
Comparative Transcriptome Analysis of the Anthers from the Cytoplasmic Male-Sterile Pepper Line HZ1A and Its Maintainer Line HZ1B

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Sample Collection and Preparation
2.2. Cytological Examination
2.3. cDNA Preparation and RNA Sequencing
2.4. Bioinformatic Analysis of the Sequencing Data
2.5. Differential Gene Expression Analysis
2.6. Functional Enrichment Analysis
2.7. Quantitative Real-Time PCR and Candidate Genes Amplification
3. Results
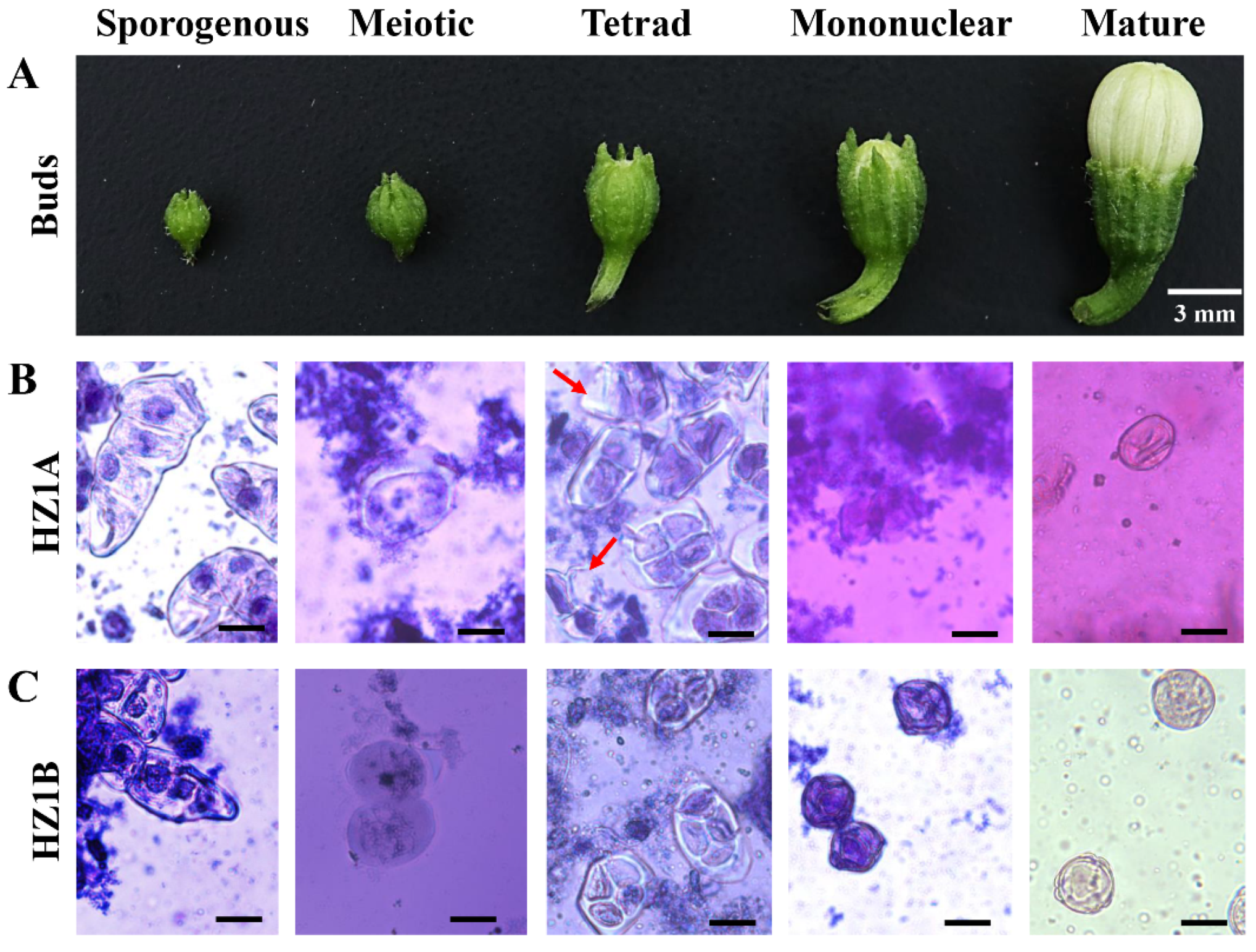
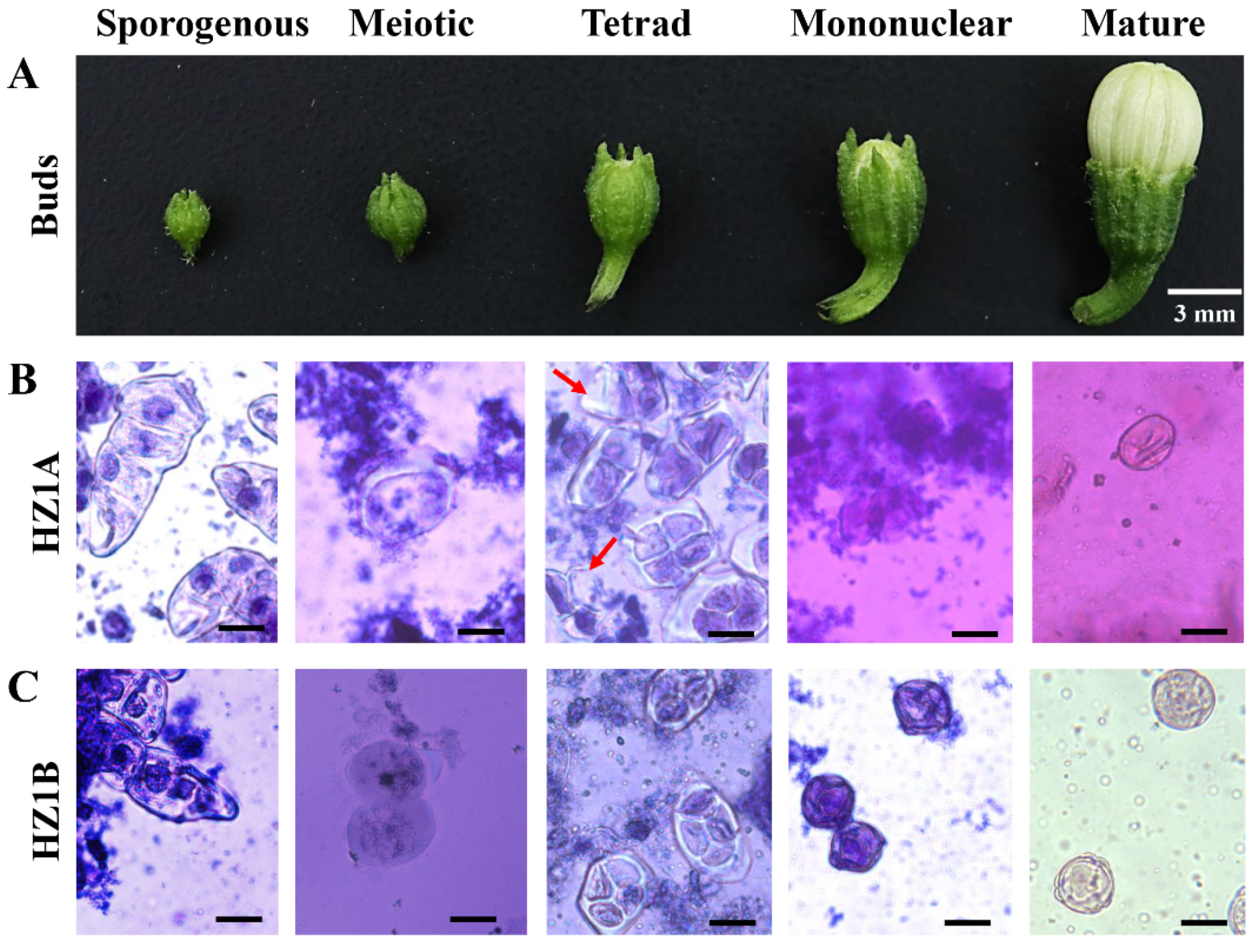
3.1. Morphological Characteristics of Male Fertility and Sterility
3.2. The Cytological Characteristics of CMS Line HZ1A and Its Maintainer Line HZ1B Microspores during the Five Developmental Events
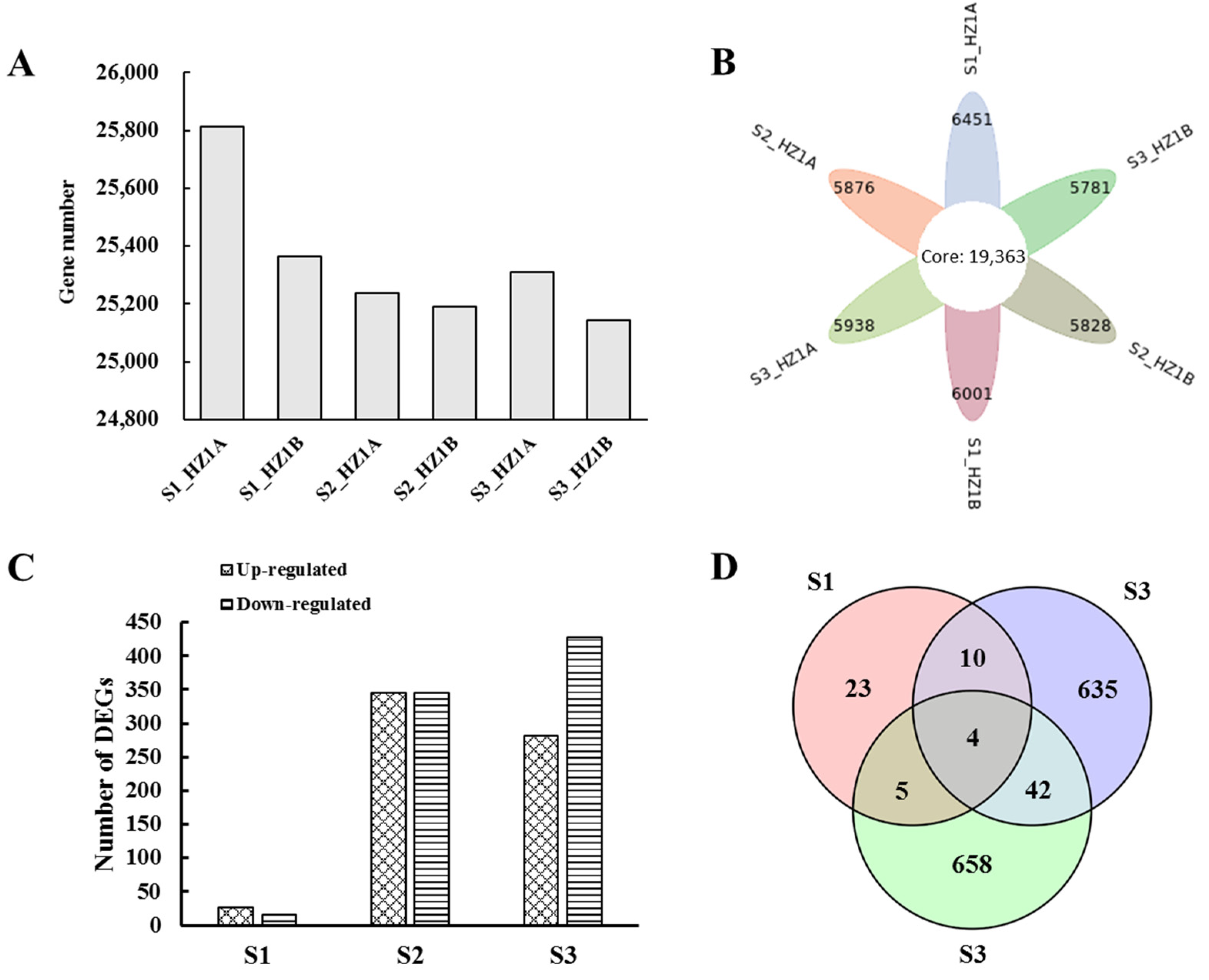
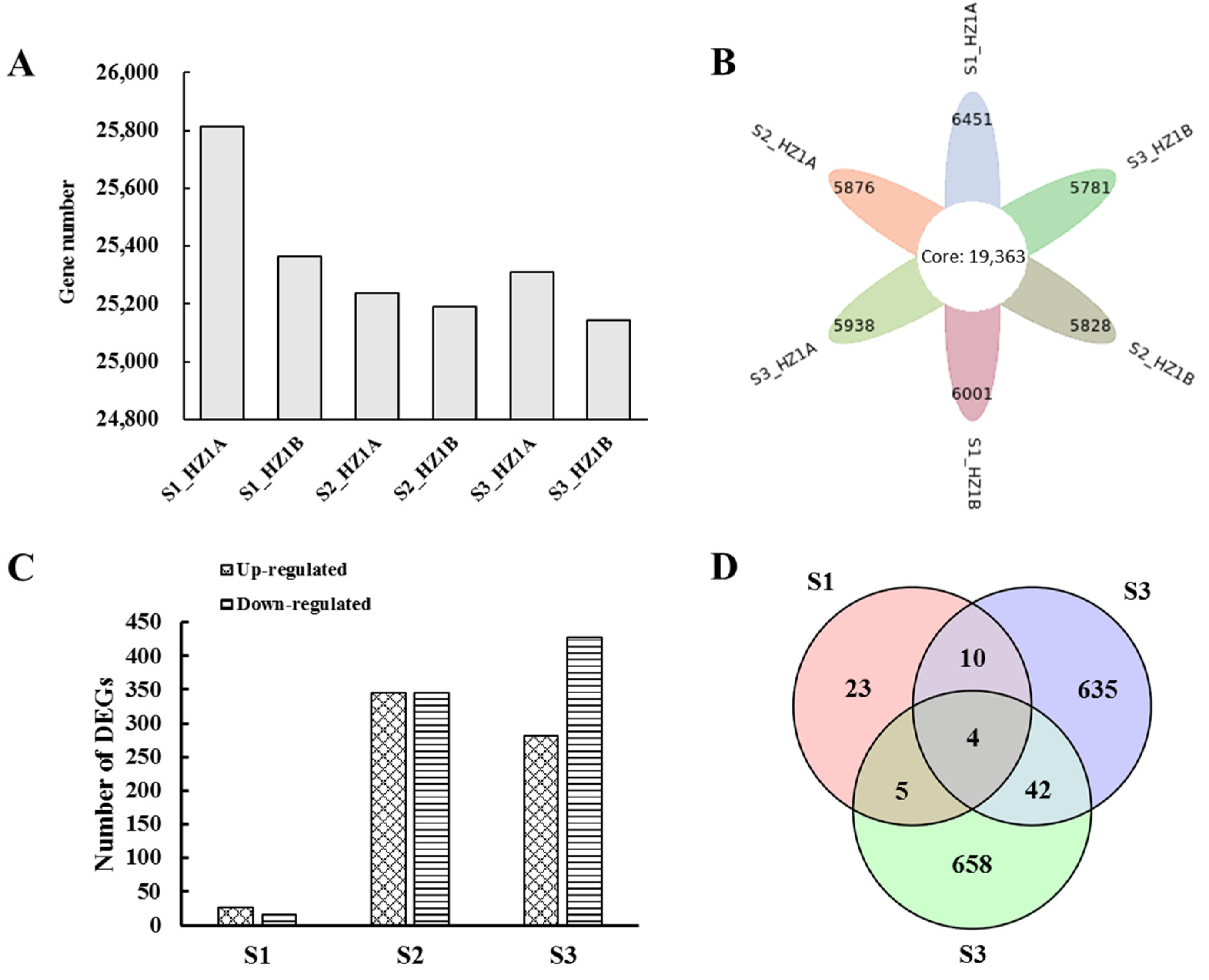
3.3. RNA Sequencing and DEG Identification
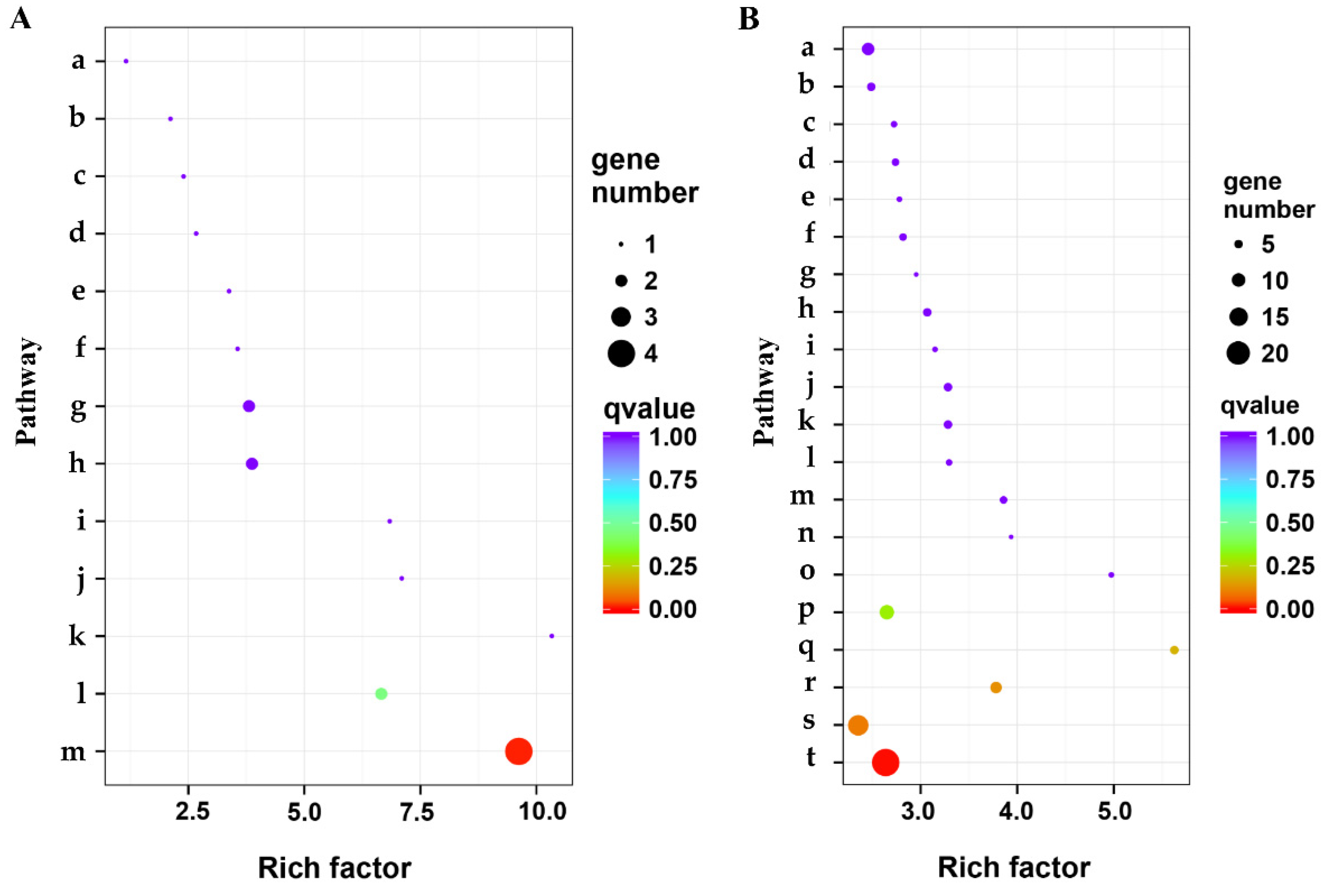
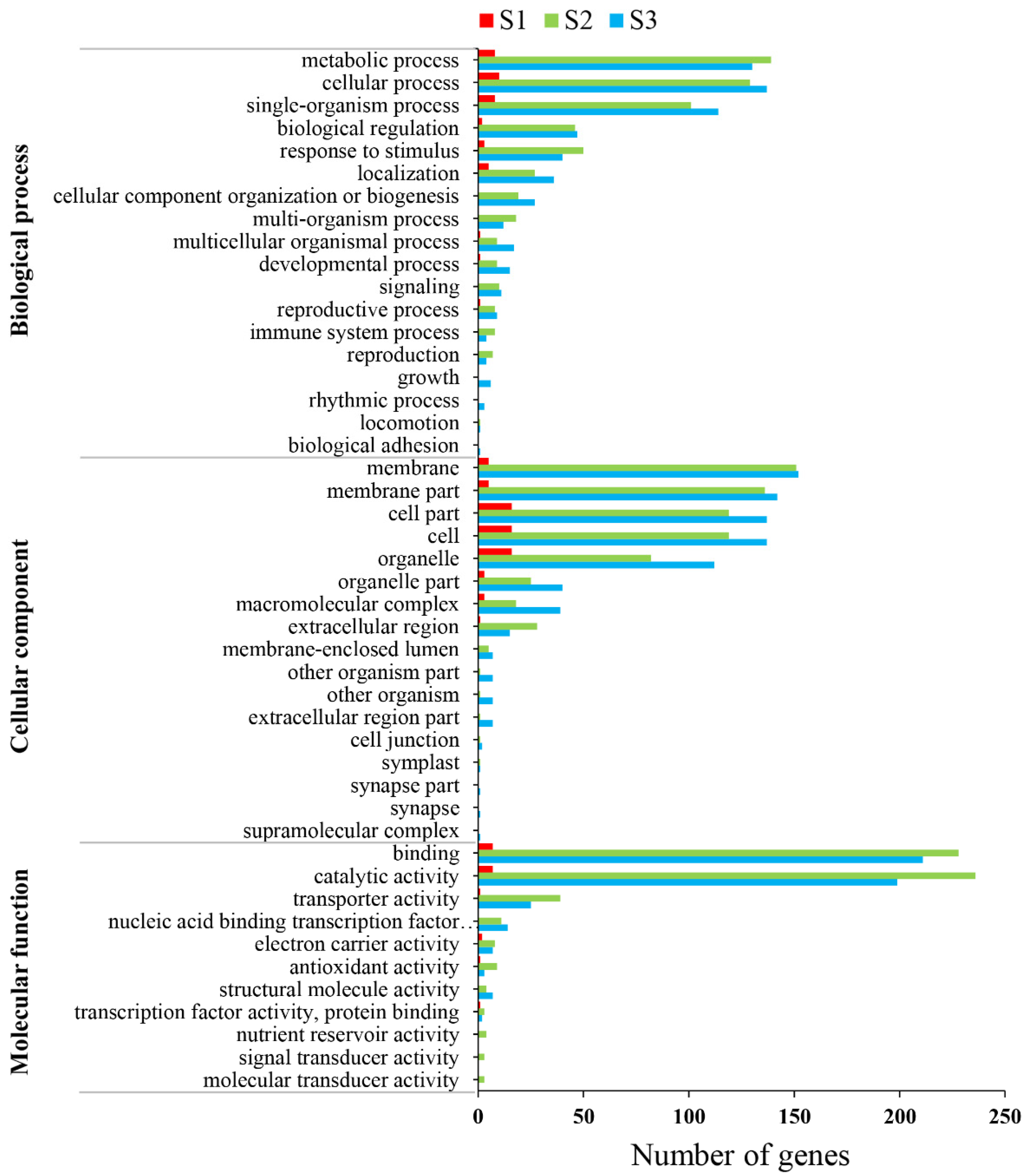
3.4. Functional Annotation by GO
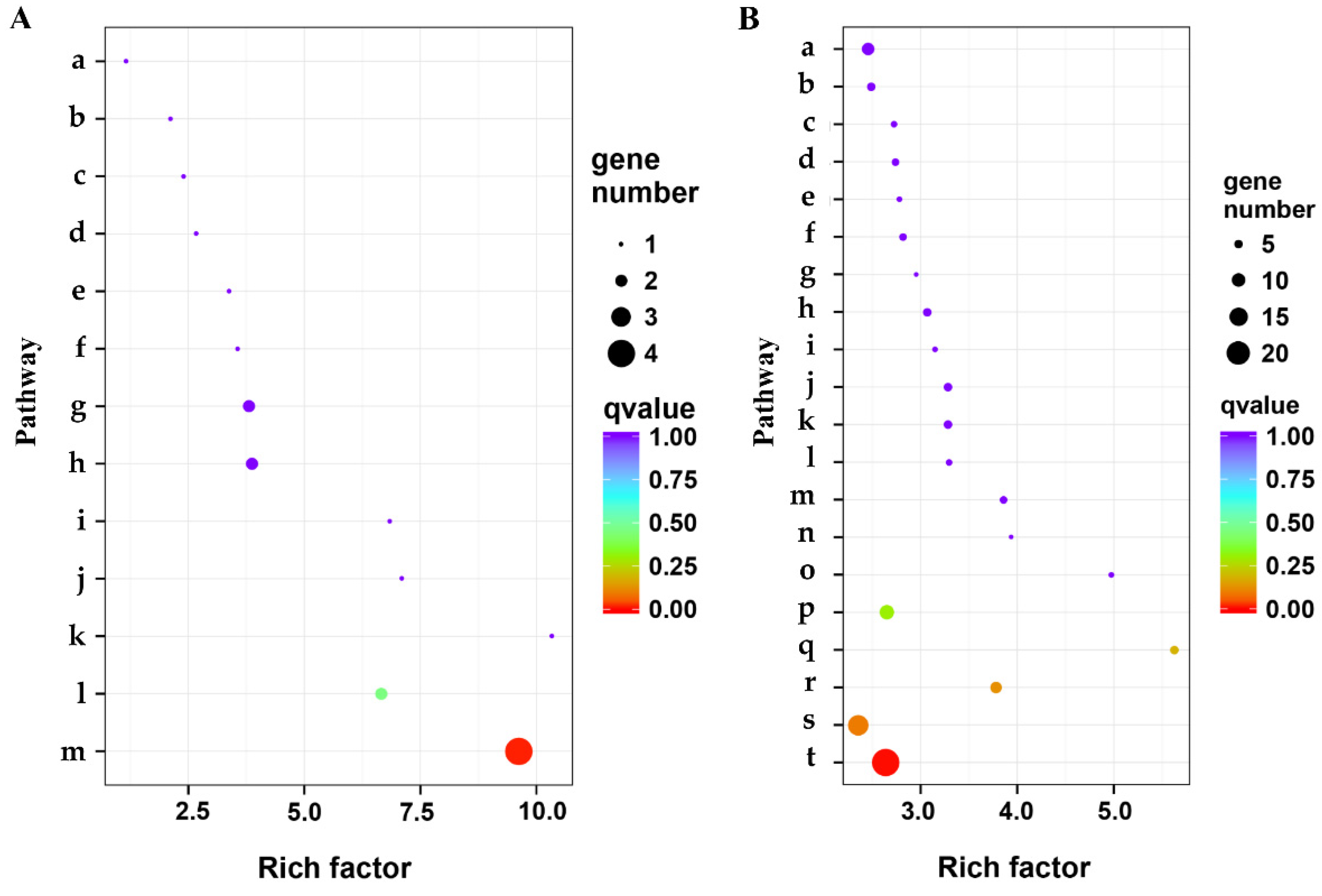
3.5. Pathway Mapping by KEGG
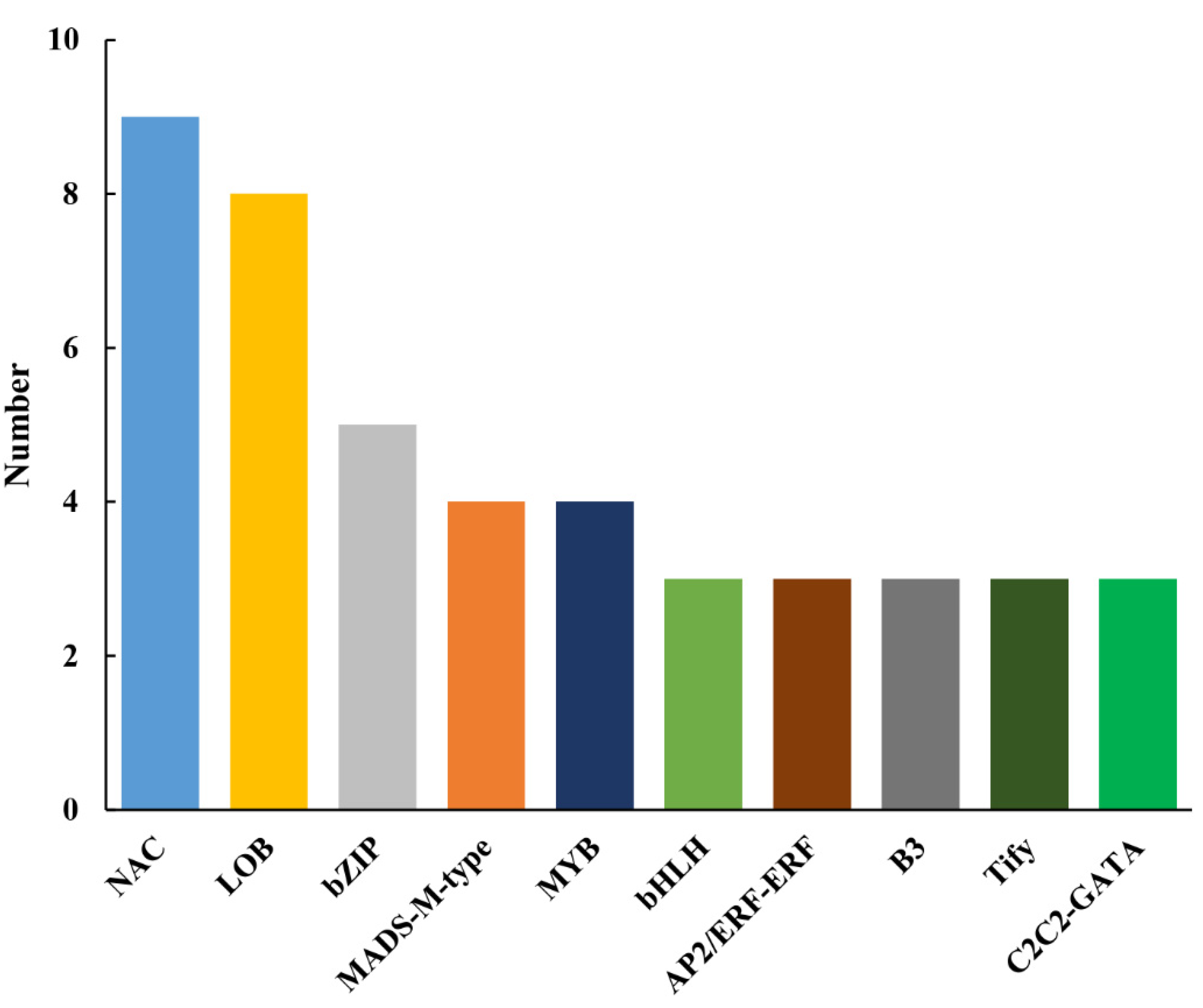
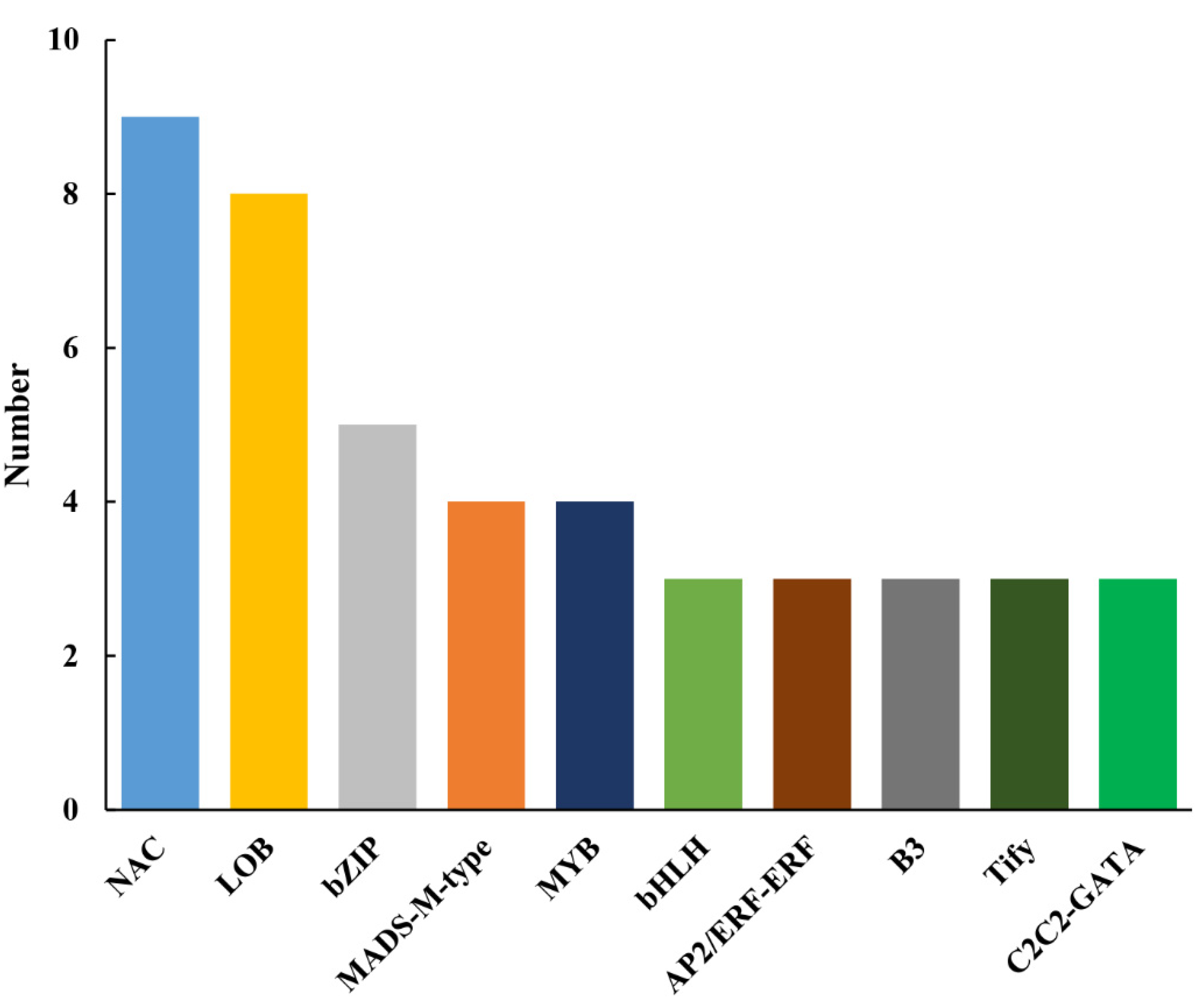
3.6. Identification of Differentially Expressed TFs
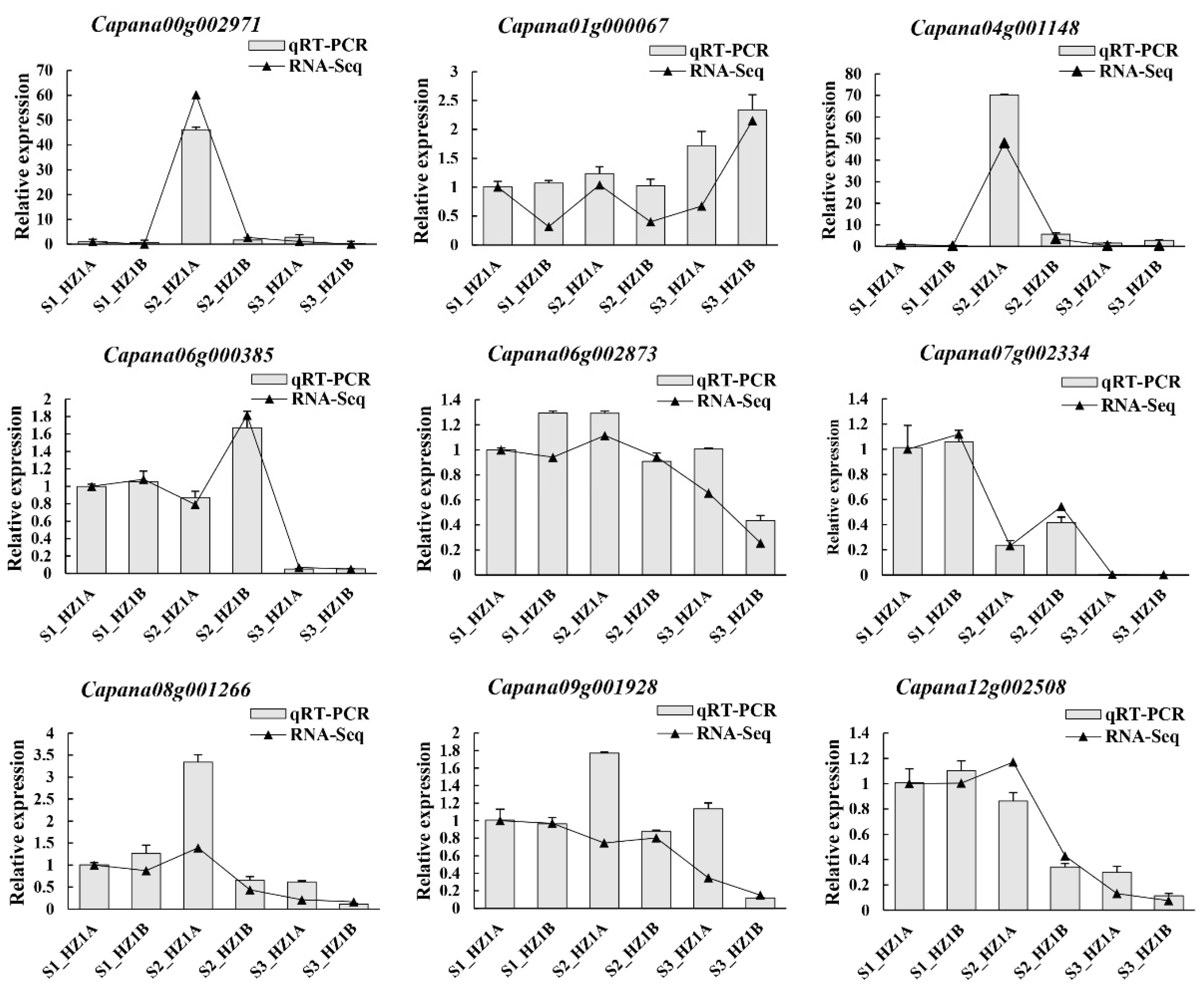
3.7. Validation of the DEGs Using Quantitative Real-Time PCR
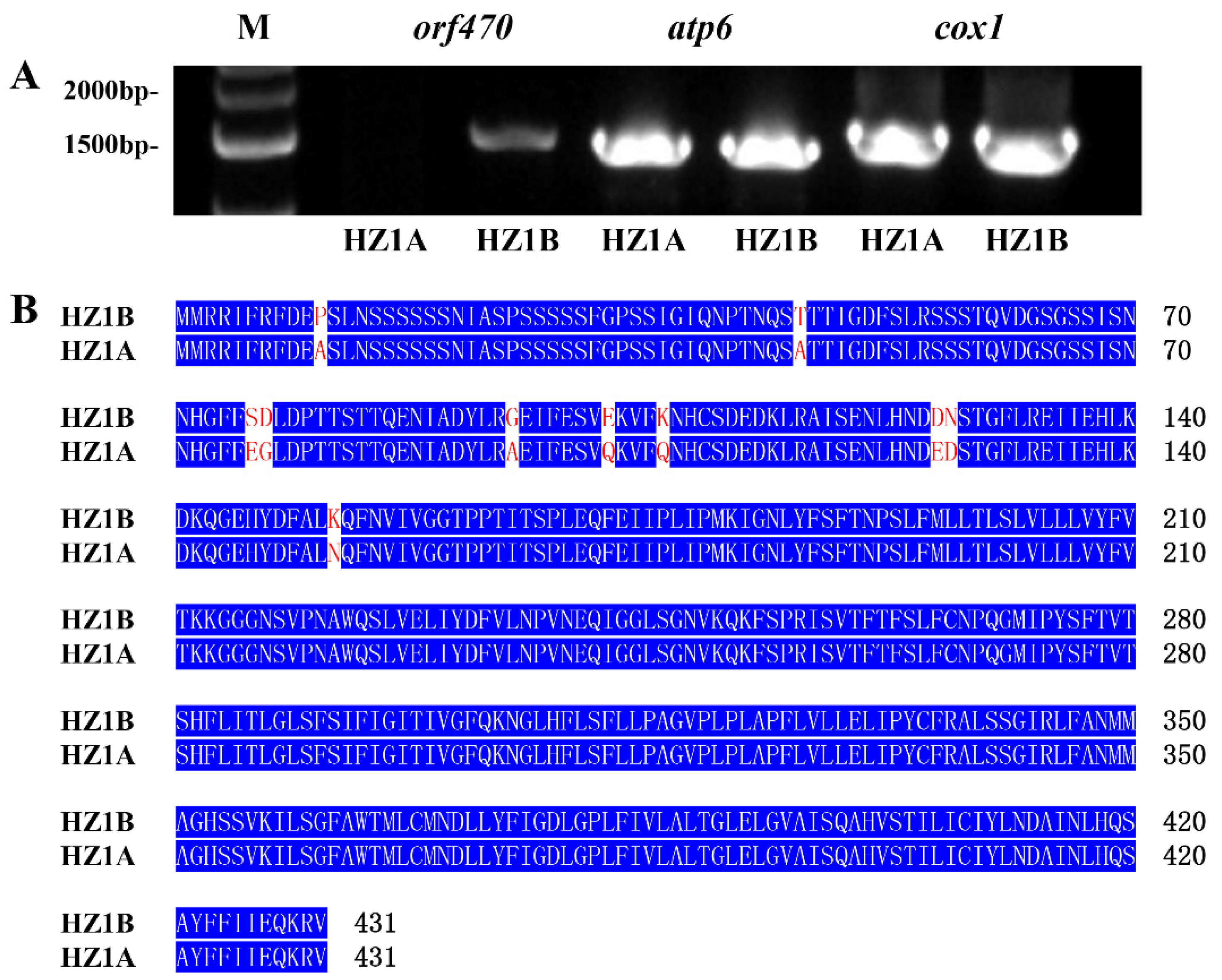
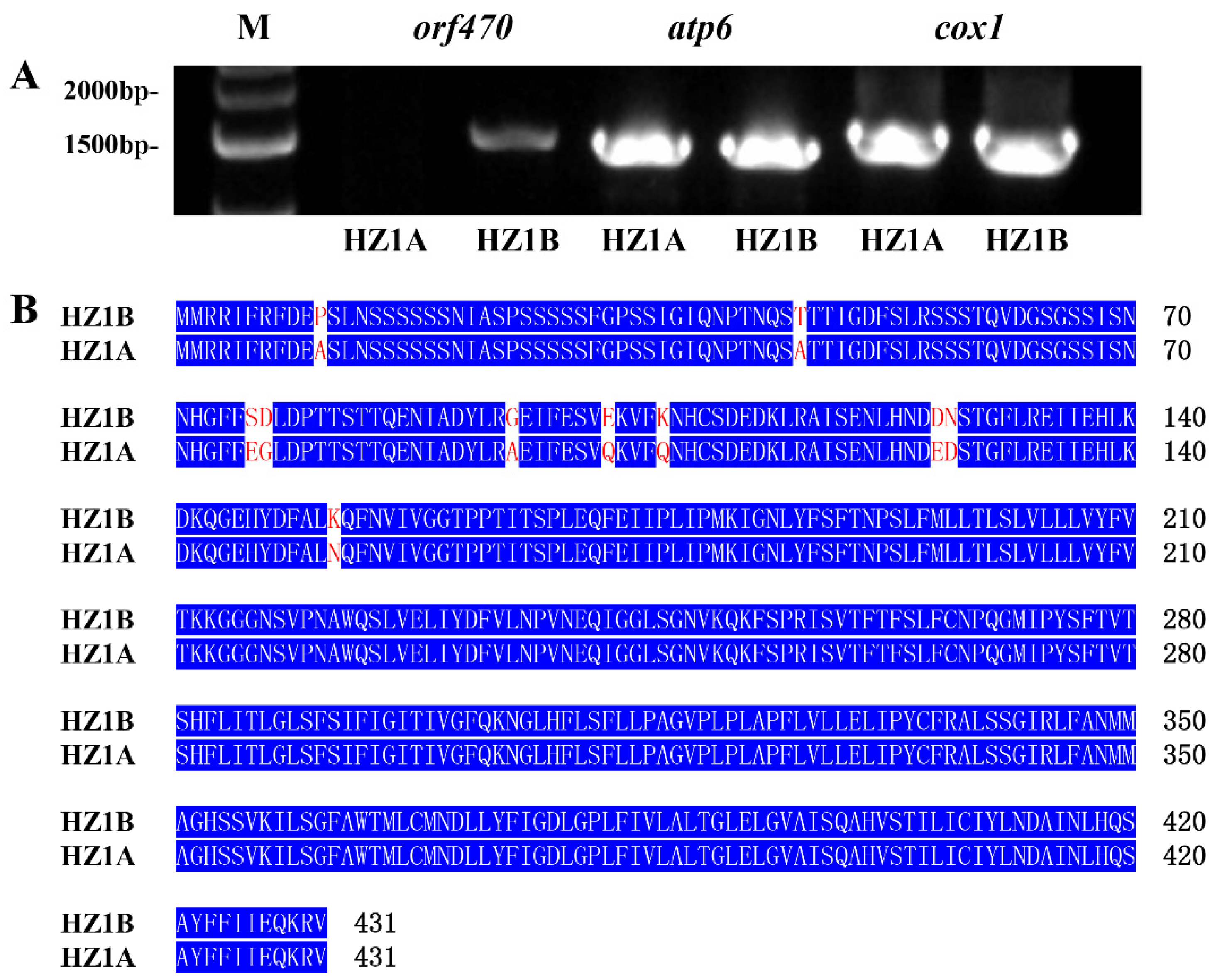
3.8. Mitochondrial Genes Involved in Energy Production Are Related to CMS
4. Discussion
4.1. The Anther Was the Best Organ for Studying Male Sterility in Pepper
4.2. Male Sterility Occurs alongside Gene Expression Level Changes in the Middle and Late Stages of Anther Development
4.3. TFs Related to the CMS in Pepper Line HZ1A
4.4. Genes Involved in Lipid Metabolism Are Related to Male Sterility
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Full definition |
| CMS | Cytoplasmic male-sterility |
| DEGs | Differentially expressed genes |
| ESCRT | Endosomal sorting complex required for transport |
| FLNC | Full-length, non-chimeric |
| GDSL | Gly-Asp-Ser-Leu |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| NCBI | National Center for Biotechnology Information |
| ONT | Oxford Nanopore Technologies |
| Rf | Restorer of male fertility |
| SEM | Scanning electron microscopy |
| TFs | Transcription factors |
References
- Wang, L.; Zhang, B.; Zhang, Z.; Cao, Y.; Yu, H.; Feng, X. Status in breeding and production of Capsicum spp. in China during ‘the thirteenth five-year plan’ period and future prospect. China Veg. 2021, 2, 21–29. [Google Scholar]
- Marame, F.; Dessalegne, L.; Fininsa, C.; Sigvald, R. Heterosis and heritability in crosses among Asian and Ethiopian parents of hot pepper genotypes. Euphytica 2009, 168, 235–247. [Google Scholar] [CrossRef]
