
Transcriptomic Insights and Quantification of Free Amino Acids in Auricularia heimuer Cultivated on Corncob Substrate

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cultivation and Sample Collection of A. heimuer
2.2. Determination of Free Amino Acids
2.3. RNA Extraction and cDNA Library Construction
2.4. Bioinformatics Analysis
2.5. RT-qPCR Validation
2.6. Data Statistics and Analysis
3. Results
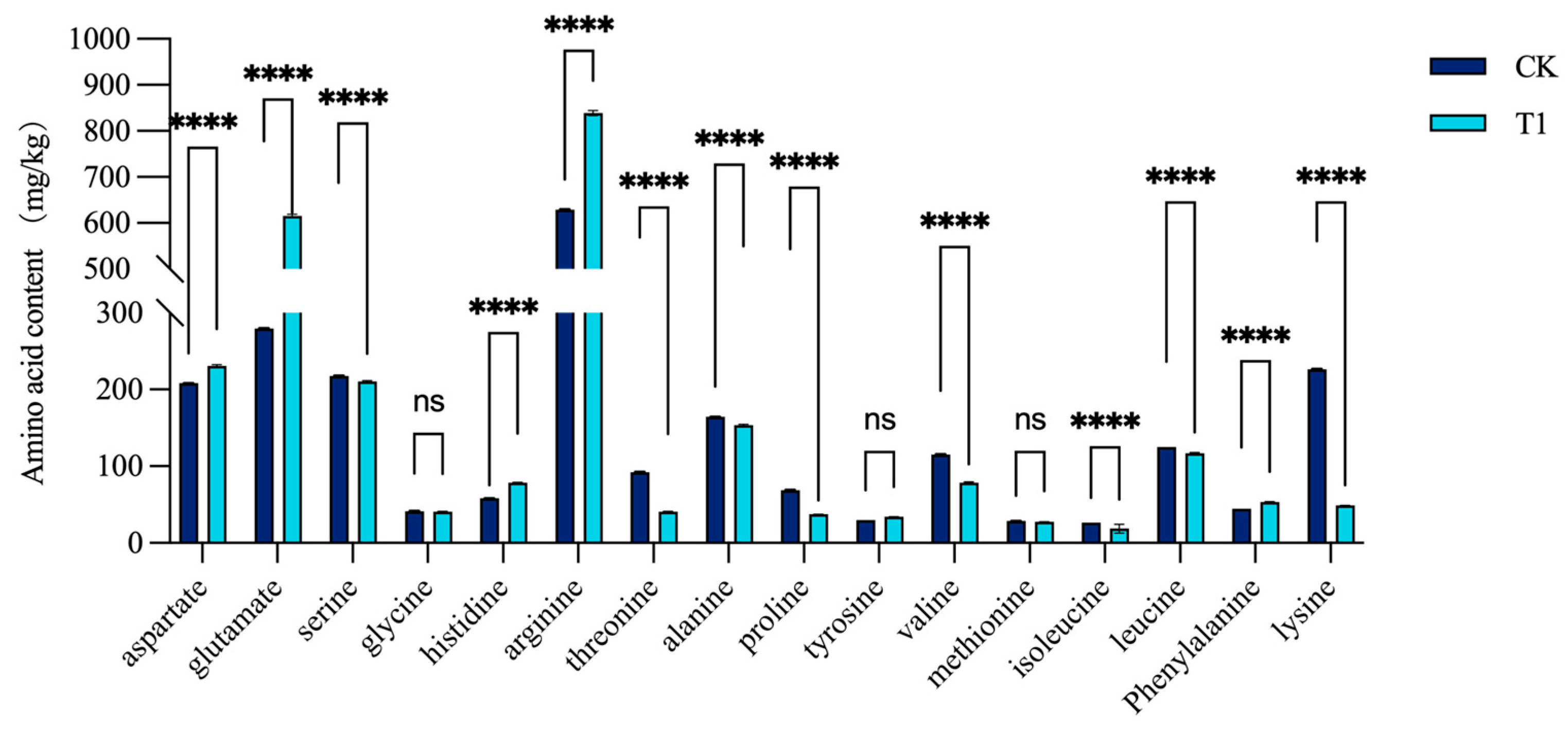
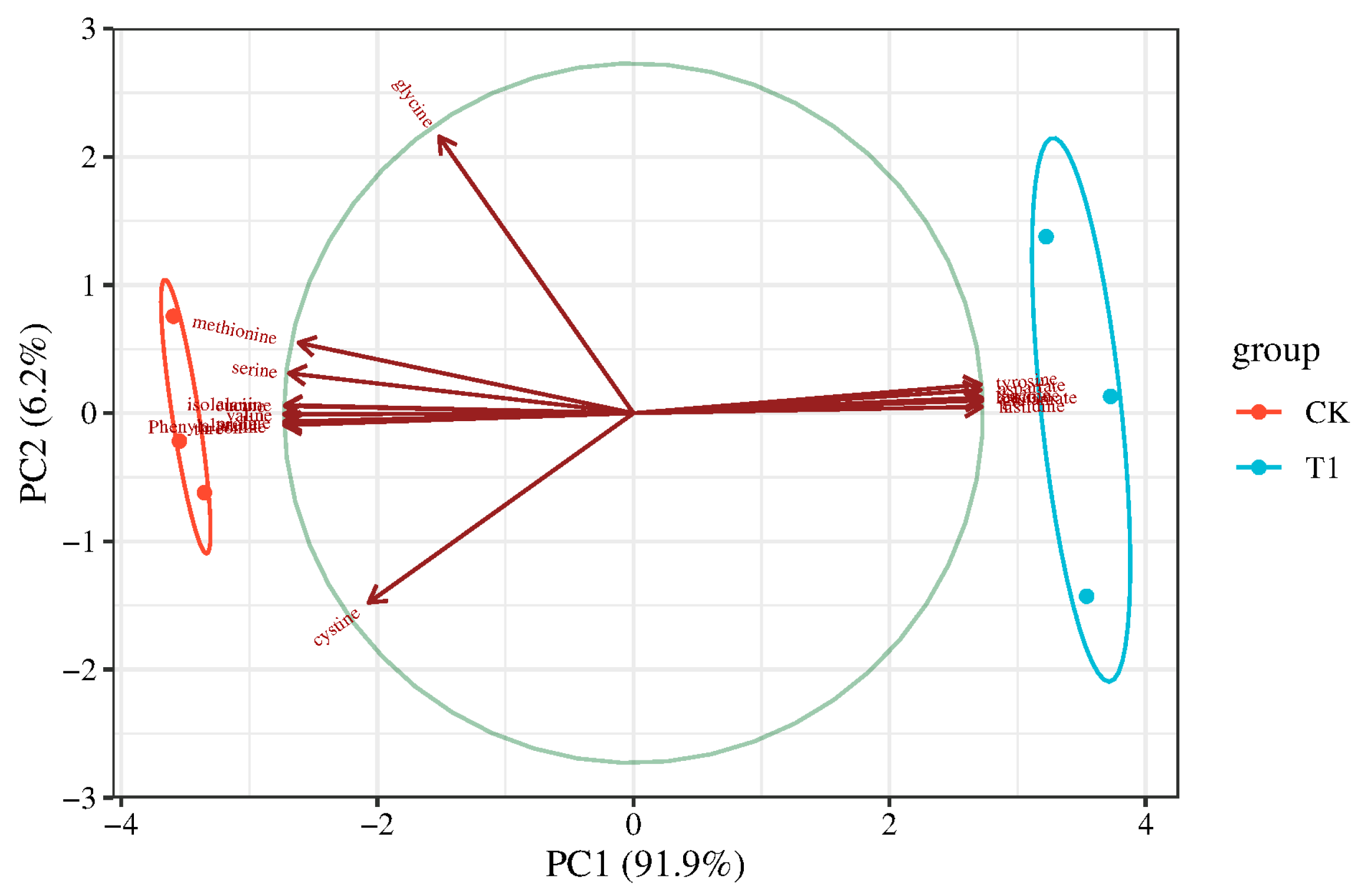
3.1. Determination of Free Amino Acid Content
3.2. Transcriptome Analysis
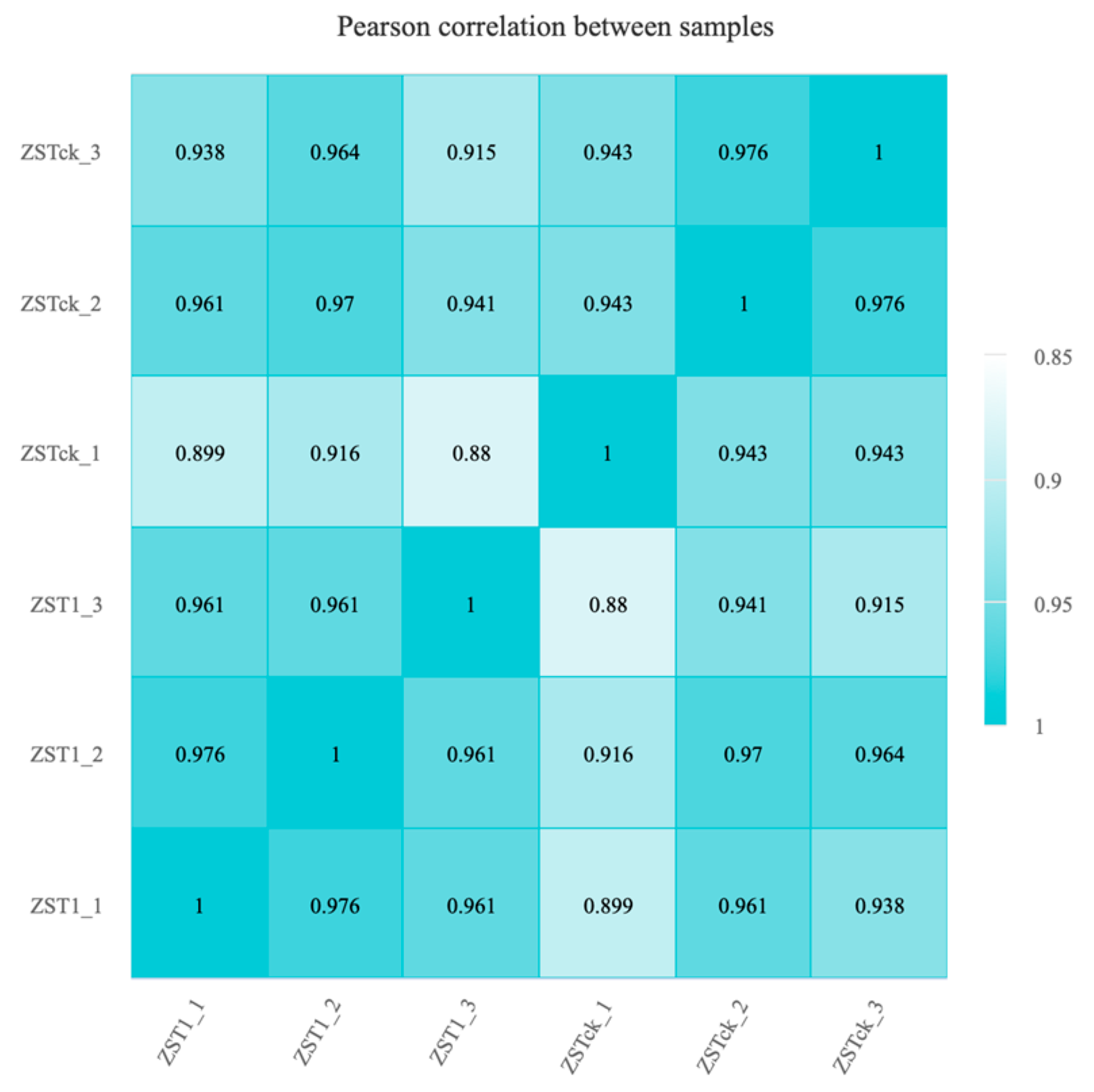
3.2.1. Quality Assessment of Sequencing Data
3.2.2. RT-qPCR Validation
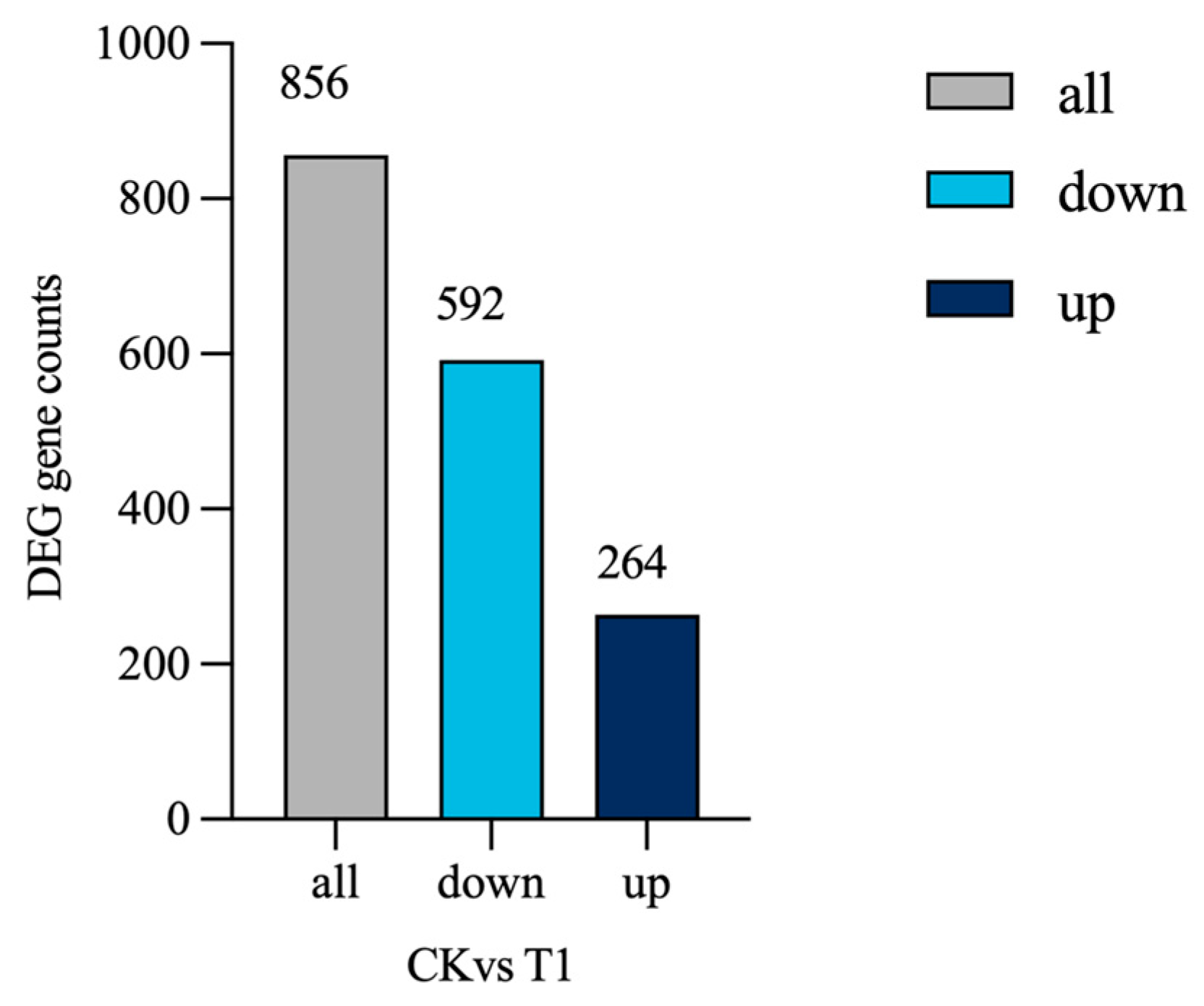
3.2.3. Differential Gene Expression
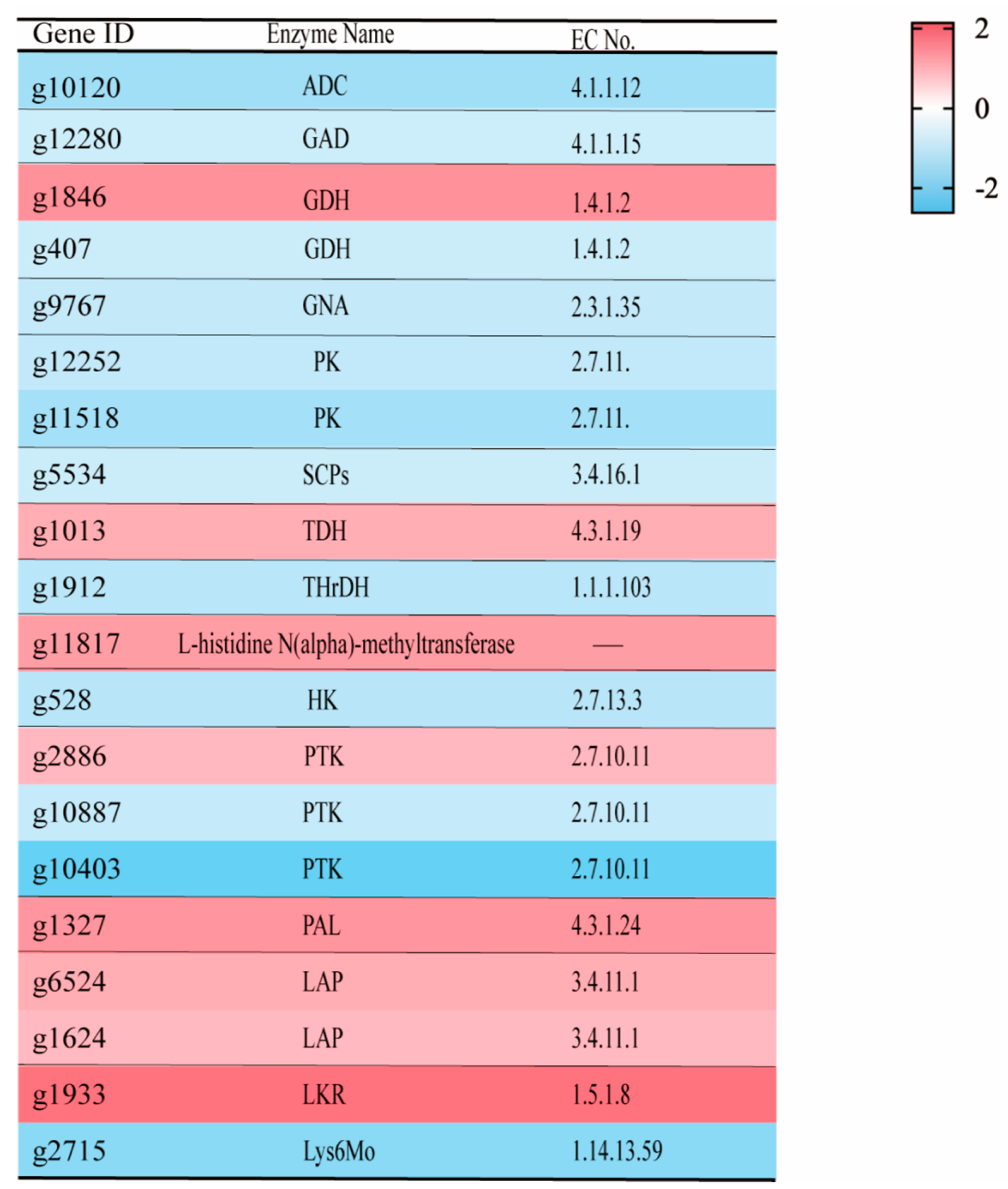
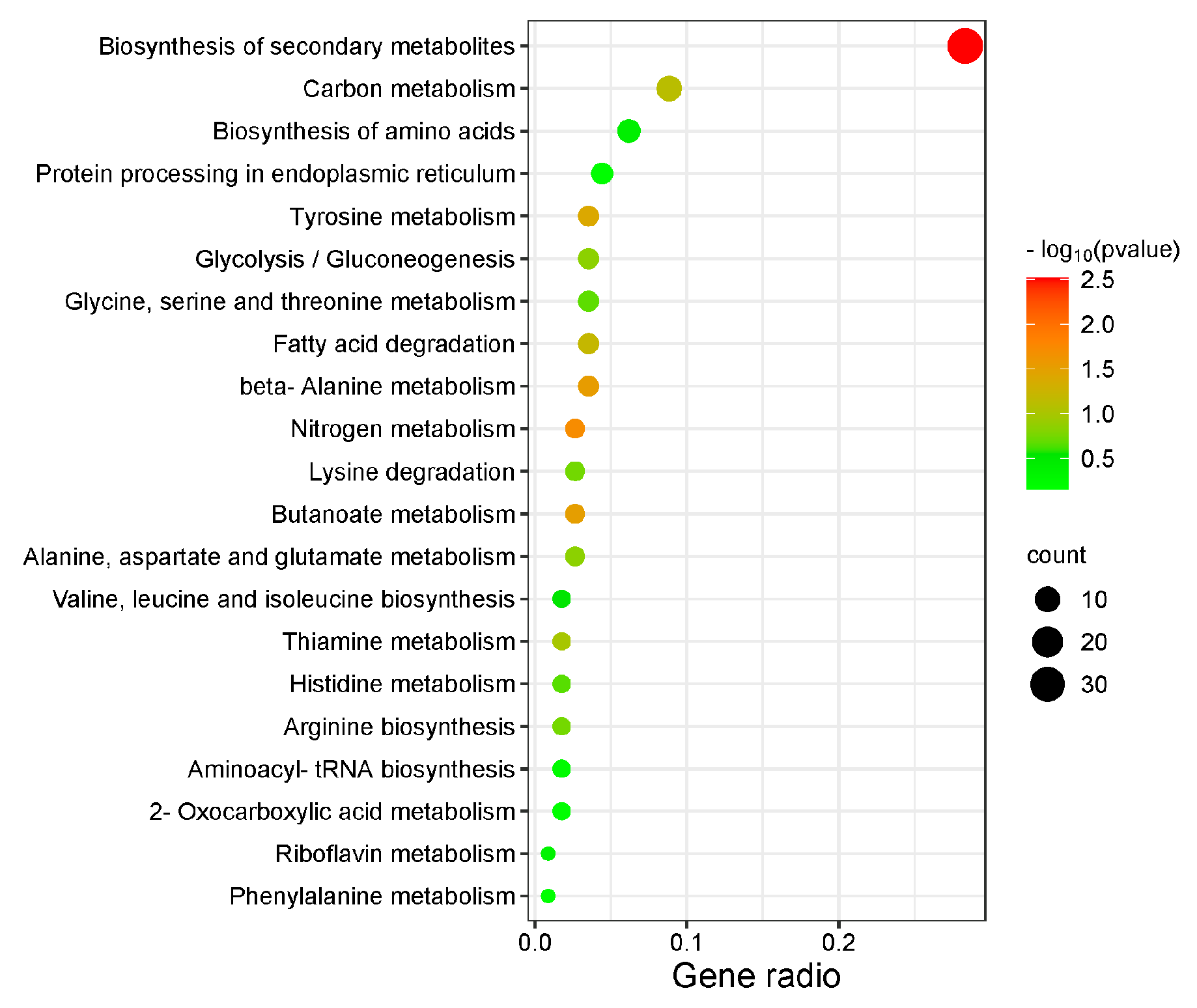
3.2.4. KEGG Pathway Enrichment Analysis
3.2.5. GO Enrichment Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, S.J.; Zhang, X.J.; Chen, W.X.; Zhang, L.Y.; Zhu, H. Production of natural edible melanin by and its physicochemical properties. Food Chem. 2016, 196, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Reis, F.S.; Martins, A.; Vasconcelos, M.H.; Morales, P.; Ferreira, I.C.F.R. Functional foods based on extracts or compounds derived from mushrooms. Trends Food Sci. Tech. 2017, 66, 48–62. [Google Scholar] [CrossRef]
- Wang, W.L.; Wang, Y.S.; Gong, Z.Q.; Yang, S.F.; Jia, F.J. Comparison of the Nutritional Properties and Transcriptome Profiling Between the Two Different Harvesting Periods of Auricularia polytricha. Front. Nutr. 2021, 8, 771757. [Google Scholar] [CrossRef] [PubMed]
- Niazi, A.R.; Ghafoor, A. Different ways to exploit mushrooms: A review. All Life 2021, 14, 450–460. [Google Scholar] [CrossRef]
