
Comparative Transcriptomic Analysis of Differentially Expressed Transcripts Associated with Flowering Time of Loquat (Eriobotya japonica Lindl.)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Endogenous Phytohormone Determination by LC-MS
2.3. Total RNA Extraction and RNA-Seq
2.4. Quality Control and de Novo Assembly
2.5. Functional Annotation
2.6. Identification of Differentially Expressed Transcripts (DETs) Involved in Flowering Time
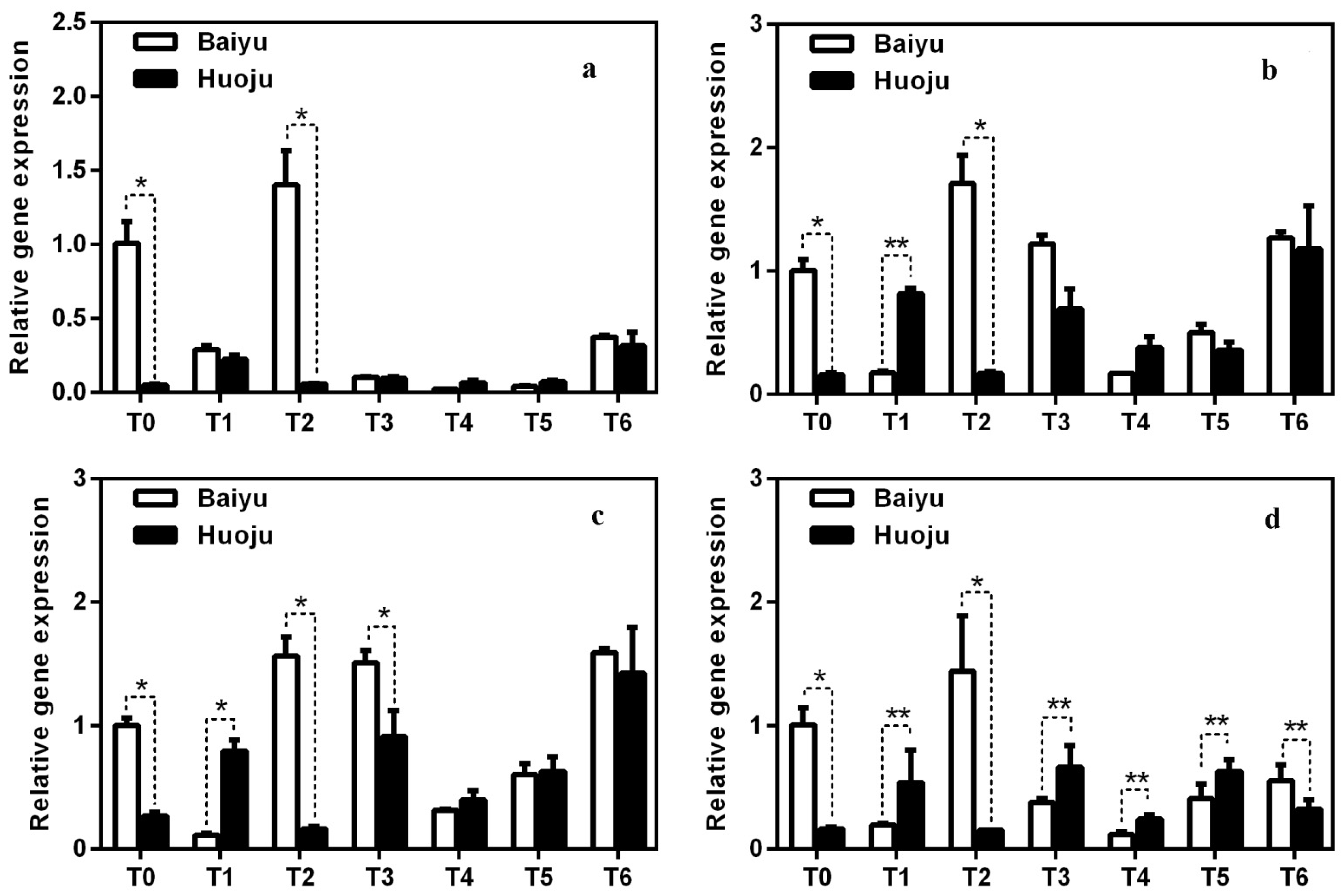
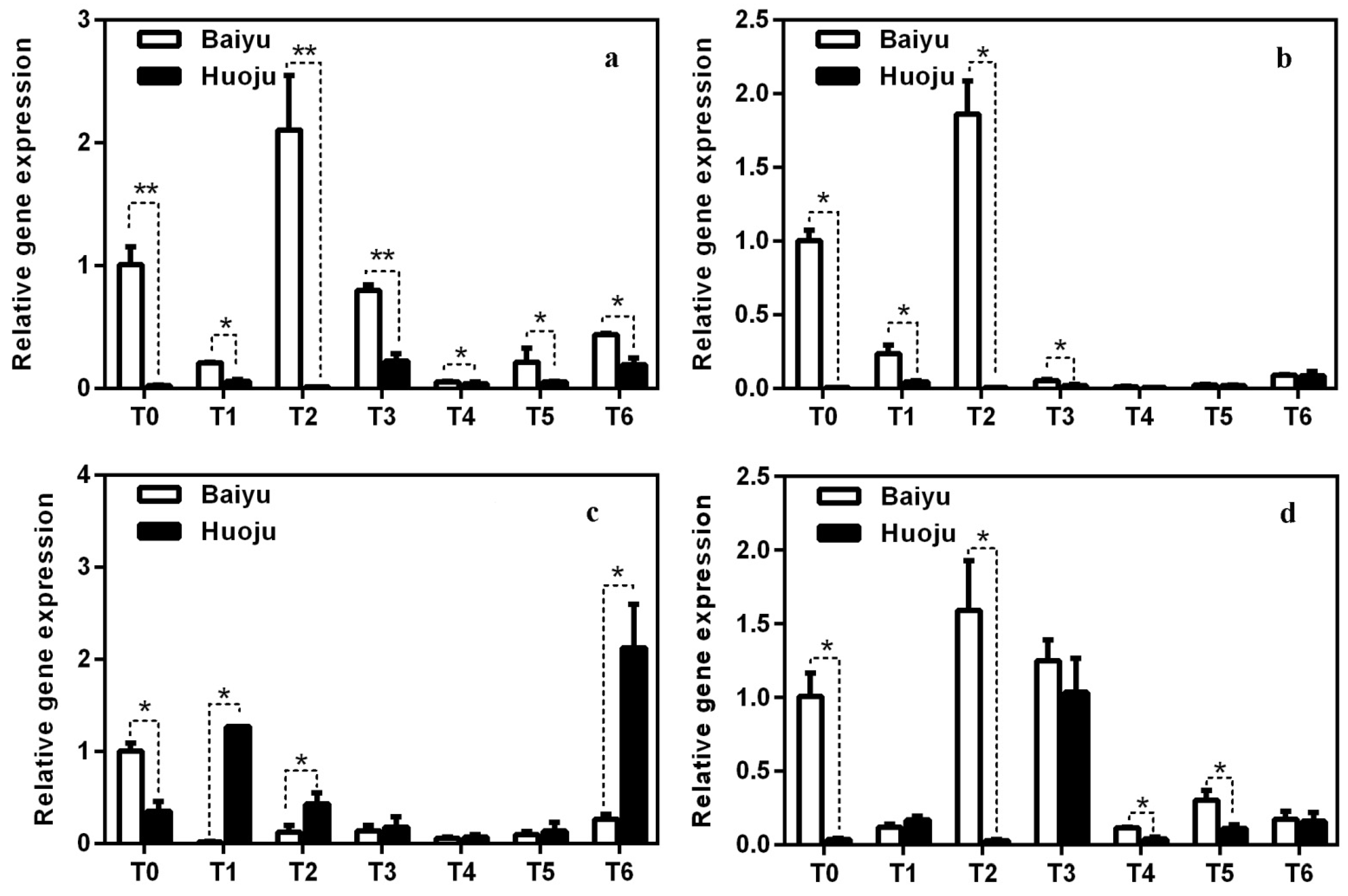
2.7. Gene Expression Analysis by qRT-PCR
2.8. Statistical Analysis
3. Results
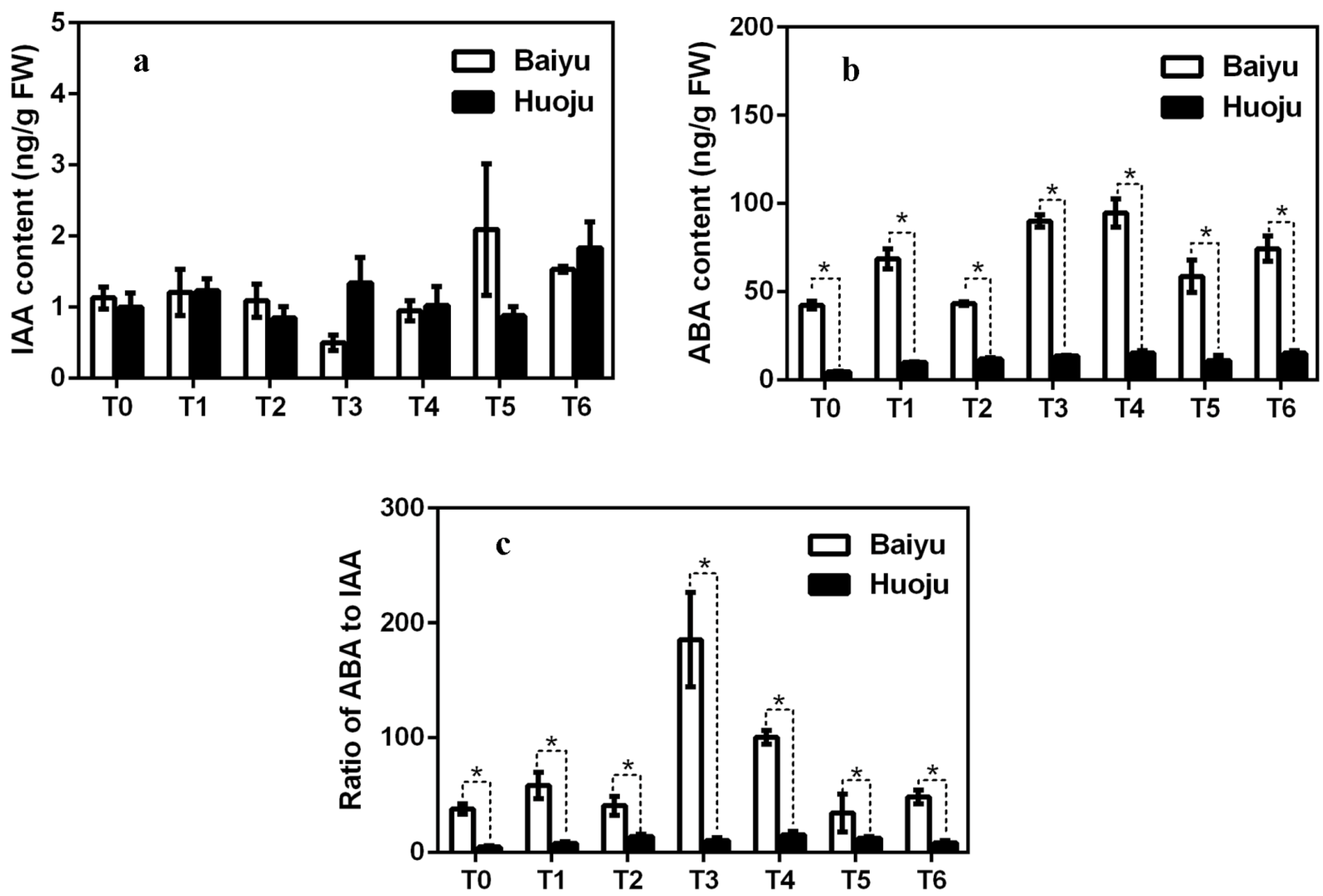
3.1. Loquat Apical Bud Phytohormone Analyses
3.2. Sequence Analysis and Assembly
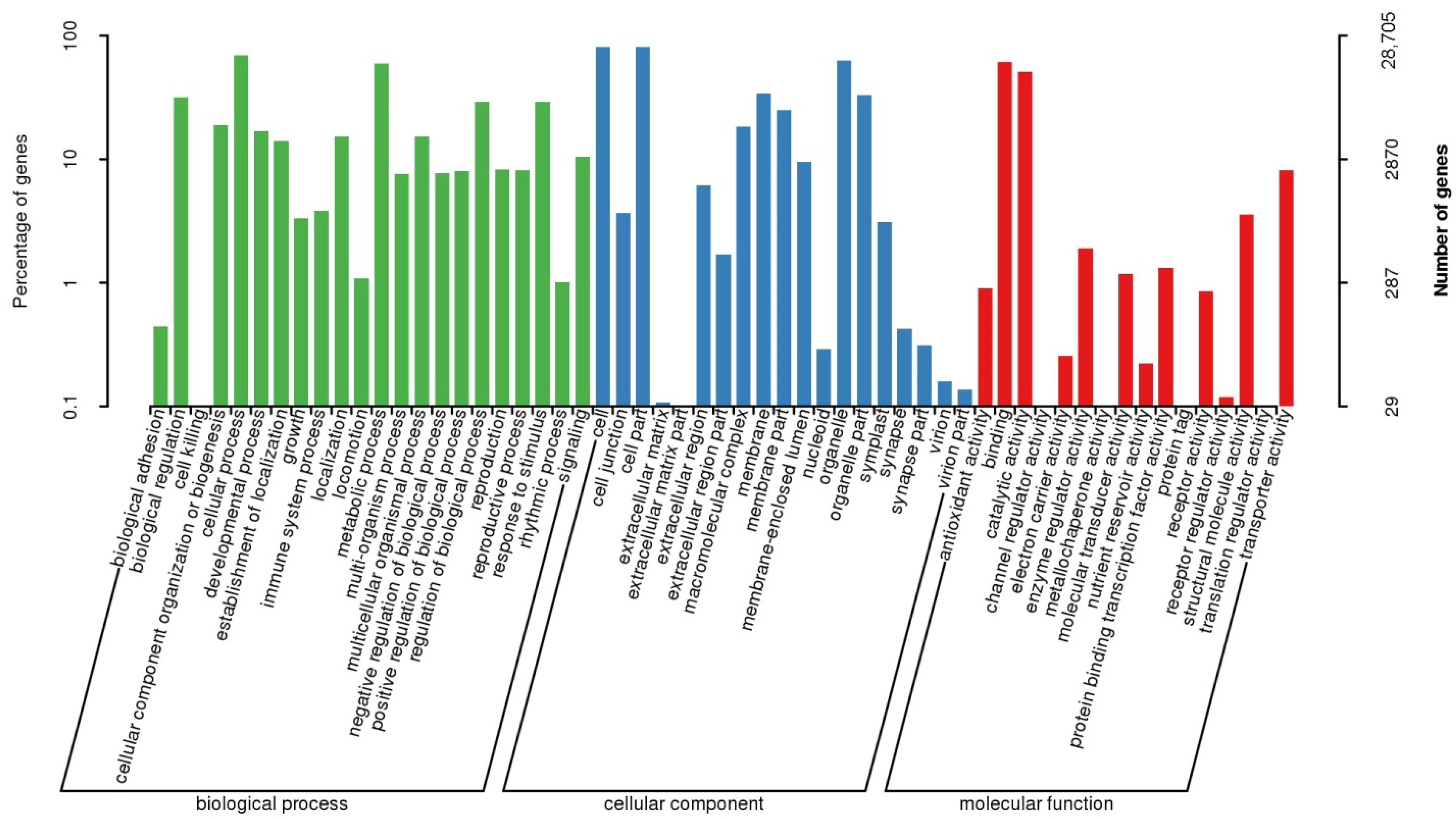
3.3. Sequence Annotation
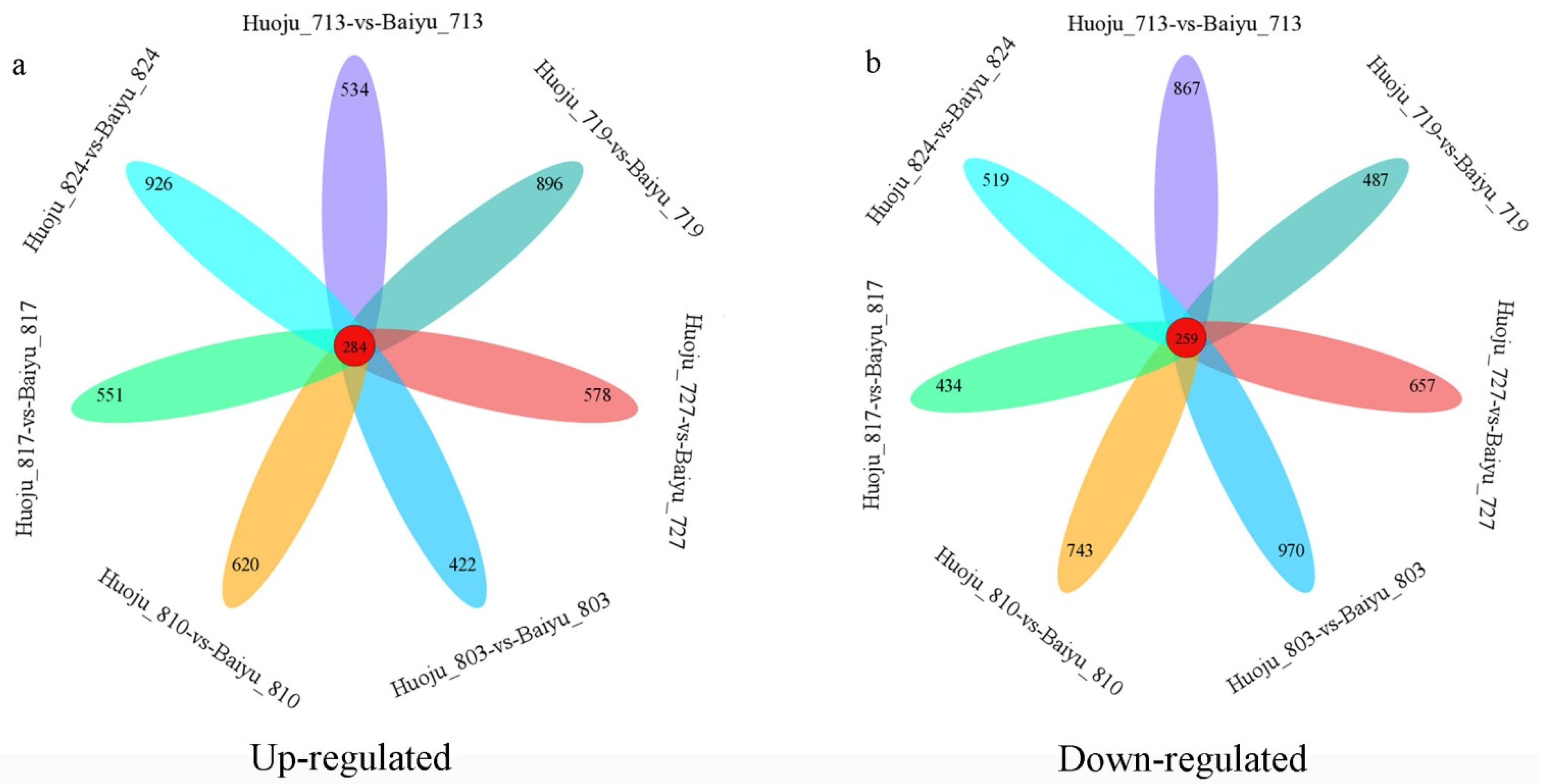
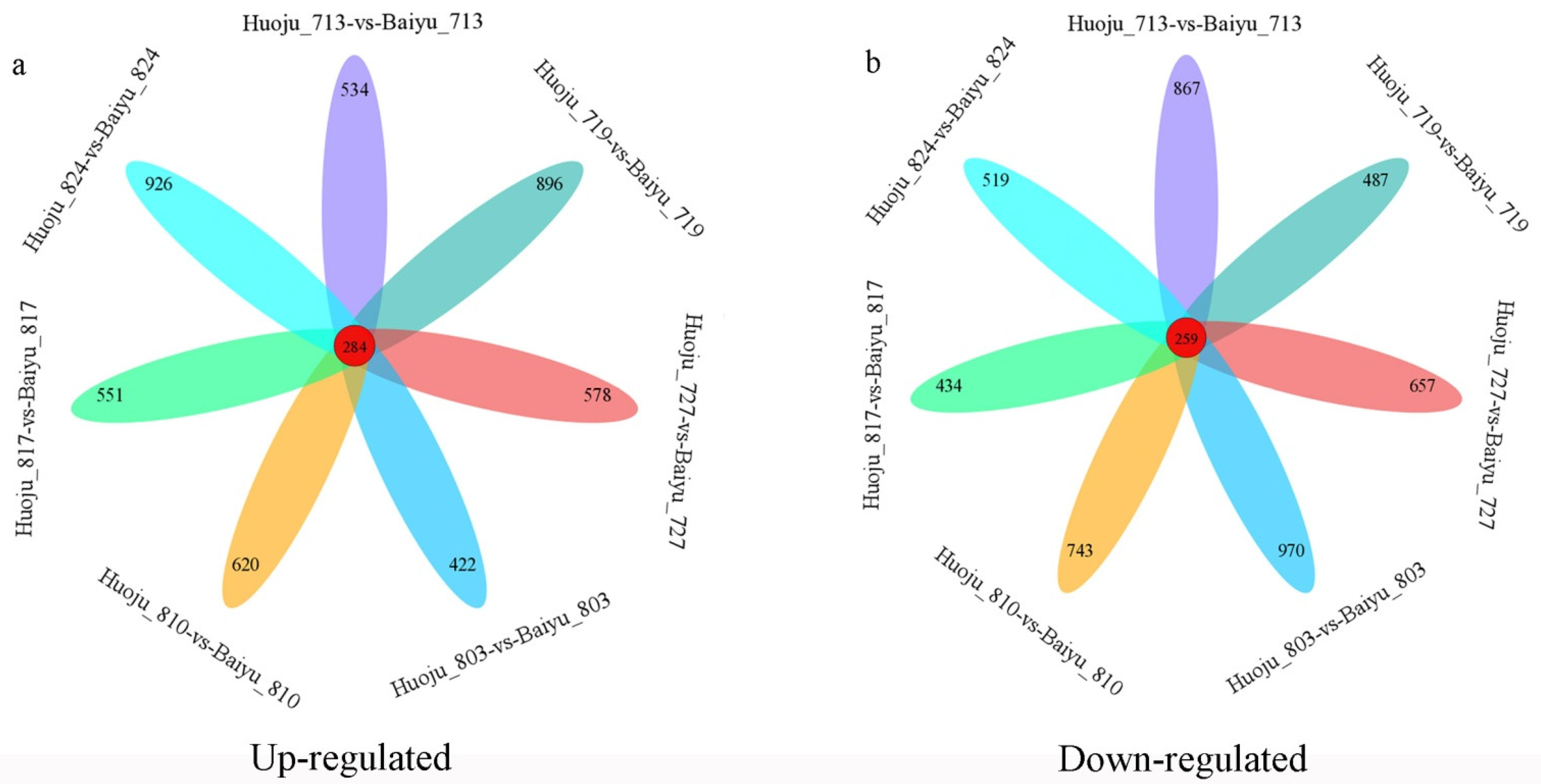
3.4. Loquat DET Identification
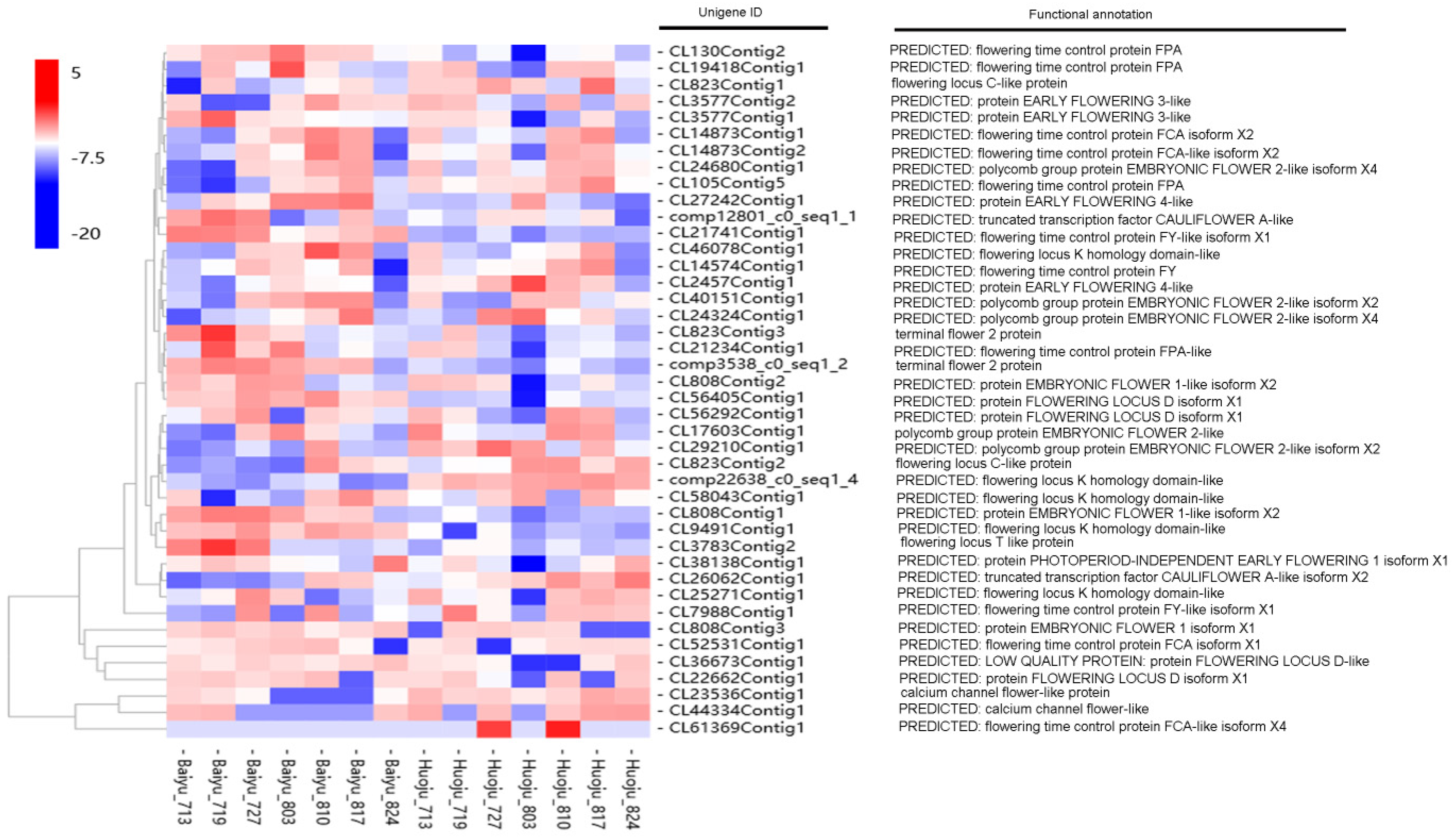
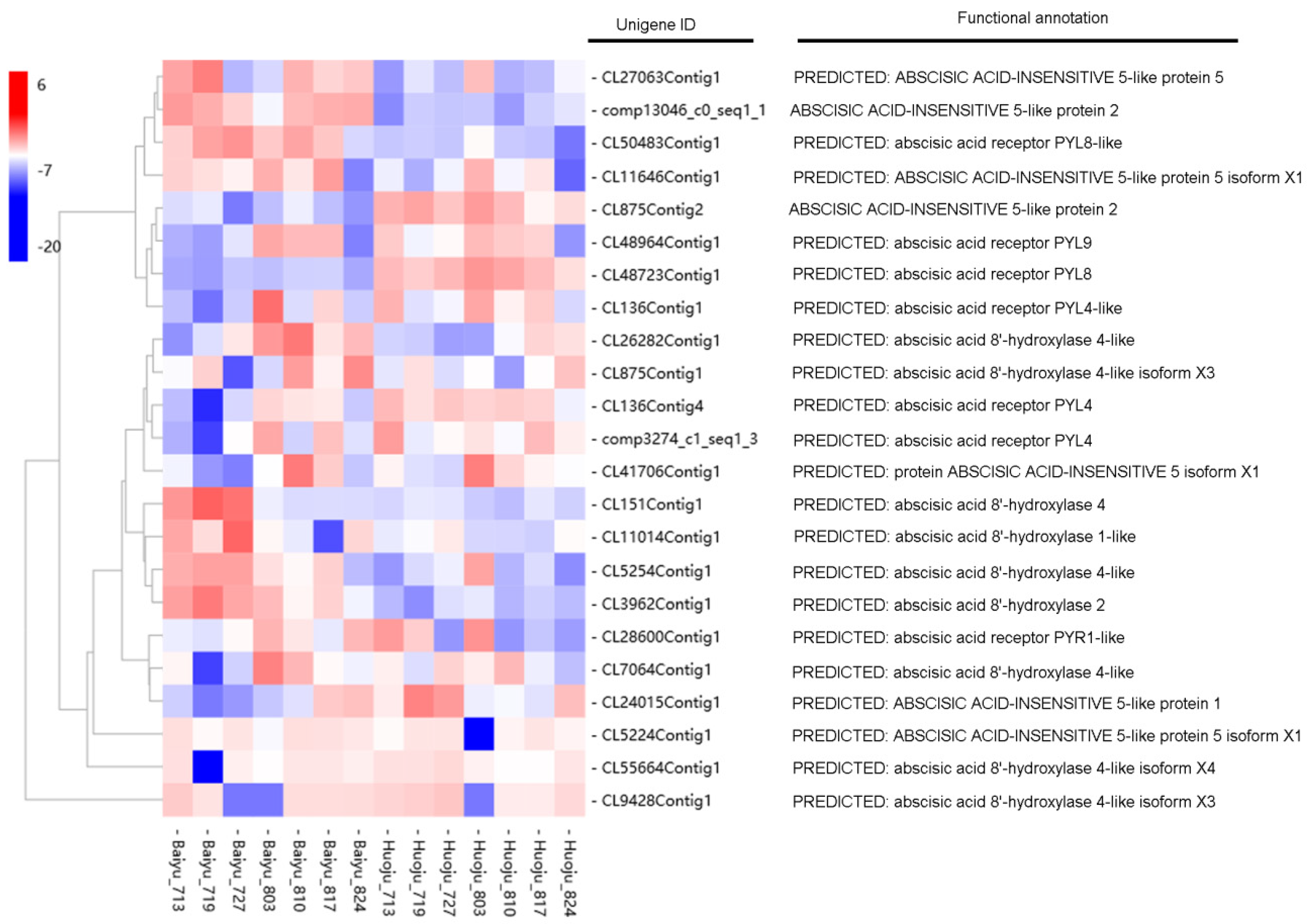
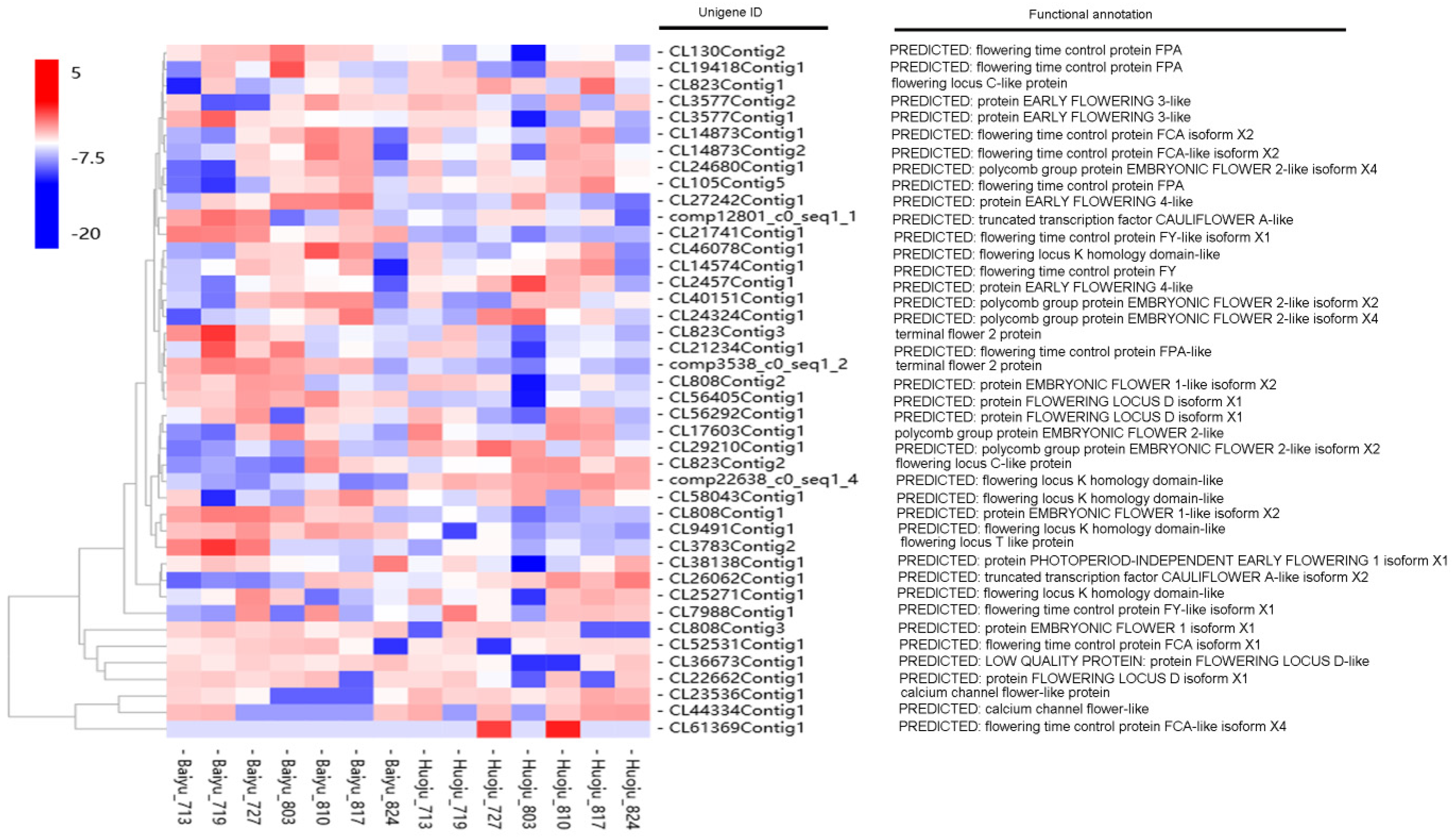
3.5. Identification of DETs Involved in Flowering Time
3.6. Identification of DETs Involved in ABA Signalling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, W.; Gu, R.; Che, G.; Cheng, Z.; Zhang, X. CsTFL1b may regulate the flowering time and inflorescence architecture in cucumber (Cucumis sativus L.). Biochem. Biophys. Res. Commun. 2018, 499, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Yano, M.; Kojima, S.; Takahashi, Y.; Lin, H.; Sasaki, T. Genetic control of flowering in rice, a short-day Plant. Plant Physiol. 2001, 127, 1425–1429. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Ai, X.Y.; Sun, L.M.; Zhang, D.L.; Guo, W.W.; Deng, X.X.; Hu, C.G. Transcriptome profile analysis of flowering molecular processes of early flowering trifoliate orange mutant and the wild type [Poncirus trifoliata (L.) Raf.] by massively parallel signature sequencing. BMC Genom. 2011, 12, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amasino, R. Seasonal and developmental timing of flowering. Plant J. 2010, 61, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, A.; Schmid, M. Regulation of flowering time: All roads leads to Rome. Cell Mol. Life Sci. 2011, 68, 2013–2037. [Google Scholar] [CrossRef] [PubMed]