- Singh, P.; Cheema, D.S.; Dhaliwal, M.S.; Garg, N. Heterosis and combining ability for earliness, plant growth, yield and fruit attributes in hot pepper (Capsicum annuum L.) involving genetic and cytoplasmic-genetic male sterile lines. Sci. Hortic.-Amst. 2014, 168, 175–188. [Google Scholar] [CrossRef]
- Li, S.; Yang, D.; Zhu, Y. Characterization and use of male sterility in hybrid rice breeding. J. Integr. Plant. Biol. 2007, 49, 791–804. [Google Scholar] [CrossRef]
- Nie, Z.X.; Zhao, T.J.; Yang, S.P.; Gai, J.Y. Development of a cytoplasmic male-sterile line NJCMS4A for hybrid soybean production. Plant Breed. 2017, 136, 516–525. [Google Scholar] [CrossRef]
- Kim, Y.J.; Zhang, D. Molecular control of male fertility for crop hybrid breeding. Trends. Plant Sci. 2018, 23, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Gaborieau, L.; Brown, G.G.; Mireau, H. The propensity of pentatricopeptide repeat genes to evolve into restorers of cytoplasmic male sterility. Front. Plant Sci. 2016, 7, 1816. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liu, Y.G. Male sterility and fertility restoration in crops. Annu. Rev. Plant Biol. 2014, 65, 579–606. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zou, Y.; Li, X.; Zhang, Q.; Chen, L.; Wu, H.; Su, D.; Chen, Y.; Guo, J.; Luo, D. Cytoplasmic male sterility of rice with boro II cytoplasm is caused by a cytotoxic peptide and is restored by two related PPR motif genes via distinct modes of mRNA silencing. Plant Cell 2006, 18, 676–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komori, T.; Ohta, S.; Murai, N.; Takakura, Y.; Kuraya, Y.; Suzuki, S.; Hiei, Y.; Imaseki, H.; Nitta, N. Map-based cloning of a fertility restorer gene, Rf-1, in rice (Oryza sativa L.). Plant J. 2004, 10, 1046–1056. [Google Scholar]
- Hu, J.; Wang, K.; Huang, W.; Liu, G.; Gao, Y.; Wang, J.; Huang, Q.; Ji, Y.; Qin, X.; Wan, L. The rice pentatricopeptide repeat protein RF5 restores fertility in Hong-Lian cytoplasmic male-sterile lines via a complex with the glycine-rich protein GRP162. Plant Cell 2012, 24, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Yu, C.; Hu, J.; Wang, L.; Dan, Z.; Zhou, W.; He, C.; Zeng, Y.; Yao, G.; Qi, J. Pentatricopeptide-repeat family protein RF6 functions with hexokinase 6 to rescue rice cytoplasmic male sterility. Proc. Natl. Acad. Sci. USA 2015, 112, 14984–14989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, D.; Mace, E.S.; Henzell, R.; Klein, P.; Klein, R. Molecular mapping and candidate gene identification of the Rf2 gene for pollen fertility restoration in sorghum [Sorghum bicolor (L.) Moench]. Theor. Appl. Genet. 2010, 120, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Koizuka, N.; Imai, R.; Fujimoto, H.; Hayakawa, T.; Kimura, Y.; Kohno-Murase, J.; Sakai, T.; Kawasaki, S.; Imamura, J. Genetic characterization of a pentatricopeptide repeat protein gene, orf687, that restores fertility in the cytoplasmic male-sterile Kosena radish. Plant J. 2003, 34, 407–415. [Google Scholar] [CrossRef]
