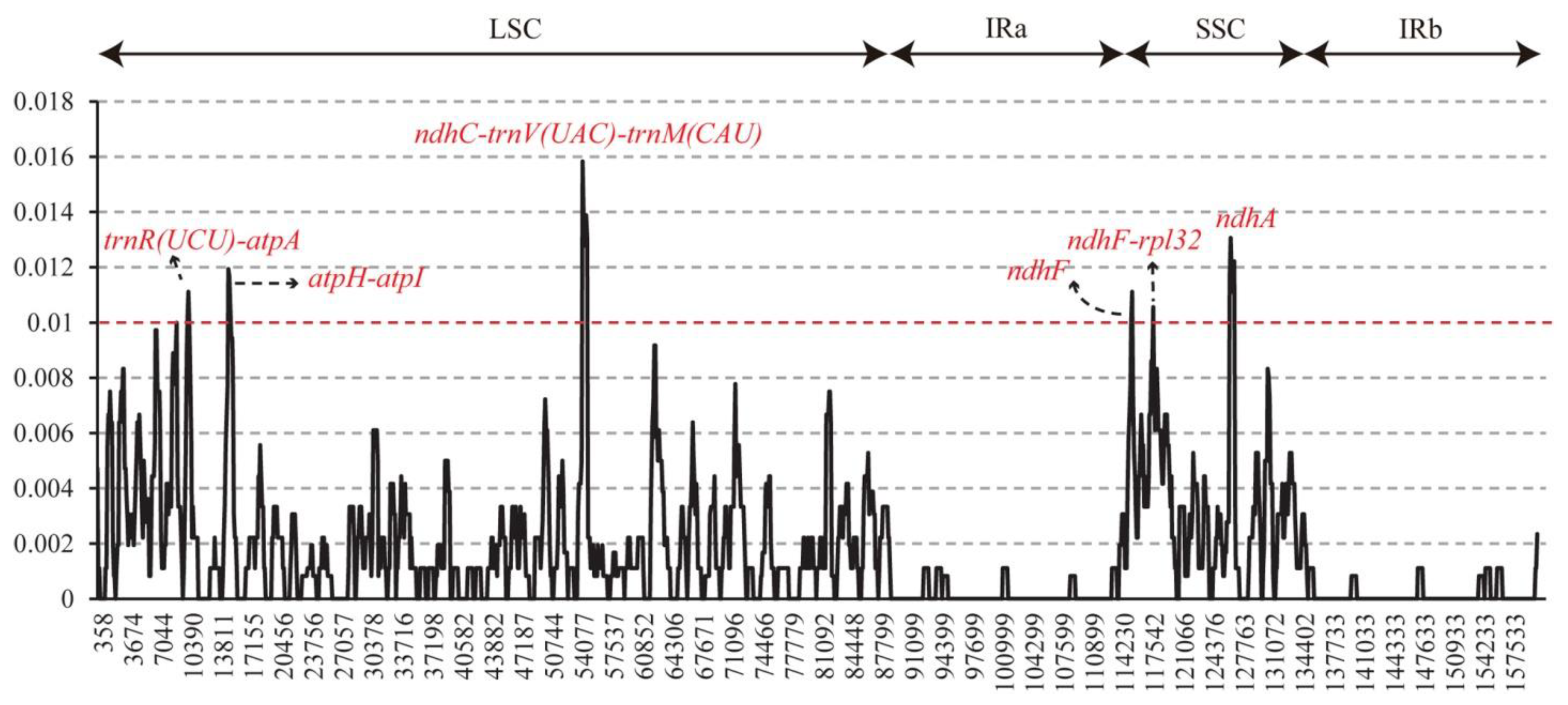
3.3. Divergence Analysis of Sequence and High Variation Region
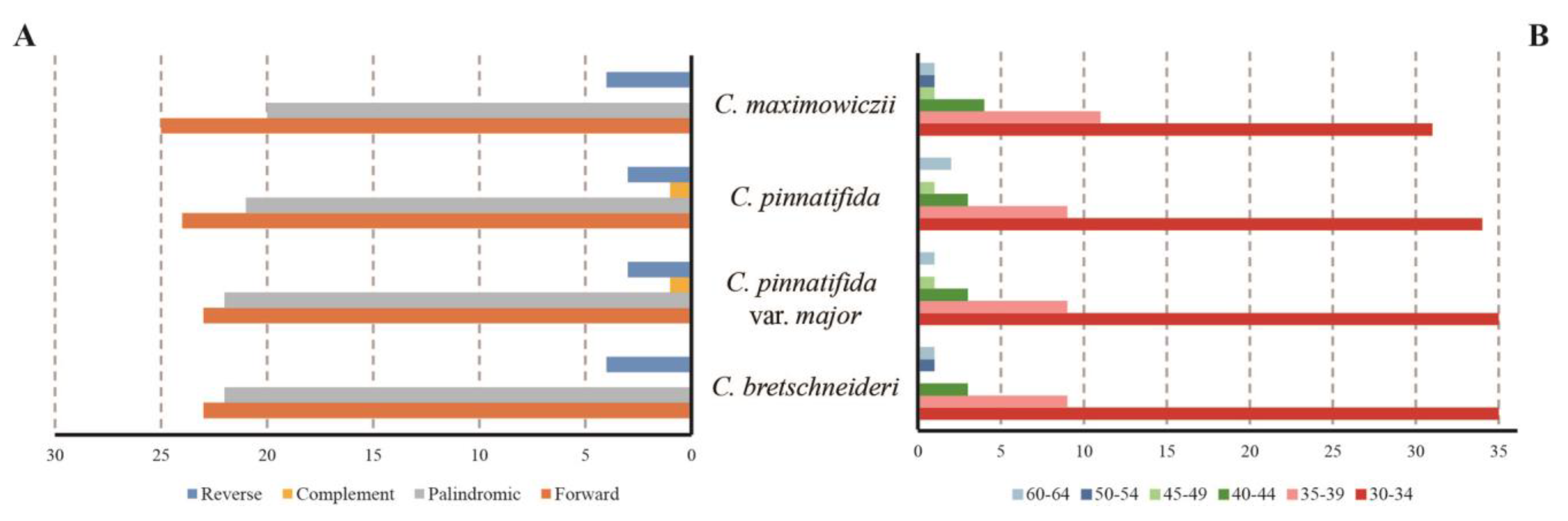
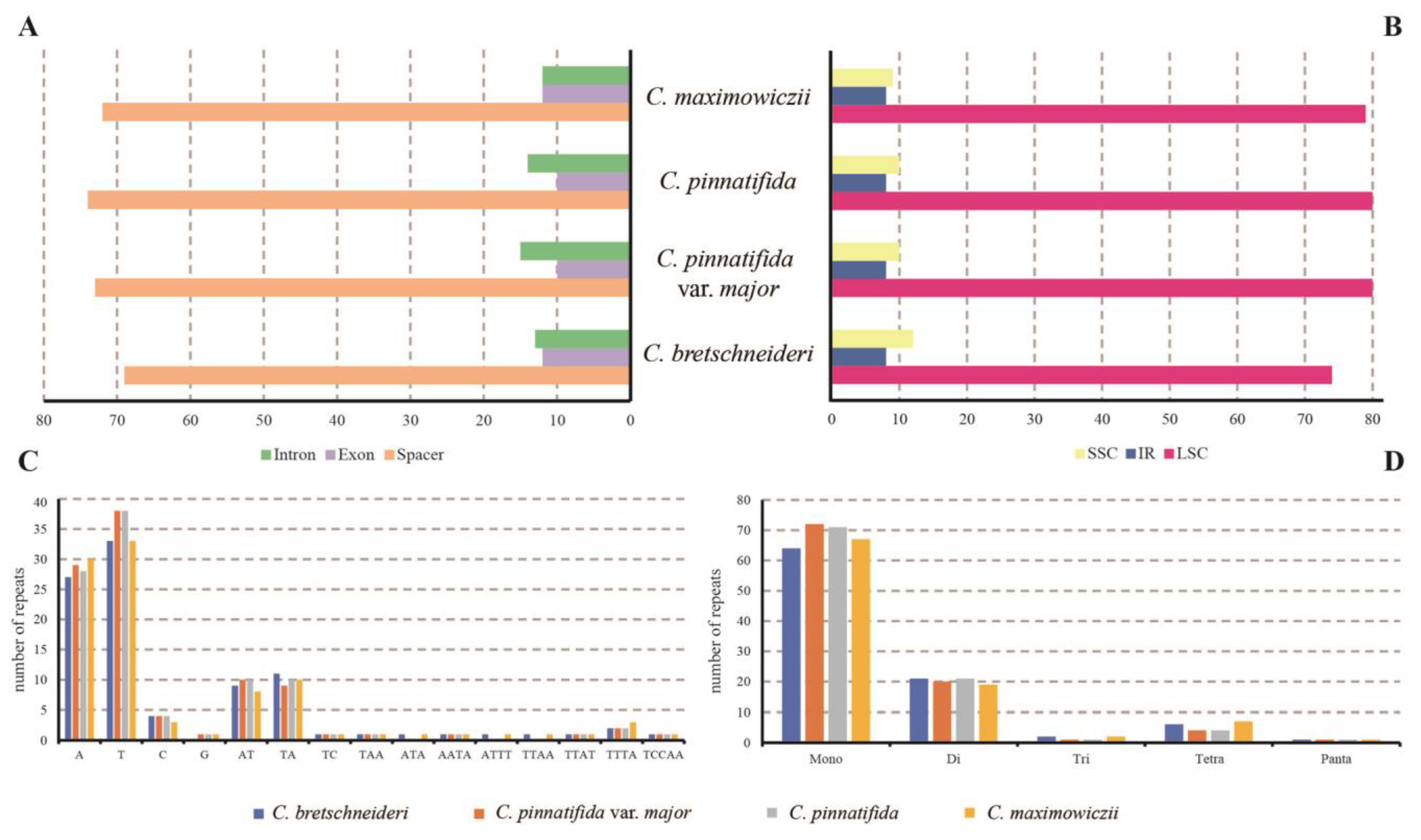
3.4. Repeat Structure and SSR Analysis
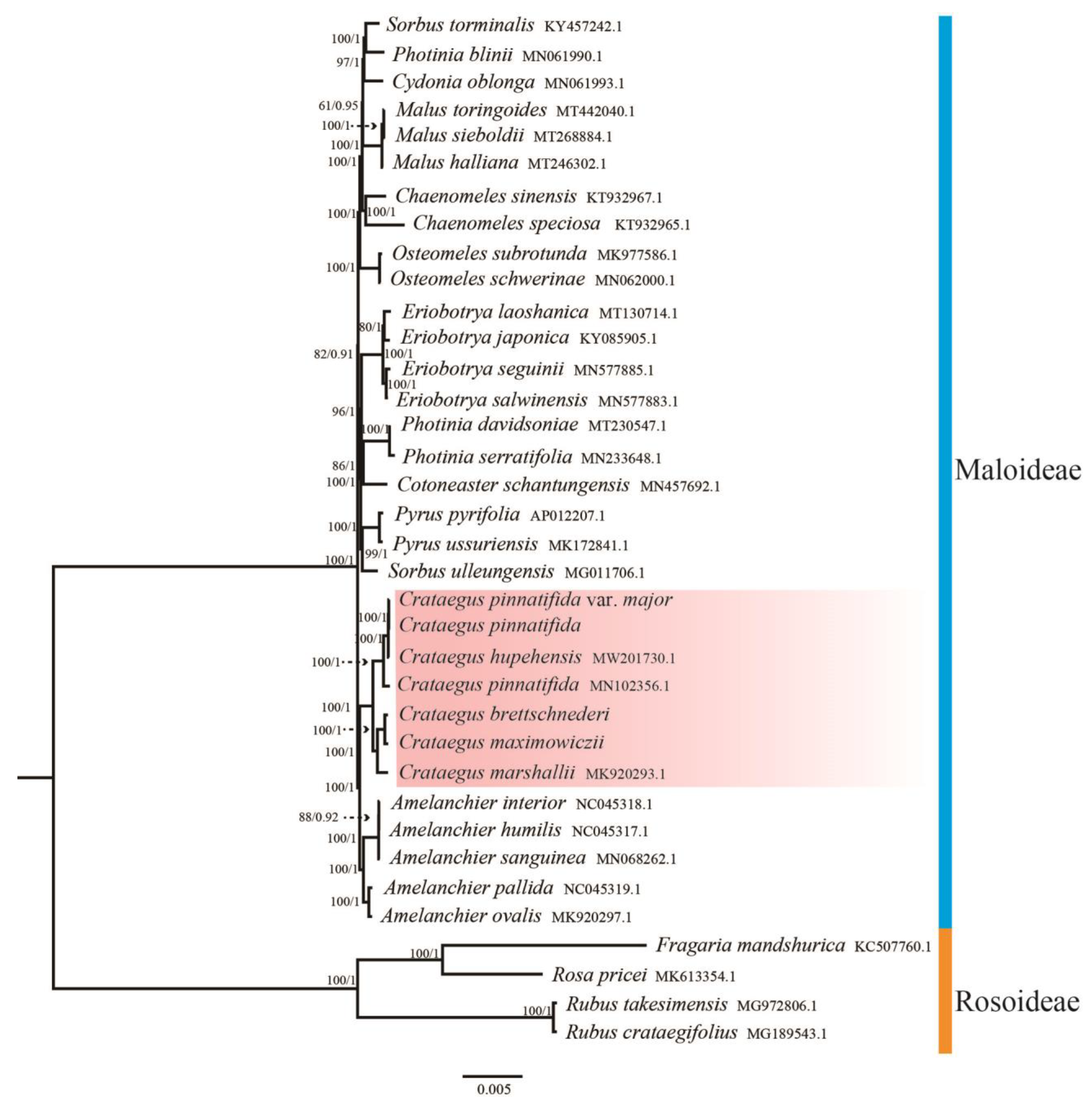
3.5. Phylogenetic Analysis
4. Discussion
4.1. Genome Features and Sequence Divergence among Crataegus Species
4.2. Repeat Structure and SSR Analysis of the Plastomes of Crataegus Species
4.3. Potential Highly Variable Chloroplast Barcodes
4.4. Phylogenetic Relationships
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Christensen, K.I. Revision of Crataegus sect. Crataegus and nothosect. Crataeguineae (Rosaceae-Maloideae) in the Old World. Syst. Bot. Monogr. 1992, 35, 1–199. [Google Scholar] [CrossRef]
- Özcan, M.; Hacıseferoğulları, H.; Marakoğlu, T.; Arslan, D. Hawthorn (Crataegus spp.) fruit: Some physical and chemical properties. J. Food Eng. 2005, 69, 409–413. [Google Scholar] [CrossRef]
- Xu, J.Y.; Zhao, Y.H.; Zhang, X.; Zhang, L.J.; Dong, W.X. Transcriptome analysis and ultrastructure observation reveal that hawthorn fruit softening is due to cellulose/hemicellulose degradation. Front. Plant Sci. 2016, 7, 1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.Z.; Yang, B.R.; Kallio, H. Characterization of phenolic compounds in Chinese hawthorn (Crataeguspinnatifida Bge. var. major) fruit by high performance liquid chromatography-electrospray ionization mass spectrometry. Food Chem. 2010, 121, 1188–1197. [Google Scholar] [CrossRef]
- Zheng, G.Q.; Deng, J.; Wen, L.R.; You, L.J.; Zhao, Z.G.; Zhou, L. Release of phenolic compounds and antioxidant capacity of Chinese hawthorn “Crataeguspinnatifida” during, in vitro, digestion. J. Funct. Foods 2018, 40, 76–85. [Google Scholar] [CrossRef]
- Wu, J.Q.; Peng, W.; Qin, R.X.; Zhou, H. Crataeguspinnatifida: Chemical constituents, pharmacology, and potential applications. Molecules 2014, 19, 1685–1712. [Google Scholar] [CrossRef] [PubMed]
- Dahmer, S.; Scott, E. Health effects of hawthorn (Complementary and Alternative Medicine). Am. Fam. Physician 2018, 81, 465–469. [Google Scholar]
- Jurikova, T.; Sochor, J.; Rop, O.; Mlcek, J.; Balla, S.; Szekeres, L.; Adam, V.; Kizek, R. Polyphenolic profile and biological activity of Chinese hawthorn (Crataegus pinnatifida BUNGE) fruits. Molecules 2012, 17, 14490–14509. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.E.; Brown, P.N.; Talent, N.; Dickinson, T.A.; Shipley, P.R. A review of the chemistry of the genus Crataegus. Phytochemistry 2012, 79, 5–26. [Google Scholar] [CrossRef]