- Andres, F.; Coupland, G. The genetic basis of flowering responses to seasonal cues. Nat. Rev. Genet. 2012, 13, 627. [Google Scholar] [CrossRef]
- Wils, C.R.; Kaufmann, K. Gene-regulatory networks controlling inflorescence and flower development in Arabidopsis thaliana. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 95–105. [Google Scholar] [CrossRef]
- Fornara, F.; de Montaigu, A.; Coupland, G. SnapShot: Control of flowering in Arabidopsis. Cell 2010, 141, 550.e1. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Lin, H.; Dubcovsky, J. Factorial combinations of protein interactions generate a multiplicity of florigen activation complexes in wheat and barley. Plant J. 2015, 84, 70–82. [Google Scholar] [CrossRef] [Green Version]
- Hoenicka, H. Over-expression of an FT-homologous gene of apple induces early flowering in annual and perennial plants. Planta 2010, 223, 1309–1324. [Google Scholar]
- Foster, T.M.; Watson, A.E.; Hooijdonk, B.M.V.; Schaffer, R.J. Key flowering genes including FT-like genes are upregulated in the vasculature of apple dwarfing rootstocks. Tree Genet. Genom. 2014, 10, 189–202. [Google Scholar] [CrossRef]
- Ito, A.; Saito, T.; Nishijima, T.; Moriguchi, T. Effect of extending the photoperiod with low-intensity red or far-red light on the timing of shoot elongation and flower-bud formation of 1-year-old Japanese pear (Pyrus pyrifolia). Tree Physiol. 2014, 34, 534–546. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Jiang, P.; Thammannagoewda, S.; Liang, H.; Wilde, H.D. Characterization of peach TFL1 and comparison with FT/TFL1 gene families of Rosaceae. J. Am. Soc. Hort. Sci. 2013, 138, 12–17. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Song, H.; Liu, Z.; Hu, G.; Lin, S. Molecular characterization of loquat EjAP1 gene in relation to flowering. Plant Growth Regul. 2013, 70, 287–296. [Google Scholar] [CrossRef]
- Reig, C.; Gil-Munoz, F.; Vera-Sirera, F.; Garcia-Lorca, A.; Martinez-Fuentes, A.; Mesejo, C.; Perez-Amador, M.A.; Agusti, M. Bud sprouting and floral induction and expression of FT in loquat [Eriobotrya japonica (Thunb.) Lindl.]. Planta 2017, 246, 915–925. [Google Scholar] [CrossRef]