- Uyttewaal, M.; Arnal, N.; Quadrado, M.; Martin-Canadell, A.; Vrielynck, N.; Hiard, S.; Gherbi, H.; Bendahmane, A.; Budar, F.; Mireau, H. Characterization of Raphanus sativus pentatricopeptide repeat proteins encoded by the fertility restorer locus for Ogura cytoplasmic male sterility. Plant Cell 2008, 20, 3331–3345. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Fu, T.; Yi, B.; Hong, D. A mitochondria-targeted PPR protein restores pol cytoplasmic male sterility by reducing orf224 transcript levels in oilseed rape. Mol. Plant 2016, 9, 1082–1084. [Google Scholar]
- Melonek, J.; Duarte, J.; Martin, J.; Beuf, L.; Murigneux, A.; Varenne, P.; Comadran, J.; Specel, S.; Levadoux, S.; Bernath-Levin, K.; et al. The genetic basis of cytoplasmic male sterility and fertility restoration in wheat. Nat. Commun. 2021, 12, 1036. [Google Scholar] [CrossRef]
- Cui, X.; Wise, R.P.; Schnable, P.S. The rf2 nuclear restorer gene of male-sterile T-cytoplasm maize. Science 1996, 272, 1334–1336. [Google Scholar] [CrossRef]
- Itabashi, E.; Iwata, N.; Fujii, S.; Kazama, T.; Toriyama, K. The fertility restorer gene, Rf2, for Lead Rice-type cytoplasmic male sterility of rice encodes a mitochondrial glycine-rich protein. Plant J. 2011, 65, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Toriyama, K. Suppressed expression of RETROGRADE-REGULATED MALE STERILITY restores pollen fertility in cytoplasmic male sterile rice plants. Proc. Natl. Acad. Sci. USA 2009, 106, 9513–9518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitazaki, K.; Arakawa, T.; Matsunaga, M.; Yui-Kurino, R.; Matsuhira, H.; Mikami, T.; Kubo, T. Post-translational mechanisms are associated with fertility restoration of cytoplasmic male sterility in sugar beet (Beta vulgaris). Plant J. 2015, 83, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Huang, W.; Huang, Q.; Qin, X.; Yu, C.; Wang, L.; Li, S.; Zhu, R.; Zhu, Y. Mitochondria and cytoplasmic male sterility in plants. Mitochondrion 2014, 19, 282–288. [Google Scholar] [CrossRef]
- Luo, D.P.; Xu, H.; Liu, Z.L.; Guo, J.X.; Li, H.Y.; Chen, L.T.; Fang, C.; Zhang, Q.Y.; Bai, M.; Yao, N.; et al. A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice. Nat. Genet. 2013, 45, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Gao, F.; Ji, Y.X.; Liu, Y.; Dan, Z.W.; Yang, P.F.; Zhu, Y.G.; Li, S.Q. ORFH79 impairs mitochondrial function via interaction with a subunit of electron transport chain complex III in Honglian cytoplasmic male sterile rice. New Phytol. 2013, 198, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Kazama, T.; Itabashi, E.; Fujii, S.; Nakamura, T.; Toriyama, K. Mitochondrial ORF79 levels determine pollen abortion in cytoplasmic male sterile rice. Plant J. 2016, 85, 707–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabaleta, E.; Mouras, A.; Hernould, M.; Suharsono, S.; Araya, A. Transgenic male-sterile plant induced by an unedited atp9 gene is restored to fertility by inhibiting its expression with antisense RNA. Proc. Natl. Acad. Sci. USA 1996, 93, 11259–11263. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Sen, S.; Chakraborty, A.; Chakraborti, P.; Maiti, M.K.; Basu, A.; Basu, D.; Sen, S.K. An unedited 1.1 kb mitochondrial orfB gene transcript in the wild abortive cytoplasmic male sterility (WA-CMS) system of Oryza sativa L. subsp indica. BMC Plant Biol. 2010, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Luo, C. Molecular and functional diversity of RNA editing in plant mitochondria. Mol. Biotechnol. 2018, 60, 935–945. [Google Scholar] [CrossRef]
- Wan, X.; Wu, S.; Li, Z.; An, X.; Tian, Y. Lipid metabolism: Critical roles in male fertility and other aspects of reproductive development in plants. Mol. Plant 2020, 13, 955–983. [Google Scholar] [CrossRef]
- Sun, L.; Sui, X.; Lucas, W.J.; Li, Y.; Feng, S.; Ma, S.; Fan, J.; Gao, L.; Zhang, Z. Down-regulation of the sucrose transporter CsSUT1 causes male sterility by altering carbohydrate supply. Plant Physiol. 2019, 180, 986–997. [Google Scholar] [CrossRef] [Green Version]
- Qian, Q.; Yang, Y.H.; Zhang, W.B.; Hu, Y.L.; Li, Y.G.; Yu, H.; Hou, X.L. A novel Arabidopsis gene RGAT1 is required for GA-mediated tapetum and pollen development. New Phytol. 2021, 231, 137–151. [Google Scholar] [CrossRef]
- Aya, K.; Ueguchi-Tanaka, M.; Kondo, M.; Hamada, K.; Yano, K.; Nishimura, M.; Matsuoka, M. Gibberellin modulates anther development in rice via the transcriptional regulation of GAMYB. Plant Cell 2009, 21, 1453–1472. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Bao, W.; Liang, W.; Yin, J.; Zhang, D. Identification of gamyb-4 and analysis of the regulatory role of GAMYB in rice anther development. J. Integr. Plant Biol. 2010, 52, 670–678. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, M.; Li, Y.; Yan, W.; Chang, Z.; Ni, H.; Chen, Z.; Wu, J.; Xu, C.; Deng, X.W.; et al. GDSL esterase/lipases OsGELP34 and OsGELP110/OsGELP115 are essential for rice pollen development. J. Integr. Plant Biol. 2020, 62, 1574–1593. [Google Scholar] [CrossRef] [Green Version]
- Huo, Y.; Pei, Y.; Tian, Y.; Zhang, Z.; Li, K.; Liu, J.; Xiao, S.; Chen, H.; Liu, J. IRREGULAR POLLEN EXINE2 encodes a GDSL lipase essential for male fertility in maize. Plant Physiol. 2020, 184, 1438–1454. [Google Scholar] [CrossRef] [PubMed]
- Goodman, K.; Paez-Valencia, J.; Pennington, J.; Sonntag, A.; Ding, X.; Lee, H.N.; Ahlquist, P.G.; Molina, I.; Otegui, M.S. ESCRT components ISTL1 and LIP5 are required for tapetal function and pollen viability. Plant Cell 2021, 33, 2850–2868. [Google Scholar] [CrossRef]