- Zhao, H.; Feng, B. China Fruit-Plant Monograph of Hawthorn (Crataegus) Flora; Zhongguo Linye Press: Beiing, China, 1996. [Google Scholar]
- Xin, X.; Zhang, Y. Chinese Hawthorn Germplasm Resources and Utilization; China Agricultural Press: Beijing, China, 1997. [Google Scholar]
- Dong, W.; Li, Z. The Science and Practice of Chinese Fruit Tree: Hawthorn; Shanxi Science Press: Xi’an, China, 2015. [Google Scholar]
- Ma, S.L.Y.; Dong, W.X.; Lyu, T.; Lyu, Y.M. An RNA sequencing transcriptome analysis and development of EST-SSR markers in Chinese hawthorn through Illumina sequencing. Forests 2019, 10, 82. [Google Scholar] [CrossRef] [Green Version]
- Dai, H.Y. Molecular Identification and Enhancement of Germplasms in Hawthorn. Ph.D. Thesis, Shenyang Agricultural University, Shenyang, China, 2007. [Google Scholar]
- Guo, T.J.; Jiao, P.J. Hawthorn (Crataegus) resources in China. HortScience 1995, 30, 1132–1134. [Google Scholar] [CrossRef]
- Han, X.Y.; Ling, Y.H.; Wang, Y.J.; Li, F.; Guo, T.J.; Xue, Y.J. Analysis of the origin and classification of C. brettschnederi by ISSR Markers. J. Jilin Agric. Univ. 2009, 31, 164–167. [Google Scholar]
- Du, X.; Zhang, X.; Bu, X.D.; Zhang, T.C.; Lao, Y.C.; Dong, W.X. Molecular analysis of evolution and origins of cultivated hawthorn (Crataegus spp.) and related species in China. Front. Plant Sci. 2019, 10, 443. [Google Scholar] [CrossRef] [Green Version]
- Neuhaus, H.E.; Emes, M.J. Nonphotosynthetic metabolism in plastids. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2000, 51, 111–140. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qi, Z.C.; Zhao, Y.P.; Fu, C.X.; Xiang, Q.Y. Complete cp DNA genome sequence of Smilaxchina and phylogenetic placement of Liliales- influences of gene partitions and taxon sampling. Mol. Phylogenet. Evol. 2012, 64, 545–562. [Google Scholar] [CrossRef]
- Allen, J.F. Why chloroplasts and mitochondria contain genomes. Comp. Funct. Genom. 2003, 4, 31–36. [Google Scholar] [CrossRef] [Green Version]
- McNeal, J.R.; Leebens-Mack, J.H.; Arumuganathan, K.; Kuehl, J.V.; Boore, J.L.; De Pamphilis, C.W. Using partial genomic fosmid libraries for sequencing complete organellar genomes. Biotechniques 2006, 41, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Wicke, S.; Schneeweiss, G.M.; De Pamphilis, C.W.; Muller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.J.; Du, L.W.; Liu, A.; Chen, J.J.; Wu, L.; Hu, W.M.; Zhang, W.; Kim, K.; Lee, S.C.; Yang, T.J.; et al. The Complete Chloroplast Genome Sequences of Five Epimedium Species: Lights into Phylogenetic and Taxonomic Analyses. Front. Plant Sci. 2016, 7, 306. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.S.; Li, P.; Qiu, Y.X. The Complete Chloroplast Genomes of Three Cardiocrinum (Liliaceae) Species: Comparative Genomic and Phylogenetic Analyses. Front. Plant Sci. 2017, 7, 2054. [Google Scholar] [CrossRef]