- Jing, D.; Chen, W.; Hu, R.; Zhang, Y.; Xia, Y.; Wang, S.; He, Q.; Guo, Q.; Liang, G. An integrative analysis of transcriptome, proteome and hormones reveals key differentially expressed genes and metabolic pathways involved in flower development in loquat. Int. J. Mol. Sci. 2020, 21, 5107. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhao, Y. A role for auxin in flower development. J. Integr. Plant Biol. 2007, 49, 99–104. [Google Scholar] [CrossRef]
- Matsoukas, I.G. Interplay between sugar and hormone signaling pathways modulate floral signal transduction. Front. Genet. 2014, 5, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, K.; Chen, Q.; Wu, Y.; Liu, R.; Zhang, H.; Wang, S.; Tang, S.; Yang, W.; Xie, Q. ABSCISIC-ACID-INSENSITIVE 4 negatively regulates flowering through directly promoting Arabidopsis FLOWERING LOCUS C transcription. J. Exp. Bot. 2016, 67, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Lin, S.; Chen, H. Time course changes of endogenous hormone levels during the floral and vegetative bud formation in loquat (Eriobotrya japonica Lindl.). Acta Horti. Sinica. 2007, 34, 339–344. (In Chinese) [Google Scholar]
- Fernandez, M.D.; Hueso, J.J.; Cuevas, J. Water stress integral for successful modification of flowering dates in ‘Algerie’ loquat. Irrig. Sci. 2010, 28, 127–134. [Google Scholar] [CrossRef]
- Lin, S.Q.; Sharpe, R.H.; Janick, J. Loquat: Botany and horticulture. Hortic. Rev. 1999, 23, 233–276. [Google Scholar]
- Demirkeser, T.H.; Caliskan, O.; Polat, A.A.; Ozgen, M.; Serce, S. Effect of natural lipid on pollen germination and pollen tube growth o loquat. Asian J. Plant Sci. 2007, 6, 304–307. [Google Scholar] [CrossRef]
- Jiang, S.; Luo, J.; Xu, F.; Zhang, X. Transcriptome analysis reveals candidate genes involved in gibberellins-induced fruit setting in tripod loquat (Eriobotrya japonica). Front. Plant Sci. 2016, 7, 1924. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Zhao, X.; Hu, W.; Wang, X.; Shen, T.; Yang, L. Comparative transcriptional analysis of loquat fruit identifies major signal networks involved in fruit development and ripening process. Int. J. Mol. Sci. 2016, 17, 1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, R.G.; Lai, J.; Liao, M.A.; Wang, Z.H.; Liang, G.L. Analysis of alterations to the transcriptome of loquat (Eriobotrya japonica Lindl.) under low temperature stress via de novo sequencing. Genet. Mol. Res. 2015, 14, 9423–9436. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Li, X.; Chen, J. Comparative transcriptome profiling of freezing stress responses in loquat (Eriobotrya japonica) fruitlets. J. Plant Res. 2017, 130, 893–907. [Google Scholar] [CrossRef] [PubMed]