- Li, X.-C.; Zhu, J.; Yang, J.; Zhang, G.R.; Xing, W.F.; Zhang, S.; Yang, Z.N. Glycerol-3-phosphate acyltransferase 6 (GPAT6) is important for tapetum development in Arabidopsis and plays multiple roles in plant fertility. Mol. Plant 2012, 5, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.F.; Zhu, J.; Lou, Y.; Guo, Z.L.; Xiong, S.X.; Wang, K.; Yang, Z.N. The functional analysis of OsTDF1 reveals a conserved genetic pathway for tapetal development between rice and Arabidopsis. Sci. Bull. 2015, 60, 1073–1082. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.H.; Han, M.J.; Lee, Y.S.; Kim, Y.W.; Hwang, I.W.; Kim, M.J.; Kim, Y.K.; Nahm, B.H.; An, G.H. Rice Undeveloped Tapetum1 is a major regulator of early tapetum development. Plant Cell 2005, 17, 2705–2722. [Google Scholar] [CrossRef] [Green Version]
- Jo, Y.D.; Ha, Y.; Lee, J.H.; Park, M.; Bergsma, A.C.; Choi, H.I.; Goritschnig, S.; Kloosterman, B.; van Dijk, P.J.; Choi, D.; et al. Fine mapping of Restorer-of-fertility in pepper (Capsicum annuum L.) identified a candidate gene encoding a pentatricopeptide repeat (PPR)-containing protein. Theor. Appl. Genet. 2016, 129, 2003–2017. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.W.; Chen, Y.J.; Hu, Y.F.; Zhou, Z.Y.; Hu, F.; Dong, J.C.; Chen, W.L.; Cui, J.J.; Wu, Z.M.; Hu, K.L. Fine mapping of restorer-of-fertility gene based on high-density genetic mapping and collinearity analysis in pepper (Capsicum annuum L.). Theor. Appl. Genet. 2020, 133, 889–902. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Zhu, Y.S.; Cao, Y.C.; Yu, H.L.; Bai, R.Q.; Zhao, H.; Zhang, B.X.; Wang, L.H. Fine mapping of the male fertility restoration gene CaRf032 in Capsicum annuum L. Theor. Appl. Genet. 2020, 133, 1177–1187. [Google Scholar] [CrossRef]
- Wei, B.Q.; Bosland, P.W.; Zhang, Z.H.; Wang, Y.F.; Zhang, G.Y.; Wang, L.L.; Yu, J.H. A predicted NEDD8 conjugating enzyme gene identified as a Capsicum candidate Rf gene using bulk segregant RNA sequencing. Hortic. Res.-Engl. 2020, 7, 210. [Google Scholar] [CrossRef]
- Wu, L.; Wang, P.; Wang, Y.H.; Cheng, Q.; Lu, Q.H.; Liu, J.Q.; Li, T.; Ai, Y.X.; Yang, W.C.; Sun, L.; et al. Genome-wide correlation of 36 agronomic traits in the 287 pepper (Capsicum) accessions obtained from the SLAF-seq-based GWAS. Int. J. Mol. Sci. 2019, 20, 5675. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.J.; Huang, W.; Li, D.W.; Yin, Y.X.; Chai, W.G.; Gong, Z.H. A CMS-related gene, ψatp6-2, causes increased ATP hydrolysis activity of the mitochondrial F1Fo-ATP synthase and induces male sterility in pepper (Capsicum annuum L.). Plant Mol. Biol. Rep. 2014, 32, 888–899. [Google Scholar] [CrossRef]
- Li, J.; Pandeya, D.; Jo, Y.D.; Liu, W.Y.; Kang, B.C. Reduced activity of ATP synthase in mitochondria causes cytoplasmic male sterility in chili pepper. Planta 2013, 237, 1097–1109. [Google Scholar] [CrossRef]