- Roy, N.S.; Jeong, U.; Na, M.; Choi, I.Y.; Cheong, E.J. Genomic analysis and a consensus chloroplast genome sequence of Prunus yedoensis for DNA marker development. Hortic. Environ. Biotechnol. 2020, 61, 859–867. [Google Scholar] [CrossRef]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.Y.; Gao, L.Z. Thirteen Camellia chloroplast tgenome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 151. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.L.; Cheng, L.L.; Huang, W.G.; Cao, Q.C.; Zhou, L.; Jia, W.S.; Lan, Y.P. Chloroplast genomes of seven species of Coryloideae (Betulaceae): Structures and comparative analysis. Genome 2020, 63, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Raman, G.; Park, K.T.; Kim, J.; Park, S. Characteristics of the completed chloroplast genome sequence of Xanthium spinosum: Comparative analyses, identification of mutational hotspots and phylogenetic implications. BMC Genom. 2020, 21, 855. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Y.; Li, C.J.; Yan, C.X.; Zhao, X.B.; Yuan, C.L.; Sun, Q.X.; Shi, C.R.; Shan, S.H. Twelve complete chloroplast genomes of wild peanuts: Great genetic resources and a better understanding of Arachis phylogeny. BMC Plant Biol. 2019, 19, 504. [Google Scholar] [CrossRef] [PubMed]
- Saina, J.K.; Gichira, A.W.; Li, Z.Z.; Hu, W.G.; Wang, Q.F.; Liao, K. The complete chloroplast genome sequence of Dodonaeaviscosa: Comparative and phylogenetic analyses. Genetica 2018, 146, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.B.; Xia, B.S.; Li, X.W. The complete chloroplast genome sequences of five pinnate-leaved Primula species and phylogenetic Analyses. Sci. Rep. 2020, 10, 20782. [Google Scholar] [CrossRef] [PubMed]
- Li, J.L.; Wang, S.; Yu, J.; Wang, L.; Zhou, S.L. A modified CTAB protocol for plant DNA extraction. Chin. Bull. Bot. 2013, 48, 72–78. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Brozynska, M.; Furtado, A.; Henry, R.J. Direct chloroplast sequencing: Comparison of sequencing platforms and analysis tools for whole chloroplast barcoding. PLoS ONE 2014, 9, e110387. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.I.; Cronk, Q. Plann: A command-line application for annotating plastome sequences. Appl. Plant Sci. 2015, 3, 1500026. [Google Scholar] [CrossRef] [Green Version]