- Niu, Q.; Zong, Y.; Qian, M.; Yang, F.; Teng, Y. Simultaneous quantitative determination of major plant hormones in pear flowers and fruit by UPLC/ESI-MS/MS. Anal. Method UK 2014, 6, 1766. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for IIlumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Trinity: Reconstructing a full-length transcriptome without genome from RNA_Seq data. Nat. Biotechnol. 2011, 2, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA_seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Robert, A.; Pachter, L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat. Methods 2013, 10, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression of RNA-Seq data at the gene level sequencing experiment. EMBL 2013, 10, f10000. [Google Scholar]
- Zhang, J.; Wu, K.; Zeng, S.; da Silva, J.A.T.; Zhao, X.; Tian, C.E.; Xia, H.; Duan, J. Transcriptome analysis of Cymbidium sinense and its application to the identification of genes associated with floral development. BMC Genom. 2013, 14, 279. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.; Guo, W.; Li, J.; Lin, H.; He, C.; Liu, Y.; Zhang, Q.; Liu, W. Comparative transcriptomic analysis of vernalization- and cytokinin-induced floral transition in Dendrobium nobile. Sci. Rep. 2017, 7, 45748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Zhang, Y.; Zhang, C.; Qi, F.; Li, X.; Mu, S.; Peng, Z. Characterization of the floral transcriptome of Moso bamboo (Phyllostachys edulis) at different flowering developmental stages by transcriptome sequencing and RNA-seq analysis. PLoS ONE 2014, 9, e98910. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, D.; Zhang, X.; Xing, L.; Fan, S.; Ma, J.; Zhao, C.; Du, L.; Han, M. A transcriptome analysis of two apple (Malus a domestica) cultivars with different flowering abilities reveals a gene network module associated with floral transitions. Sci. Hortic. 2018, 239, 269–281. [Google Scholar] [CrossRef]