- Yan, X.; Li, S.; Tu, T.Y.; Li, Y.Q.; Niu, M.S.; Tong, Y.Q.; Yang, Y.; Xu, T.; Zhao, J.; Shen, C.H.; et al. Free amino acids identification and process optimization in greengage wine fermentation and flavor formation. J. Food Sci. 2023, 88, 988–1003. [Google Scholar] [CrossRef]
- Jeong, S.; Jeon, Y.; Mun, J.; Jeong, S.M.; Liang, H.L.; Chung, K.Y.W.; Yi, P.I.; An, B.S.; Seo, S. Ninhydrin Loaded Microcapsules for Detection of Natural Free Amino Acid. Chemosensors 2023, 11, 49. [Google Scholar] [CrossRef]
- Menchini, R.J.; Chaudhry, F.A. Multifaceted regulation of the system A transporter Slc38a2 suggests nanoscale regulation of amino acid metabolism and cellular signaling. Neuropharmacology 2019, 161, 107789. [Google Scholar] [CrossRef]
- Han, X.; Guan, Q.; Zhou, X. Analysis of volatile flavors and non-volatile flavor compounds of 7 common edible fungi. Food Technol. 2024, 49, 133–140. [Google Scholar]
- Li, J.H.; Xie, J.C.; Huang, Z.N.; Yang, P.L.; Li, D.; Chen, L.D.; Sun, S.J. Metabolomics-Based Analysis on the Effect and Metabolic Response of Mycelia by Sawdust Addition from Hypsizygus marmoreus. Foods 2024, 13, 867. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, X.; Wang, D.; Cui, C.; Gao, C.; Wang, L.; Wang, Y.; Bi, Y. Estimation and utilization of corncob resources in China. Agric. Resour. Zoning China 2016, 37, 1–8. [Google Scholar]
- Aggarangsi, N.S.P. Physical and chemical characteristics of carbonized corncob through hydrothermal and pyrolysis conversion. IOP Conf. Ser. Mater. Sci. Eng. 2021, 1137, 012004. [Google Scholar] [CrossRef]
- Yu, H.L.; Zhang, D.; Zhang, L.J.; Li, Q.Z.; Song, C.Y.; Shang, X.D.; Bao, D.P.; Tan, Q.; Chen, H.Y.; Lv, B.B. Corncob as a Substrate for the Cultivation of Lentinula edodes. Waste Biomass Valori 2022, 13, 929–939. [Google Scholar] [CrossRef]