- Conant, G.C.; Wolfe, K.H. GenomeVx: Simple web-based creation of editable circular chromosome maps. Bioinformatics 2008, 24, 861–862. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Albert, F.M.; Juan Carlos, S.D.; Sara, G.R.; Pablo, L.; Ramos-Onsins, S.E.; Alejandro, S.G. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, 273–279. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Oliver, M.J.; Murdock, A.G.; Mishler, B.D.; Kuehl, J.V.; Boore, J.L.; Mandoli, D.F.; Everett, K.D.; Wolf, P.G.; Duffy, A.M.; Karol, K.G. Chloroplast genome sequence of the moss Tortularuralis: Gene content, polymorphism, and structural arrangement relative to other green plant chloroplast genomes. BMC Genom. 2010, 11, 143. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Leebens-Mack, J.; Muller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.Q.; Liu, Y.L.; Yang, Y.; Xie, X.M.; Lu, Y.Z.; Yang, Z.R.; Jin, X.B.; Dong, W.P.; Suo, Z.L. Interspecific chloroplast genome sequence diversity and genomic resources in Diospyros. BMC Plant Biol. 2018, 18, 210. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Dong, W.P.; Li, W.Q.; Lu, Y.Z.; Xie, X.M.; Jin, X.B.; Shi, J.P.; He, K.H.; Suo, Z.L. Comparative analysis of six Lagerstroemia complete chloroplast genomes. Front. Plant Sci. 2017, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Dong, W.P.; Liu, B.; Xu, C.; Yao, X.; Gao, J.; Corlett, R.T. Comparative analysis of complete chloroplast genome sequences of two tropical trees Machilusyunnanensis and Machilusbalansae in the family Lauraceae. Front. Plant Sci. 2015, 6, 662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.H.; Wang, S.; Liu, Y.L.; Yuan, Q.J.; Sun, J.H.; Guo, L.P. Chloroplast genome variation and phylogenetic relationships of Atractylodes species. BMC Genom. 2021, 22, 103. [Google Scholar]
- Perry, A.S.; Wolfe, K.H. Nucleotide substitution rates in legume chloroplast DNA depend on the presence of the inverted repeat. J. Mol. Evol. 2002, 55, 501–508. [Google Scholar] [CrossRef]
- Zhu, A.D.; Guo, W.H.; Gupta, S.; Fan, W.S.; Mower, J.P. Evolutionary dynamics of the plastid inverted repeat: The effects of expansion, contraction, and loss on substitution rates. New Phytol. 2016, 209, 1747–1756. [Google Scholar] [CrossRef] [Green Version]