- Liu, H.; Sun, M.; Du, D.; Pan, H.; Cheng, T.; Wang, J. Whole-transcriptome analysis of differentially expressed genes in the vegetative buds, floral buds, and buds of Chrysanthemum morifolium. PLoS ONE 2015, 10, e0128009. [Google Scholar] [CrossRef]
- Ren, L.; Liu, T.; Cheng, Y.; Sun, J.; Gao, J.; Dong, B.; Chen, S.; Chen, F.; Jiang, J. Transcriptomic analysis of differentially expressed genes in the floral transition of the summer flowering chrysanthemum. BMC Genom. 2016, 17, 673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, P.; Dong, B.; Wang, H.; Cao, P.; Liu, T.; Liu, Y.; Gao, J.; Liao, Y.; Fang, W.; Chen, S.; et al. A transcriptomic analysis targeting genes involved in the floral transition of winter-flowering chrysanthemum. J. Plant Growth Regul. 2018, 37, 220–232. [Google Scholar] [CrossRef]
- Jiang, Y.; Peng, J.; Zhu, Y.; Su, W.; Zhang, L.; Jing, Y.; Lin, S.; Gao, Y. The role of EjSOC1s in flower initiation in Eriobotrya japonica. Front. Plant Sci. 2019, 10, 253. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Jiang, Y.; Zhu, Y.; Su, W.; Long, T.; Huang, T.; Peng, J.; Yu, H.; Lin, S.; Gao, Y. Functional characterization of GI and CO homologs from Eriobotrya deflexa Nakai forma koshunensis. Plant Cell Rep. 2019, 38, 533–543. [Google Scholar] [CrossRef]
- Michaels, S.D.; Amasino, R.M. Loss of FLOWERING LOCUS C activity eliminates the late-flowering phenotype of FRIGIDA and autonomous pathway mutations but not responsiveness to vernalization. Plant Cell 2001, 13, 934–941. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhao, Q.; Meng, N.; Song, H.; Li, C.; Hu, G.; Wu, J.; Lin, S.; Zhang, Z. Over-expression of EjLFY-1 leads to an early flowering habit in strawberry (Fragaria × ananassa) and its asexual progeny. Front. Plant Sci. 2017, 8, 496. [Google Scholar] [PubMed] [Green Version]
- Mouradov, A.; Cremer, F.; Coupland, G. Control of flowering time: Interacting pathways as a basis for diversity. Plant Cell 2002, 14, S111–S130. [Google Scholar] [CrossRef] [Green Version]
- Campos-Rivero, G.; Osorio-Montalvo, P.; Sanchez-Borges, R.; Us-Camas, R.; Duarte-Ake, F.; De-la-Pena, C. Plant hormone signaling in flowering: An epigenetic point of view. J. Plant Physiol. 2017, 214, 16–27. [Google Scholar] [CrossRef]
