
Comparative Transcriptome Profiling Reveals Differential Defense Responses of Resistant and Susceptible Phalaenopsis to Dickeya fangzhongdai


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials, Bacterial Strain, and Plant Infection
2.2. Determination of Physiological and Biochemical Indicators
2.3. Isolation of Total RNA, Library Preparation, and Sequencing
2.4. Differentially Expressed Gene (DEG) Analysis
2.5. GO and KEGG Enrichment Analysis
2.6. Transcription Factor Family Analysis
2.7. Quantitative Real-Time PCR Analysis
2.8. Statistical Analysis
3. Results
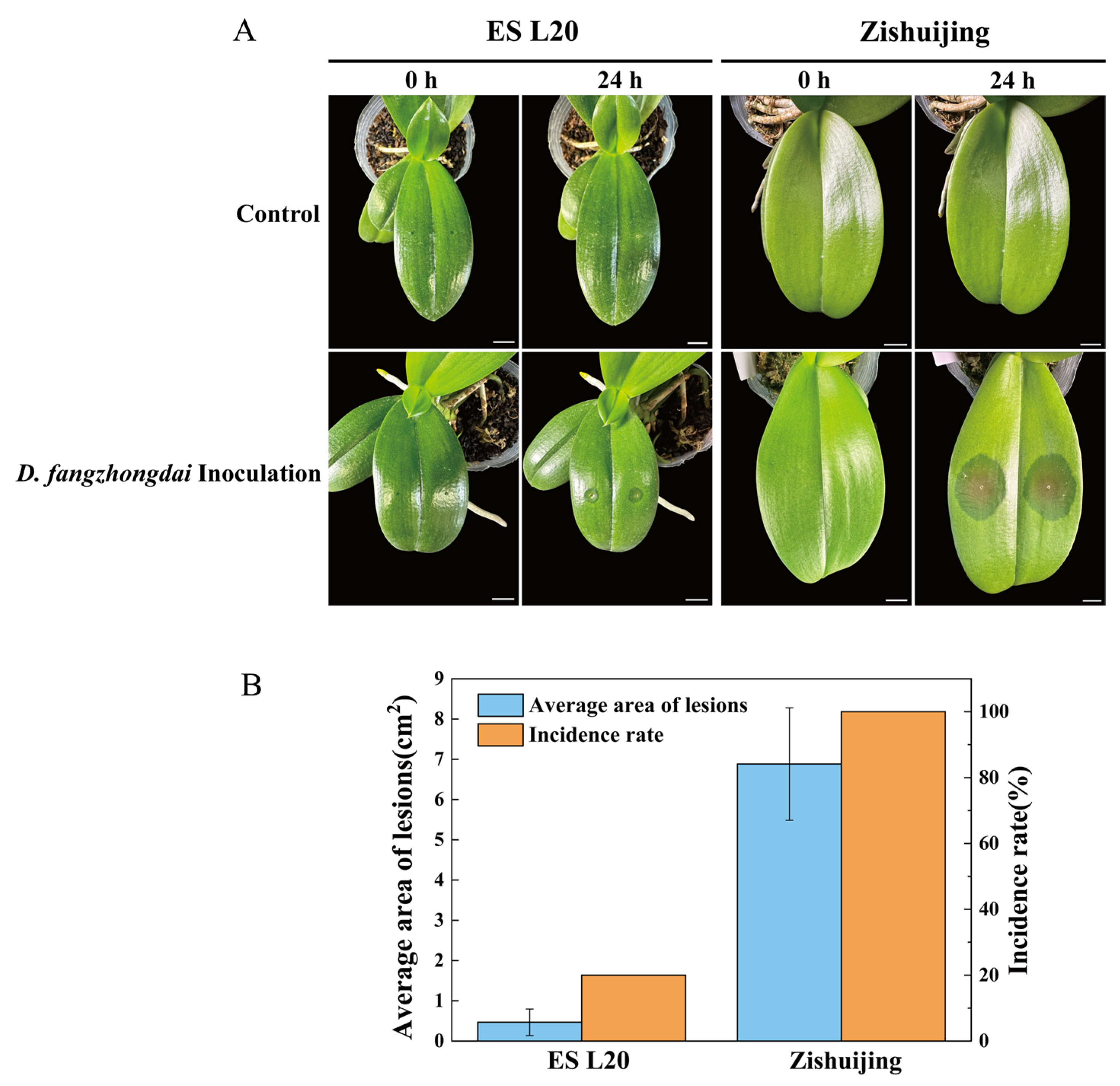