- Li, F.W.; Kuo, L.Y.; Pryer, K.M.; Rothfels, C.J. Genes translocated into the plastid inverted repeat show decelerated substitution rates and elevated GC content. Genome Biol. Evol. 2016, 8, 2452–2458. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.S.; Chaw, S.M. Evolutionary stasis in cycad plastomes and the first case of plastome GC-biased gene conversion. Genome Biol. Evol. 2015, 7, 2000–2009. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean ginseng (Panaxschinseng Nees) and comparative analysis of sequence evolution among17vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar]
- Asaf, S.; Waqas, M.; Khan, A.L.; Khan, M.A.; Kang, S.M.; Imran, Q.M.; Shahzad, R.; Bilal, S.; Yun, B.W.; Lee, I.J. The complete chloroplast genome of wild rice (Oryza minuta) and its comparison to related species. Front. Plant Sci. 2017, 8, 304. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.S.; Park, T.H. Complete chloroplast genome sequence of Solanum nigrum and development of markers for the discrimination of S. nigrum. Horticult. Environ. Biotechnol. 2016, 57, 69–78. [Google Scholar] [CrossRef]
- Hu, Y.; Woeste, K.E.; Zhao, P. Completion of the chloroplast genomes of five chinesejuglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 2016, 7, 1955. [Google Scholar]
- Asaf, S.; Khan, A.L.; Khan, A.; Khan, G.; Lee, I.J.; Al-Harrasi, A. Expanded inverted repeat region with large scale inversion in the first complete plastid genome sequence of Plantago ovata. Sci. Rep. 2020, 10, 3881. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.Y.; Cheon, K.S.; Yoo, K.O.; Lee, H.O.; Cho, K.S.; Suh, J.T.; Kim, S.J.; Nam, J.H.; Sohn, H.B.; Kim, Y.H. Complete Chloroplast Genome Sequences and Comparative Analysis of Chenopodium quinoa and C. album. Front. Plant Sci. 2017, 8, 1696. [Google Scholar] [CrossRef]
- Nie, X.J.; Lv, S.Z.; Zhang, Y.X.; Du, X.H.; Wang, L.; Biradar, S.S.; Tan, X.F.; Wan, F.H.; Song, W.N. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratinaadenophora). PLoS ONE 2012, 7, e36869. [Google Scholar] [CrossRef] [Green Version]
- Xie, D.F.; Yu, Y.; Deng, Y.Q.; Li, J.; Liu, H.Y.; Zhou, S.D.; He, X.J. Comparative analysis of the chloroplast genomes of the Chinese endemic genus Urophysa and their contribution to chloroplast phylogeny and adaptive evolution. Int. J. Mol. Sci. 2018, 19, 1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.L.; Xiong, Y.; He, J.; Yu, Q.Q.; Zhao, J.M.; Lei, X.; Dong, Z.X.; Yang, J.; Peng, Y.; Zhang, X.Q.; et al. The Complete Chloroplast Genome of Two Important Annual Clover Species, Trifoliumalexandrinum and T. resupinatum: Genome Structure, Comparative Analyses and Phylogenetic Relationships with Relatives in Leguminosae. Plants 2020, 9, 478. [Google Scholar] [CrossRef] [Green Version]