- Domagalska, M.A.; Sarnowska, E.; Nagy, F.; Davis, S.J. Genetic analysis of interactions among gibberellins, abscisic acid and brassinosteroids in the control of flowering time in Arabidopsis thaliana. PLoS ONE 2010, 5, e14012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankowski, K.; Wilmowicz, E.; Kucko, A.; Kesy, J.; Swiezawska, B.; Kopcewicz, J. Ethylene, auxin, and abscisic acid interactions in the control of photoperiodic flower induction in Pharbitis nil. Biol. Plantar. 2014, 58, 305–310. [Google Scholar] [CrossRef]
- Brocard, I.M.; Lynch, T.J.; Finkelstein, R.R. Regulation and role of the Arabidopsis abscisic acid-insensitive 5 in abscisic acid, sugar, and stress response. Plant Physiol. 2002, 129, 1533–1543. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, L.; Ye, T.; Chen, X.; Wu, Y. The inhibitory effect of ABA on floral transition is mediated by ABI5 in Arabidopsis. J. Exp. Bot. 2013, 64, 675–684. [Google Scholar] [CrossRef]
- Sherrard, M.E.; Maherali, H. The adaptive significance of drought escape in Avena barbata, an annual grass. Evolution 2006, 60, 2478–2489. [Google Scholar] [CrossRef]
- Su, Z.; Ma, X.; Guo, H.; Sukiran, N.L.; Guo, B.; Assmann, S.M.; Ma, H. Flower development under drought stress: Morphological and transcriptomic analyses reveal acute responses and long-term acclimation in Arabidopsis. Plant Cell 2013, 25, 3785–3807. [Google Scholar] [CrossRef] [Green Version]
- Tsao, T.H.; Wang, H.W.; Jiao, J.P.; Tan, Z.Y. Changes in endogenous ABA and GA contents during floral induction of Lemna aequinoctialis. Act. Botan. Neeriand. 1986, 35, 443–448. [Google Scholar] [CrossRef]
- Leng, P.; Zhang, Y.; Du, Y.; Wang, J.; Jiang, L.; Kai, W.; Liang, B.; Zhai, X.; Sun, Y.; Liu, H.; et al. Expression pattern of ABA metabolic and signaling genes during floral development and fruit set in sweet cherry. Plant Growth Regul. 2018, 84, 71–80. [Google Scholar] [CrossRef]
- Takeno, K. Stress-induced flowering: The third category of flowering response. J. Exp. Bot. 2016, 67, 4925–4934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riboni, M.; Galbiati, M.; Tonelli, C.; Conti, L. GIGANTEA enables drought escape response via ABA-dependent activation of the florigens and SUPPERSSOR OF OVEREXPRESSION OF CONSTANS1. Plant Physiol. 2013, 162, 1706–1719. [Google Scholar] [CrossRef] [Green Version]