- Kim, D.H.; Kang, J.G.; Kim, B.D. Isolation and characterization of the cytoplasmic male sterility-associated orf456 gene of chili pepper (Capsicum annuum L.). Plant Mol. Biol. 2007, 63, 519–532. [Google Scholar] [CrossRef]
- Wang, P.; Lu, Q.; Ai, Y.; Wang, Y.; Li, T.; Wu, L.; Liu, J.; Cheng, Q.; Sun, L.; Shen, H. Candidate gene selection for cytoplasmic male sterility in pepper (Capsicum annuum L.) through whole mitochondrial genome sequencing. Int. J. Mol. Sci. 2019, 20, 578. [Google Scholar] [CrossRef] [Green Version]
- Peterson, R.; Slovin, J.P.; Chen, C. A simplified method for differential staining of aborted and non-aborted pollen grains. Int. J. Plant Biol. 2010, 1, e13. [Google Scholar] [CrossRef] [Green Version]
- Nie, Z.; Zhao, T.; Liu, M.; Dai, J.; He, T.; Lyu, D.; Zhao, J.; Yang, S.; Gai, J. Molecular mapping of a novel male-sterile gene msNJ in soybean [Glycine max (L.) Merr.]. Plant Reprod. 2019, 32, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Teng, L.; Feng, Y.C.; Guo, S.T.; Wang, P.L.; Qi, T.F.; Yue, Y.M.; Wang, S.X.; Zhang, S.N.; Tang, C.X.; La, T.; et al. The pan-cancer lncRNA PLANE regulates an alternative splicing program to promote cancer pathogenesis. Nat. Commun. 2021, 12, 3734. [Google Scholar] [CrossRef]
- Qin, C.; Yu, C.; Shen, Y.; Fang, X.; Chen, L.; Min, J.; Cheng, J.; Zhao, S.; Xu, M.; Luo, Y.; et al. Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc. Natl. Acad. Sci. USA 2014, 111, 5135–5140. [Google Scholar] [CrossRef] [Green Version]
- Jo, Y.D.; Choi, Y.; Kim, D.H.; Kim, B.D.; Kang, B.C. Extensive structural variations between mitochondrial genomes of CMS and normal peppers (Capsicum annuum L.) revealed by complete nucleotide sequencing. BMC Genom. 2014, 15, 561. [Google Scholar] [CrossRef] [Green Version]
- Jo, Y.D.; Park, J.; Kim, J.; Song, W.; Hur, C.G.; Lee, Y.H.; Kang, B.C. Complete sequencing and comparative analyses of the pepper (Capsicum annuum L.) plastome revealed high frequency of tandem repeats and large insertion/deletions on pepper plastome. Plant Cell Rep. 2011, 30, 217–229. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Yi, Z.; Chen, J.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Goseq: Gene ontology testing for RNA-seq datasets. R. Bioconduct. 2012, 8, 1–25. [Google Scholar]
- Mao, X.Z.; Cai, T.; Olyarchuk, J.G.; Wei, L.P. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Lv, J.H.; Liu, Z.B.; Liu, Y.H.; Ou, L.J.; Deng, M.H.; Wang, J.; Song, J.S.; Ma, Y.Q.; Chen, W.C.; Zhang, Z.Q.; et al. Comparative transcriptome analysis between cytoplasmic male-sterile line and its maintainer during the floral bud development of pepper. Hortic. Plant J. 2020, 6, 89–98. [Google Scholar] [CrossRef]
- Qiu, Y.; Liao, L.; Jin, X.; Mao, D.; Liu, R. Analysis of the meiotic transcriptome reveals the genes related to the regulation of pollen abortion in cytoplasmic male-sterile pepper (Capsicum annuum L.). Gene 2018, 641, 8–17. [Google Scholar] [CrossRef]