- Castorina, G.; Cappa, C.; Negrini, N.; Criscuoli, F.; Casiraghi, M.C.; Marti, A.; Rollini, M.; Consonni, G.; Erba, D. Characterization and nutritional valorization of agricultural waste corncobs from Italian maize landraces through the growth of medicinal mushrooms. Sci. Rep. 2023, 13, 21148. [Google Scholar] [CrossRef]
- Ariyanti, D.; Rimantho, D.; Leonardus, M.; Ardyani, T.; Fiviyanti, S.; Sarwana, W.; Fiviyanti, S.; Sarwana, W.; Hanifah, Y.; Agustian, E.; et al. Valorization of corn cob waste for furfural production: A circular economy approach. Biomass Bioenergy 2025, 194, 107665. [Google Scholar] [CrossRef]
- Asonja, A.; Desnica, E.; Radovanovic, L. Energy efficiency analysis of corn cob used as a fuel. Energy Source Part B 2017, 12, 1–7. [Google Scholar] [CrossRef]
- Fan, Y.F.; Ji, H.R. Corncob Fractionations Toward Two Purposes: Furfural Production and Papermaking. Bioenergy Res. 2024, 17, 359–368. [Google Scholar] [CrossRef]
- Li, X.R.; Luo, L.; Wang, X.Y.; Zhu, M. Further insights into the molecular mechanisms underlying tobacco straw cultivation of Pleurotus ostreatus by comparative transcriptome analyses. Genomics 2025, 117, 110992. [Google Scholar] [CrossRef]
- Xu, S.; Wang, F.; Fu, Y.P.; Li, D.; Sun, X.Z.; Li, C.T.; Song, B.; Li, Y. Effects of mixed agro-residues (corn crop waste) on lignin-degrading enzyme activities, growth, and quality of Lentinula edodes. Rsc Adv. 2020, 10, 9798–9807. [Google Scholar] [CrossRef]
- Lu, J.; Lu, L.X.; Yao, F.J.; Fang, M.; Ma, X.X.; Meng, J.J.; Shao, K.S. Dicarboxylic Amino Acid Permease 7219 Regulates Fruiting Body Type of Auricularia heimuer. J. Fungi 2023, 9, 876. [Google Scholar] [CrossRef]
- Xu, A.R.; Yang, D.; Jacob, M.S.; Qian, K.Q.; Yang, X.Y.; Zhang, B.; Li, X. Comprehensive evaluation of agronomic traits and mineral elements of Auricularia heimuer cultivated on corncob substrates. Sci. Hortic. 2023, 314, 111942. [Google Scholar] [CrossRef]