- Riboni, M.; Test, A.R.; Galbiati, M.; Tonelli, C.; Conti, L. Environmental stress and flowering time. Plant Signal. Behav. 2014, 9, e29036. [Google Scholar] [CrossRef] [Green Version]
- Southwick, S.M.; Davenport, T.L. Characterization of water stress and low temperature effects on flower induction in Citrus. Plant Physiol. 1986, 81, 26–29. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.G.; Kim, S.Y.; Park, C.M. A membrane-associated NAC transcription factor regulates salt-responsive flowering via FLOWERING LOCUS T in Arabidopsis. Planta 2007, 226, 647–654. [Google Scholar] [CrossRef]
- Ryu, J.Y.; Lee, H.J.; Seo, P.J.; Jung, J.H.; Ahn, J.H.; Park, C.M. The Arabidopsis floral repressor BFT delays flowering by competing with FT for FD binding under high salinity. Mol. Plant 2013, 7, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Wang, Y.; Han, C.; Zhang, W.; Jia, H.; Li, X. GA signaling and CO/FT regulatory module mediate salt-induced late flowering in Arabidopsis thaliana. Plant Growth Regul. 2007, 53, 195–206. [Google Scholar] [CrossRef]
- Kohli, A.; Sreenivasulu, N.; Lakshmanan, P.; Kumar, P.P. The phytohormone crosstalk paradigm takes center stage in understanding how plants respond to abiotic stresses. Plant Cell Rep. 2013, 32, 945–957. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cultivar | Sampling Date | Raw Reads | Clean Reads | Q30 (%) | GC (%) |
|---|---|---|---|---|---|
| ‘Baiyu’ | T0 | 48,991,094 | 46,581,732 | 93.30 | 47.44 |
| T1 | 46,842,090 | 44,358,410 | 93.02 | 47.14 | |
| T2 | 49,193,824 | 46,596,186 | 93.12 | 47.02 | |
| T3 | 47,387,894 | 40,851,014 | 87.95 | 47.36 | |
| T4 | 49,068,496 | 46,912,890 | 93.54 | 47.02 | |
| T5 | 48,987,770 | 46,583,754 | 93.22 | 47.22 | |
| T6 | 48,997,596 | 46,632,968 | 93.14 | 46.92 | |
| ‘Huoju’ | T0 | 49,150,924 | 47,013,524 | 93.70 | 46.93 |
| T1 | 49,784,092 | 47,560,900 | 93.54 | 46.99 | |
| T2 | 49,889,156 | 47,546,378 | 93.35 | 47.13 | |
| T3 | 49,605,682 | 47,133,928 | 93.29 | 47.57 | |
| T4 | 48,971,664 | 46,264,188 | 93.00 | 47.23 | |
| T5 | 49,348,500 | 46,926,020 | 93.17 | 47.14 | |
| T6 | 49,333,148 | 47,048,570 | 93.23 | 46.71 |