- Browne, R.G.; Li, S.F.; Iacuone, S.; Dolferus, R.; Parish, R.W. Differential responses of anthers of stress tolerant and sensitive wheat cultivars to high temperature stress. Planta 2021, 254, 4. [Google Scholar] [CrossRef]
- Nie, Z. Discovery of a New Cytoplasmic Male-Sterile Source, Detection of a Nuclear Male-Sterile Gene and QTL-Allele Constitutions of Heterosis in Soybean [Glycine max (L.) Merr.]. Ph.D. Thesis, Nanjing Agricultural University, Nanjing, China, 2017. [Google Scholar]
- Yang, Y.; Bao, S.Y.; Zhou, X.H.; Liu, J.; Zhuang, Y. The key genes and pathways related to male sterility of eggplant revealed by comparative transcriptome analysis. BMC Plant Biol. 2018, 18, 209. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Park, S.W.; Hong, W.J.; Silva, J.; Liang, W.Q.; Zhang, D.B.; Jung, K.H.; Kim, Y.J. Genome-wide analysis of RopGEF gene family to identify genes contributing to pollen tube growth in rice (Oryza sativa). BMC Plant Biol. 2020, 20, 95. [Google Scholar] [CrossRef] [Green Version]
- Fernández Gómez, J.; Wilson, Z.A. A barley PHD finger transcription factor that confers male sterility by affecting tapetal development. Plant Biotechnol. J. 2014, 12, 765–777. [Google Scholar] [CrossRef]
- Xiao, S.; Zang, J.; Pei, Y.; Liu, J.; Liu, J.; Song, W.; Shi, Z.; Su, A.; Zhao, J.; Chen, H. Activation of mitochondrial orf355 gene expression by a nuclear-encoded DREB transcription factor causes cytoplasmic male sterility in maize. Mol. Plant 2020, 13, 1270–1283. [Google Scholar] [CrossRef]
- Jung, Y.J.; Kim, D.H.; Lee, H.J.; Nam, K.H.; Bae, S.; Nou, I.S.; Cho, Y.G.; Kim, M.K.; Kang, K.K. Knockout of SlMS10 Gene (Solyc02g079810) encoding bHLH transcription factor using CRISPR/Cas9 system confers male sterility phenotype in tomato. Plants 2020, 9, 1189. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.M.; Kong, X.D.; Chen, F.; Huang, J.X.; Lou, X.Y.; Zhao, J.Y. Transcriptome analysis of Brassica napus pod using RNA-Seq and identification of lipid-related candidate genes. BMC Genom. 2015, 16, 858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lannert, H.; Gorgas, K.; Meißner, I.; Wieland, F.T.; Jeckel, D. Functional organization of the Golgi apparatus in glycosphingolipid biosynthesis: Lactosylceramide and subsequent glycosphingolipids are formed in the lumen of the late Golgi. J. Biol. Chem. 1998, 273, 2939–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobsen, M.K. Pollen development and fertilization in arabidopsis is dependent on the male gametogenesis impaired anthers gene encoding a type v p-type ATPase. Genes Dev. 2005, 19, 2757–2769. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SampleID | Clean Data (Gb) | Clean Data ReadNum | Mean Length (bp) | Max Length (bp) | Number of Full-Length Reads | Full-Length Ratio | Mapped Reads | Mapped Rates Ratio |