- Chen, Y.; Yao, F.; Liu, G.; Wang, H.; Liang, Y. The Announcements and Suggestions of Auricularia auricula Cultivation Using Substitute Media. Edible Fungi China 2010, 29, 55–58. [Google Scholar] [CrossRef]
- Wang, P.; Liang, N.; Han, S.; Yu, D.; Xin, X. Simultaneous Determination of 17 Free Amino Acids in Bee Products by UHPLC-MS/MS Method. China Port Sci. Technol. 2025, 7, 70–83. [Google Scholar]
- Mortazavi, A.; Williams, B.A.; Mccue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Fangjie, Y.; Wenjuan, S.; Ming, F.; Chunshuang, W. Screening of reference genes for qRT-PCR amplification in Auricularia heimuer. Mycosystema 2020, 39, 1510–1519. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Hoa, H.T.; Wang, C.L.; Wang, C.H. The Effects of Different Substrates on the Growth, Yield, and Nutritional Composition of Two Oyster Mushrooms (Pleurotus ostreatus and Pleurotus cystidiosus). Mycobiology 2015, 43, 423–434. [Google Scholar] [CrossRef]
- Chilanti, G.; da Rosa, L.O.; Poleto, L.; Branco, C.S.; Camassola, M.; Fontana, R.C.; Dillon, A.J.P. Effect of different substrates on Pleurotus spp. cultivation in Brazil-Ergothioneine and lovastatin. J. Food Compos. Anal. 2022, 107, 104367. [Google Scholar] [CrossRef]
- Huang, L.; Sun, N.; Ban, L.T.; Wang, Y.; Yang, H.P. Ability of different edible fungi to degrade crop straw. Amb Express 2019, 9, 4. [Google Scholar] [CrossRef]
- Sun, L.P.; Liu, Q.M.; Bao, C.J.; Fan, J. Comparison of Free Total Amino Acid Compositions and Their Functional Classifications in 13 Wild Edible Mushrooms. Molecules 2017, 22, 350. [Google Scholar] [CrossRef]
- Tagkouli, D.; Kaliora, A.; Bekiaris, G.; Koutrotsios, G.; Christea, M.; Zervakis, G.I.; Kalogeropoulos, N. Free Amino Acids in Three Pleurotus Species Cultivated on Agricultural and Agro-Industrial By-Products. Molecules 2020, 25, 4015. [Google Scholar] [CrossRef]