- Tong, W.; Kim, T.S.; Park, Y.J. Rice chloroplast genome variation architecture and phylogenetic dissection in diverse Oryza species assessed by whole-genome resequencing. Rice 2016, 9, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdereau, A.; Klaas, M.; Barth, S.; Hodkinson, T.R. Plastid genome sequencing reveals biogeographical structure and extensive population genetic variation in wild populations of Phalarisarundinacea L. in north-western Europe. Glob. Chang. Biol. Bioenergy 2017, 9, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Borsch, T.; Quandt, D. Mutational dynamics and phylogenetic utility of noncoding chloroplast DNA. Plant Syst. Evol. 2009, 282, 169–199. [Google Scholar] [CrossRef]
- Piedra-Malagón, E.M.; Albarrán-Lara, A.L.; Rull, J.; Piero, D.; Sosa, V. Using multiple sources of characters to delimit species in the genus Crataegus (Rosaceae): The case of the Crataegusrosei complex. Syst. Biodivers. 2016, 14, 244–260. [Google Scholar] [CrossRef]
- Brown, J.A.; Beatty, G.E.; Finlay, C.; Montgomery, I.; Tosh, D.G.; Provan, J. Genetic analyses reveal high levels of seed and pollen flow in hawthorn (Crataegusmonogyna, Jacq.), a key component of hedgerows. Tree Genet. Genomes 2016, 12, 58. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.J.; Ding, C.H.; He, J.; Cheng, J.; Pei, L.Y.; Xie, L. Complete chloroplast genomes of Archiclematis, Naravelia and Clematis (Ranunculaceae), and their phylogenetic implications. Phytotaxa 2018, 343, 214–226. [Google Scholar] [CrossRef]
- Pauwels, M.; Vekemans, X.; Gode, C.; Frerot, H.; Castric, V.; Saumitou-Laprade, P. Nuclear and chloroplast DNA phylogeography reveals vicariance among European populations of the model species for the study of metal tolerance, Arabidopsis halleri (Brassicaceae). New Phytol. 2012, 193, 916–928. [Google Scholar] [CrossRef]
- Li, X.W.; Gao, H.H.; Wang, Y.T.; Song, J.Y.; Henry, R.; Wu, H.Z.; Hu, Z.G. Complete chloroplast genome sequence of Magnolia grandiflora and comparative analysis with related species. Sci. China Life Sci. 2013, 56, 189–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khakhlova, O.; Bock, R. Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 2006, 46, 85–94. [Google Scholar] [CrossRef]