|---|---|---|---|---|---|---|---|---|
| S1_HZ1A_1 | 2.39 | 2,838,187 | 840 | 30,800 | 2,416,304 | 86.52% | 2,122,768 | 87.85% |
| S1_HZ1A_2 | 2.05 | 2,886,667 | 711 | 664,652 | 2,366,701 | 83.69% | 1,888,008 | 79.77% |
| S1_HZ1A_3 | 2.14 | 2,772,877 | 771 | 135,578 | 2,347,619 | 86.18% | 1,959,880 | 83.48% |
| S2_HZ1A_1 | 1.97 | 2,380,595 | 825 | 41,246 | 2,039,897 | 87.33% | 1,565,821 | 76.76% |
| S2_HZ1A_2 | 2.13 | 2,315,185 | 921 | 93,710 | 1,982,909 | 87.13% | 1,656,220 | 83.52% |
| S2_HZ1A_3 | 1.94 | 2,589,626 | 750 | 41,295 | 2,188,560 | 86.17% | 1,593,667 | 72.82% |
| S3_HZ1A_1 | 2.04 | 2,668,696 | 766 | 22,338 | 2,205,049 | 84.24% | 1,928,150 | 87.44% |
| S3_HZ1A_2 | 2.16 | 2,871,550 | 750 | 33,736 | 2,366,822 | 84.01% | 2,042,014 | 86.28% |
| S3_HZ1A_3 | 2.38 | 2,576,099 | 923 | 165,639 | 2,122,088 | 83.87% | 1,880,618 | 88.62% |
| S1_HZ1B_1 | 2.38 | 3,026,268 | 787 | 17,426 | 2,547,867 | 85.76% | 2,156,677 | 84.65% |
| S1_HZ1B-2 | 2.07 | 2,776,136 | 744 | 418,861 | 2,347,047 | 86.04% | 1,990,528 | 84.81% |
| S1_HZ1B_3 | 2.19 | 2,715,765 | 806 | 30,900 | 2,295,265 | 86.10% | 2,024,787 | 88.22% |
| S2_HZ1B_1 | 1.92 | 2,130,396 | 903 | 143,803 | 1,811,522 | 86.44% | 1,420,948 | 78.44% |
| S2_HZ1B_2 | 2.09 | 2,914,952 | 718 | 37,501 | 2,518,478 | 87.85% | 1,858,513 | 73.80% |
| S2_HZ1B_3 | 2.12 | 2,398,923 | 882 | 46,612 | 2,035,351 | 86.46% | 1,592,519 | 78.24% |
| S3_HZ1B_1 | 2.04 | 2,709,718 | 753 | 12,610 | 2,330,314 | 87.69% | 1,803,068 | 77.37% |
| S3_HZ1B_2 | 2.09 | 2,730,251 | 764 | 163,019 | 2,368,407 | 88.33% | 1,890,532 | 79.82% |
| S3_HZ1B_3 | 2.01 | 2,644,302 | 760 | 78,489 | 2,272,739 | 87.72% | 1,814,061 | 79.82% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nie, Z.; Chen, J.; Song, Y.; Fu, H.; Wang, H.; Niu, Q.; Zhu, W. Comparative Transcriptome Analysis of the Anthers from the Cytoplasmic Male-Sterile Pepper Line HZ1A and Its Maintainer Line HZ1B. Horticulturae 2021, 7, 580. https://doi.org/10.3390/horticulturae7120580
Nie Z, Chen J, Song Y, Fu H, Wang H, Niu Q, Zhu W. Comparative Transcriptome Analysis of the Anthers from the Cytoplasmic Male-Sterile Pepper Line HZ1A and Its Maintainer Line HZ1B. Horticulturae. 2021; 7(12):580. https://doi.org/10.3390/horticulturae7120580
Chicago/Turabian StyleNie, Zhixing, Jianying Chen, Yunpeng Song, Hongfei Fu, Hong Wang, Qingliang Niu, and Weimin Zhu. 2021. "Comparative Transcriptome Analysis of the Anthers from the Cytoplasmic Male-Sterile Pepper Line HZ1A and Its Maintainer Line HZ1B" Horticulturae 7, no. 12: 580. https://doi.org/10.3390/horticulturae7120580
APA StyleNie, Z., Chen, J., Song, Y., Fu, H., Wang, H., Niu, Q., & Zhu, W. (2021). Comparative Transcriptome Analysis of the Anthers from the Cytoplasmic Male-Sterile Pepper Line HZ1A and Its Maintainer Line HZ1B. Horticulturae, 7(12), 580. https://doi.org/10.3390/horticulturae7120580