- Davila, M.; Muniz, A.; Du, X.F. The impact of roasting and steaming on savory flavors contributed by amino acids, 5′-nucleotides, and volatiles in Agaricus bisporus mushrooms. Int. J. Gastron. Food Sci. 2022, 30, 100590. [Google Scholar] [CrossRef]
- Kim, M.Y.; Chung, M.; Lee, S.J.; Ahn, J.K.; Kim, E.H.; Kim, M.J.; Kim, S.L.; Moon, H.I.; Ro, H.M.; Kang, E.Y.; et al. Comparison of free amino acid, carbohydrates concentrations in Korean edible and medicinal mushrooms. Food Chem. 2008, 113, 386–393. [Google Scholar] [CrossRef]
- Bachmanov, A.A.; Bosak, N.P.; Glendinning, J.I.; Inoue, M.; Li, X.; Manita, S.; McCaughey, S.A.; Murata, Y.; Reed, D.R.; Tordoff, M.G.; et al. Genetics of amino acid taste and appetite. Chem. Senses 2016, 41, E270–E271. [Google Scholar] [CrossRef]
- Wang, Z.C.; Li, M.R.; Fan, J.D.; Bao, Y.T.; Chen, Q. Cultivation and Nutritional Evaluation of Agaricus bisporus with Tea Residue as Culture Medium. Foods 2023, 12, 2440. [Google Scholar] [CrossRef]
- Wang, Q.Y.; Zhao, M.R.; Wang, Y.Y.; Xie, Z.F.; Zhao, S.Y.; You, S.N.; Chen, Q.J.; Zhang, W.W.; Qin, Y.; Zhang, G.Q. Microbial Inoculation during the Short-Term Composting Process Enhances the Nutritional and Functional Properties of Oyster Mushrooms (Pleurotus ostreatus). Life 2024, 14, 201. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, Y.P.; Zhang, Z.J.; Liu, X.M.; Li, C.; Ma, F.W. Arginine Increases Tolerance to Nitrogen Deficiency in Malus hupehensis via Alterations in Photosynthetic Capacity and Amino Acids Metabolism. Front. Plant Sci. 2022, 12, 772086. [Google Scholar] [CrossRef]
- Zimmermann, S.E.; Benstein, R.M.; Flores-Tornero, M.; Blau, S.; Anoman, A.D.; Rosa-Téllez, S.; Gerlich, S.C.; Salem, M.A.; Alseekh, S.; Kopriva, S.; et al. The phosphorylated pathway of serine biosynthesis links plant growth with nitrogen metabolism. Plant Physiol. 2021, 186, 1487–1506. [Google Scholar] [CrossRef]