| Term | ≥300 bp | ≥500 bp | ≥1000 bp | Total Number | Total Length (nt) | Average Length | N50 |
|---|---|---|---|---|---|---|---|
| Unigene | 78,599 | 55,189 | 28,292 | 162,080 | 87,192,678 | 1109.34 | 1625 |
| Anno_Database | Annotated Number | 300 ≤ Length < 1000 | Length ≥ 1000 |
|---|---|---|---|
| NR | 45,107 (57.39%) | 20,034 (25.49%) | 25,073 (31.90%) |
| Swissport | 31,795 (40.45%) | 11,273 (14.34%) | 20,522 (26.11%) |
| KEGG | 15,890 (20.22%) | 6774 (8.62%) | 9116 (11.60%) |
| KOG | 23,886 (30.39%) | 8853 (11.26%) | 15,033 (19.13) |
| eggNOG | 38,848 (49.43%) | 15,125 (19.24%) | 23,723 (30.18%) |
| GO | 28,705 (36.52%) | 10,314 (13.12%) | 18,391 (23.40%) |
| Pfam | 57 (0.07%) | 50 (0.06%) | 7 (0.01%) |
| Item | Upregulated | Downregulated | Total |
|---|---|---|---|
| Huoju_713-vs-Baiyu_713 | 1885 | 2224 | 4109 |
| Huoju_719-vs-Baiyu_719 | 2628 | 1707 | 4335 |
| Huoju_727-vs-Baiyu_727 | 2341 | 1986 | 4327 |
| Huoju_803-vs-Baiyu_803 | 1339 | 1913 | 3252 |
| Huoju_810-vs-Baiyu_810 | 2039 | 2121 | 4160 |
| Huoju_817-vs-Baiyu_817 | 2420 | 1692 | 4112 |
| Huoju_824-vs-Baiyu_824 | 2731 | 1816 | 4547 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, H.; Jiang, S.; Zhang, J.; Xu, F.; Zhang, X. Comparative Transcriptomic Analysis of Differentially Expressed Transcripts Associated with Flowering Time of Loquat (Eriobotya japonica Lindl.). Horticulturae 2021, 7, 171. https://doi.org/10.3390/horticulturae7070171
An H, Jiang S, Zhang J, Xu F, Zhang X. Comparative Transcriptomic Analysis of Differentially Expressed Transcripts Associated with Flowering Time of Loquat (Eriobotya japonica Lindl.). Horticulturae. 2021; 7(7):171. https://doi.org/10.3390/horticulturae7070171
Chicago/Turabian StyleAn, Haishan, Shuang Jiang, Jiaying Zhang, Fangjie Xu, and Xueying Zhang. 2021. "Comparative Transcriptomic Analysis of Differentially Expressed Transcripts Associated with Flowering Time of Loquat (Eriobotya japonica Lindl.)" Horticulturae 7, no. 7: 171. https://doi.org/10.3390/horticulturae7070171
APA StyleAn, H., Jiang, S., Zhang, J., Xu, F., & Zhang, X. (2021). Comparative Transcriptomic Analysis of Differentially Expressed Transcripts Associated with Flowering Time of Loquat (Eriobotya japonica Lindl.). Horticulturae, 7(7), 171. https://doi.org/10.3390/horticulturae7070171