- Dong, W.P.; Liu, J.; Yu, J.; Wang, L.; Zhou, S.L. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Tan, Y.H.; Liu, Y.Y.; Song, Y.; Yang, J.B.; Corlett, R.T. Chloroplast genome structure in Ilex (Aquifoliaceae). Sci. Rep. 2016, 6, 28559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Zhao, T.T.; Ma, Q.H.; Liang, L.S.; Wang, G.X. Comparative genomics and phylogenetic analysis revealed the chloroplast genome variation and interspecific relationships of Corylus (Betulaceae) species. Front. Plant Sci. 2018, 9, 927. [Google Scholar] [CrossRef] [PubMed]
- Fehrer, J.; Gemeinholzer, B.; Chrtek, J.; Bräutigam, S. Incongruent plastid and nuclear DNA phylogenies reval ancient intergeneric hybridization in Pilosellahawkweeda (Hieracium, Cichorieae, Asteraceae). Mol. Phylogenet. Evol. 2007, 42, 347–361. [Google Scholar] [CrossRef]
- Du, F.K.; Peng, X.L.; Liu, J.Q.; Lascoux, M.; Hu, F.S.; Petit, R.J. Direction and extent of organelle DNA introgression between two spruce species in the Qinghai-Tibetan Plateau. New Phytol. 2011, 192, 1024–1033. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxon | Identification Code | Biogeographic Region | Collection Site |
|---|---|---|---|
| C. bretschneideri | ZF1H | Northeast, China | Shenyang |
| C. pinnatifida | CZSLH | Northeast, China | Shenyang |
| C. pinnatifida var. major | JD1H | North, China | Shenyang |
| C. maximowiczii | MSZ1H | Northeast, China | Shenyang |
| Characteristics | C. bretschneideri | C. pinnatifida | C. pinnatifida var. Major | C. maximowiczii |
|---|---|---|---|---|
| Total size(bp) | 159,607 | 159,656 | 159,676 | 159,875 |
| LSC length (bp) | 87,601 | 87,749 | 87,744 | 87,874 |
| SSC length (bp) | 19,312 | 19,139 | 19,164 | 19,233 |
| IR length (bp) | 26,347 | 26,384 | 26,384 | 26,384 |
| Overall GC content(%) | 36.6% | 36.7% | 36.6% | 36.6% |
| GC in LSC (%) | 34.4% | 34.4% | 34.4% | 34.3% |
| GC in IR (%) | 42.7% | 42.6% | 42.6% | 42.6% |
| GC in SSC (%) | 30.3% | 30.6% | 30.5% | 30.4% |
| Total number of genes | 113 | 113 | 113 | 113 |
| Protein genes | 79 | 79 | 79 | 79 |
| rRNA genes | 30 | 30 | 30 | 30 |
| tRNA genes | 4 | 4 | 4 | 4 |
| Duplicated genes | 19 | 19 | 19 | 19 |
| Accession number | MW963339 | MZ494514 | MZ494513 | MZ494512 |
| Gene Category | Gene Group | Names of Gene |
|---|---|---|
| Photosynthetic | Subunit of rubisco | rbcL |
| Photosystem I | psaA, psaB, psaC, psaI, psaJ | |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Subunit of synthase | atpA, atpB, atpE, atpF *, atpH, atpI | |
| Cytochromecompelx | petA, petB *, petD *, petG, petL, petN | |