- Ma, H.L.; Xu, X.H.; Feng, L.J. Responses of antioxidant defenses and membrane damage to drought stress in fruit bodies of Auricularia auricula-judae. World J. Microbiol. Biotechnol. 2014, 30, 119–124. [Google Scholar] [CrossRef]
- Lin, T.; Li, Z.Y.; Fan, G.J.; Xie, C.Y. Enhancing the nutritional value and antioxidant properties of foxtail millet by solid-state fermentation with edible fungi. Food Sci. Nutr. 2024, 12, 6660–6672. [Google Scholar] [CrossRef]
- Bonvini, A.; Coqueiro, A.Y.; Tirapegui, J.; Calder, P.C.; Rogero, M.M. Immunomodulatory role of branched-chain amino acids. Nutr. Rev. 2018, 76, 840–856. [Google Scholar] [CrossRef]
- Li, J.; Ma, J.M.; Fan, S.F.; Mi, S.Q.; Zhang, Y. Comparison of the Nutritional and Taste Characteristics of 5 Edible Fungus Powders Based on the Composition of Hydrolyzed Amino Acids and Free Amino Acids. J. Food Qual. 2022, 2022, 3618002. [Google Scholar] [CrossRef]
- Bao, X.Y.; Feng, Z.M.; Yao, J.M.; Li, T.J.; Yin, Y.L. Roles of Dietary Amino Acids and Their Metabolites in Pathogenesis of Inflammatory Bowel Disease. Mediat. Inflamm. 2017, 2017, 6869259. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, G.; Pasini, E.; Scarabelli, T.M.; Romano, C.; Singh, A.; Scarabelli, C.C.; Dioguardi, F.S. Importance of Energy, Dietary Protein Sources, and Amino Acid Composition in the Regulation of Metabolism: An Indissoluble Dynamic Combination for Life. Nutrients 2024, 16, 2417. [Google Scholar] [CrossRef]
- Ye, Z.H.; Wang, S.Y.; Zhang, C.M.; Zhao, Y. Coordinated Modulation of Energy Metabolism and Inflammation by Branched-Chain Amino Acids and Fatty Acids. Front. Endocrinol. 2020, 11, 617. [Google Scholar] [CrossRef]
- Li, D.; Wang, D.; Fang, Y.D.; Belwal, T.; Li, L.; Lin, X.Y.; Xu, Y.Q.; Chen, H.J.; Zhu, M.; Luo, Z.S. Involvement of energy metabolism and amino acid metabolism in quality attributes of postharvest treated with a novel phase change material. Postharvest Biol. Technol. 2021, 173, 111427. [Google Scholar] [CrossRef]
- El-Azaz, J.; de la Torre, F.; Pascual, M.B.; Debille, S.; Canlet, F.; Harvengt, L.; Trontin, J.F.; Avila, C.; Cánovas, F.M. Transcriptional analysis of arogenate dehydratase genes identifies a link between phenylalanine biosynthesis and lignin biosynthesis. J. Exp. Bot. 2020, 71, 3080–3093. [Google Scholar] [CrossRef]
- Moura, C.S.; Lollo, P.C.B.; Morato, P.N.; Risso, E.M.; Amaya-Farfan, J. Modulatory effects of arginine, glutamine and branched-chain amino acids on heat shock proteins, immunity and antioxidant response in exercised rats. Food Funct. 2017, 8, 3228–3238. [Google Scholar] [CrossRef]
- Xu, B.; Feng, X.Y.; Piechatzek, A.; Zhang, S.Q.; Konrad, K.R.; Kromdijk, J.; Hedrich, R.; Gilliham, M. The GABA shunt contributes to ROS homeostasis in guard cells of Arabidopsis. New Phytol. 2024, 241, 73–81. [Google Scholar] [CrossRef]
- Benidickson, K.H.; Raytek, L.M.; Hoover, G.J.; Flaherty, E.J.; Shelp, B.J.; Snedden, W.A.; Plaxton, W.C. Glutamate decarboxylase-1 is essential for efficient acclimation of Arabidopsis thaliana to nutritional phosphorus deprivation. New Phytol. 2023, 240, 2372–2385. [Google Scholar] [CrossRef]
- Moreira, T.B.; Williams, T.C.R. Inferring catabolism through analysis of amino acid balance in Vicia faba L. seedlings. Braz. J. Bot. 2021, 44, 859–868. [Google Scholar] [CrossRef]
- Hu, G.Q.; He, H.B.; Zhang, W.; Zhao, J.S.; Cui, J.H.; Li, B.; Zhang, X.D. The transformation and renewal of soil amino acids induced by the availability of extraneous C and N. Soil Biol. Biochem. 2016, 96, 86–96. [Google Scholar] [CrossRef]