| Subunits of NADPH dehydrogenase | ndhA *, ndhB *, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Self-replication | Transfer RNA | trnA-UGC *, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-UCC, trnG-GCC *, trnH-GUG, trnI-CAU, trnI-GAU *, trnK-UUU *, trnL-CAA, trnL-UAA *, trnL-UAG, trnfM-CAUI, trnM-CAU, trnN-GUU, trnP-UGG, trnQ-UUG, trnR-ACG, trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC, trnV-UAC *, trnW-CCA, trnY-GUA |
| Ribosomal RNA | rrn5, rrn4.5, rrn16, rrn23 | |
| Proteins of large ribosomal subunit | rpl2 *, rpl14, rpl16 *, rpl20, rpl22, rpl23, rpl32, rpl33, rpl36 | |
| Proteins of small ribosomal subunit | rps2, rps3, rps4, rps7, rps8, rps11, rps12 *, rps14, rps15, rps16, rps18, rps19, | |
| RNA polymerase | rpoA, rpoB, rpoC1, rpoC2 | |
| Biosynthesis | Maturase | matK |
| Carbon metabolism | cemA | |
| Protease | clpP * | |
| Fatty acid synthesis | accD | |
| Cytochrome synthesis gene | ccsA | |
| Translation initiation factor | infA | |
| Unknown function | Conserved open reading frames | ycf1, ycf2, ycf3 *, ycf4 |
| Length | Variable Sites | Parsimony-Informative Sites | Nucleotide Diversity | |||
|---|---|---|---|---|---|---|
| (bp) | Number | % | Number | % | ||
| LSC region | 88,705 | 316 | 0.3562 | 228 | 0.257 | 0.0022 |
| IR | 26,383 | 22 | 0.0834 | 16 | 0.0606 | 0.0003 |
| SSC region | 19,435 | 107 | 0.5506 | 87 | 0.4476 | 0.0036 |
| Total | 160,906 | 445 | 0.2766 | 331 | 0.2057 | 0.0017 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, G.; Wang, Y.; Wang, Y.; Zheng, S.; Dong, W.; Dong, N. New Insight into the Phylogeny and Taxonomy of Cultivated and Related Species of Crataegus in China, Based on Complete Chloroplast Genome Sequencing. Horticulturae 2021, 7, 301. https://doi.org/10.3390/horticulturae7090301
Hu G, Wang Y, Wang Y, Zheng S, Dong W, Dong N. New Insight into the Phylogeny and Taxonomy of Cultivated and Related Species of Crataegus in China, Based on Complete Chloroplast Genome Sequencing. Horticulturae. 2021; 7(9):301. https://doi.org/10.3390/horticulturae7090301
Chicago/Turabian StyleHu, Guanglong, Yiheng Wang, Yan Wang, Shuqi Zheng, Wenxuan Dong, and Ningguang Dong. 2021. "New Insight into the Phylogeny and Taxonomy of Cultivated and Related Species of Crataegus in China, Based on Complete Chloroplast Genome Sequencing" Horticulturae 7, no. 9: 301. https://doi.org/10.3390/horticulturae7090301
APA StyleHu, G., Wang, Y., Wang, Y., Zheng, S., Dong, W., & Dong, N. (2021). New Insight into the Phylogeny and Taxonomy of Cultivated and Related Species of Crataegus in China, Based on Complete Chloroplast Genome Sequencing. Horticulturae, 7(9), 301. https://doi.org/10.3390/horticulturae7090301