- Fuentes-Lemus, E.; Reyes, J.S.; Figueroa, J.D.; Davies, M.J.; López-Alarcón, C. The enzymes of the oxidative phase of the pentose phosphate pathway as targets of reactive species consequences for NADPH production. Biochem. Soc. Trans. 2023, 51, 2173–2187. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.S.; Fuentes-Lemus, E.; Romero, J.; Arenas, F.; Fierro, A.; Davies, M.J.; López-Alarcón, C. Peroxyl radicals modify 6-phosphogluconolactonase from Escherichia coli via oxidation of specific amino acids and aggregation which inhibits enzyme activity. Free Radic. Bio Med. 2023, 204, 118–127. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Yao, F.; Lai, F.; Fang, M.; Lu, L.; Ma, X.; Wang, W.; Meng, J.; Shao, K. Transcriptomic Insights and Quantification of Free Amino Acids in Auricularia heimuer Cultivated on Corncob Substrate. Horticulturae 2025, 11, 563. https://doi.org/10.3390/horticulturae11060563
Sun X, Yao F, Lai F, Fang M, Lu L, Ma X, Wang W, Meng J, Shao K. Transcriptomic Insights and Quantification of Free Amino Acids in Auricularia heimuer Cultivated on Corncob Substrate. Horticulturae. 2025; 11(6):563. https://doi.org/10.3390/horticulturae11060563
Chicago/Turabian StyleSun, Xu, Fangjie Yao, Fanchao Lai, Ming Fang, Lixin Lu, Xiaoxu Ma, Wei Wang, Jingjing Meng, and Kaisheng Shao. 2025. "Transcriptomic Insights and Quantification of Free Amino Acids in Auricularia heimuer Cultivated on Corncob Substrate" Horticulturae 11, no. 6: 563. https://doi.org/10.3390/horticulturae11060563
APA StyleSun, X., Yao, F., Lai, F., Fang, M., Lu, L., Ma, X., Wang, W., Meng, J., & Shao, K. (2025). Transcriptomic Insights and Quantification of Free Amino Acids in Auricularia heimuer Cultivated on Corncob Substrate. Horticulturae, 11(6), 563. https://doi.org/10.3390/horticulturae11060563