3.1. Evaluation of Phalaenopsis Varieties After Soft Rot Infection
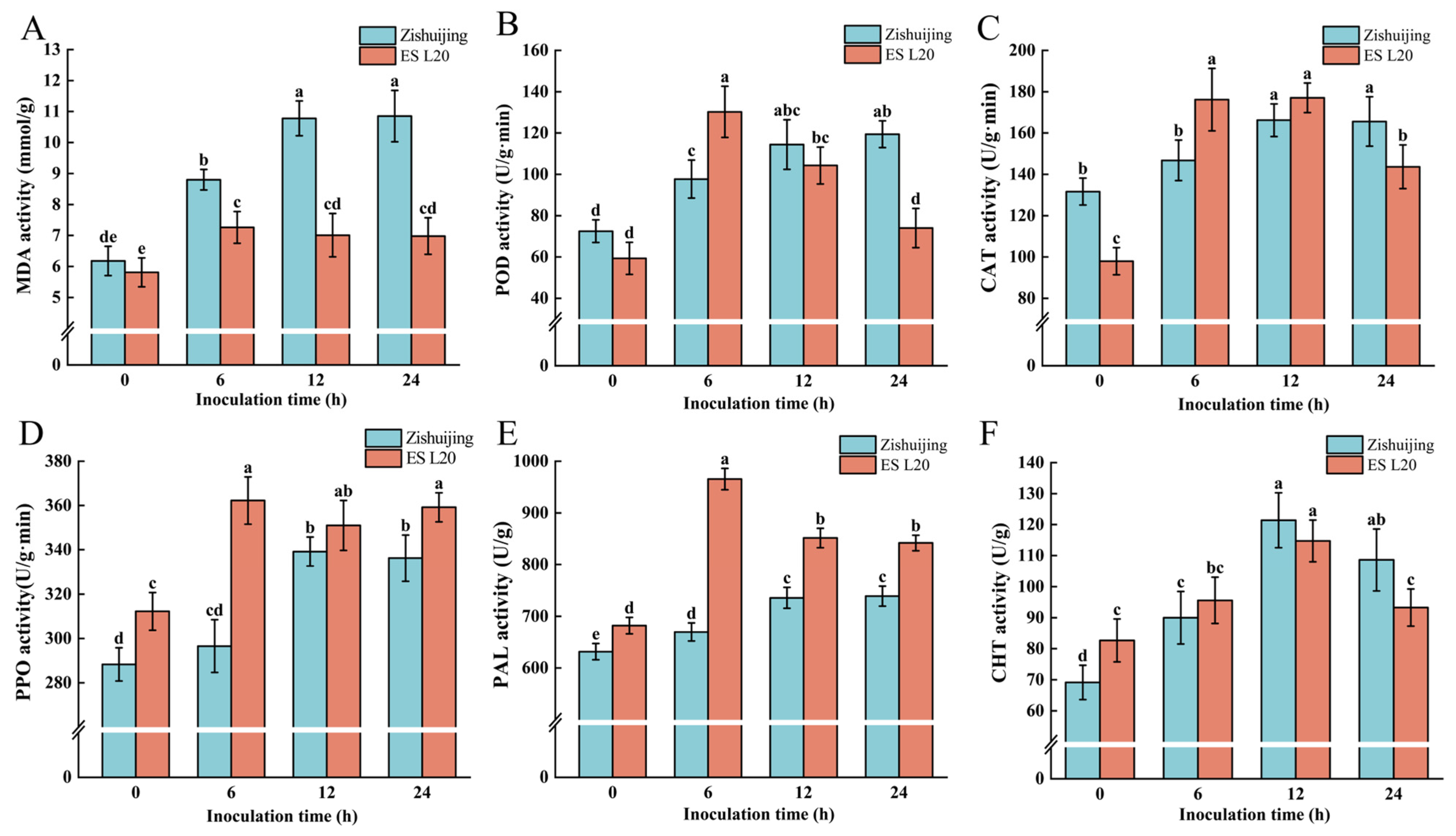
3.2. Physiological Indexes of the Two Varieties After D. fangzhongdai Infection
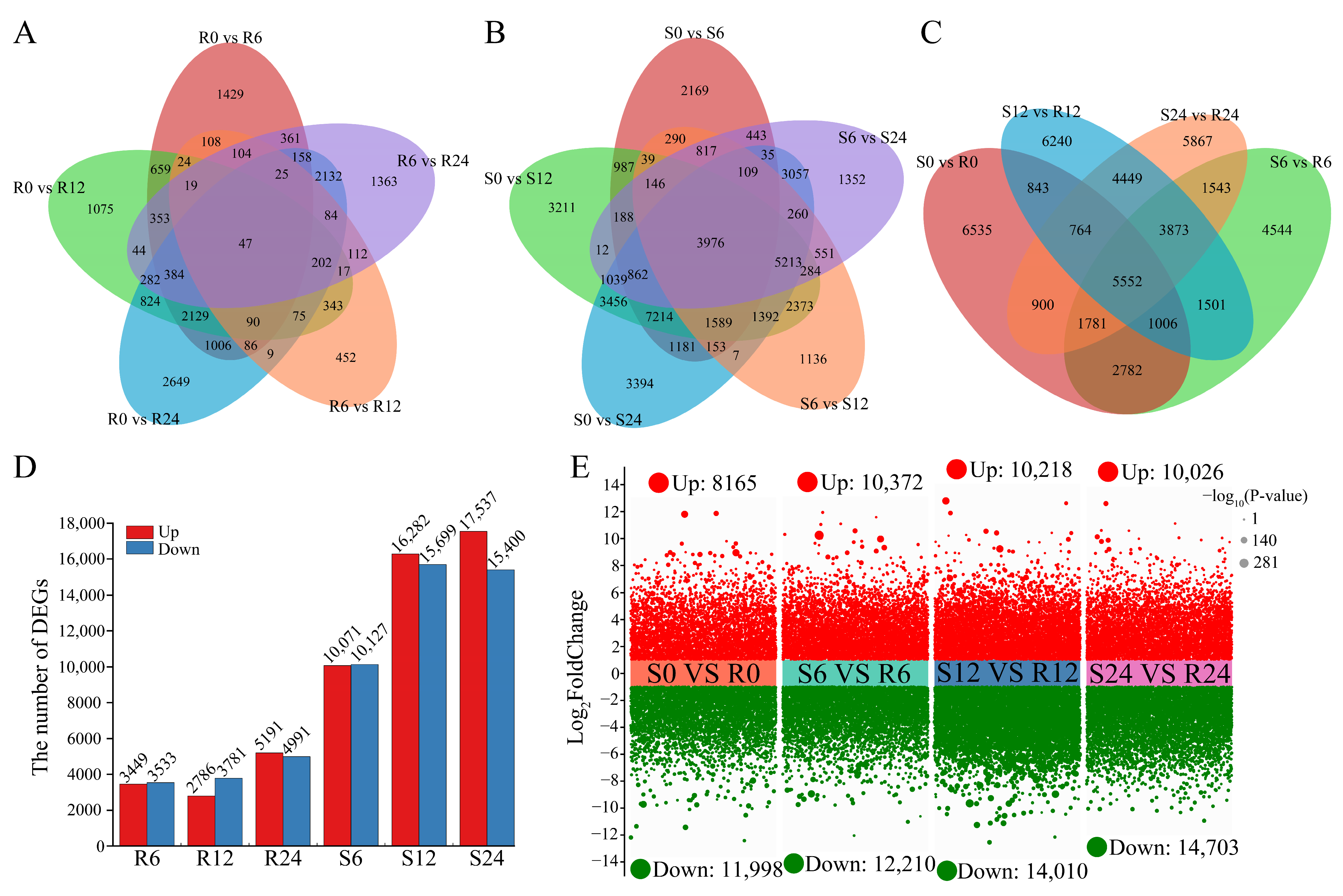
3.3. Transcriptome Sequencing Analysis
3.4. DEGs Between R (‘ES L20’) and S (‘Zishuijing’) at Four Time Points
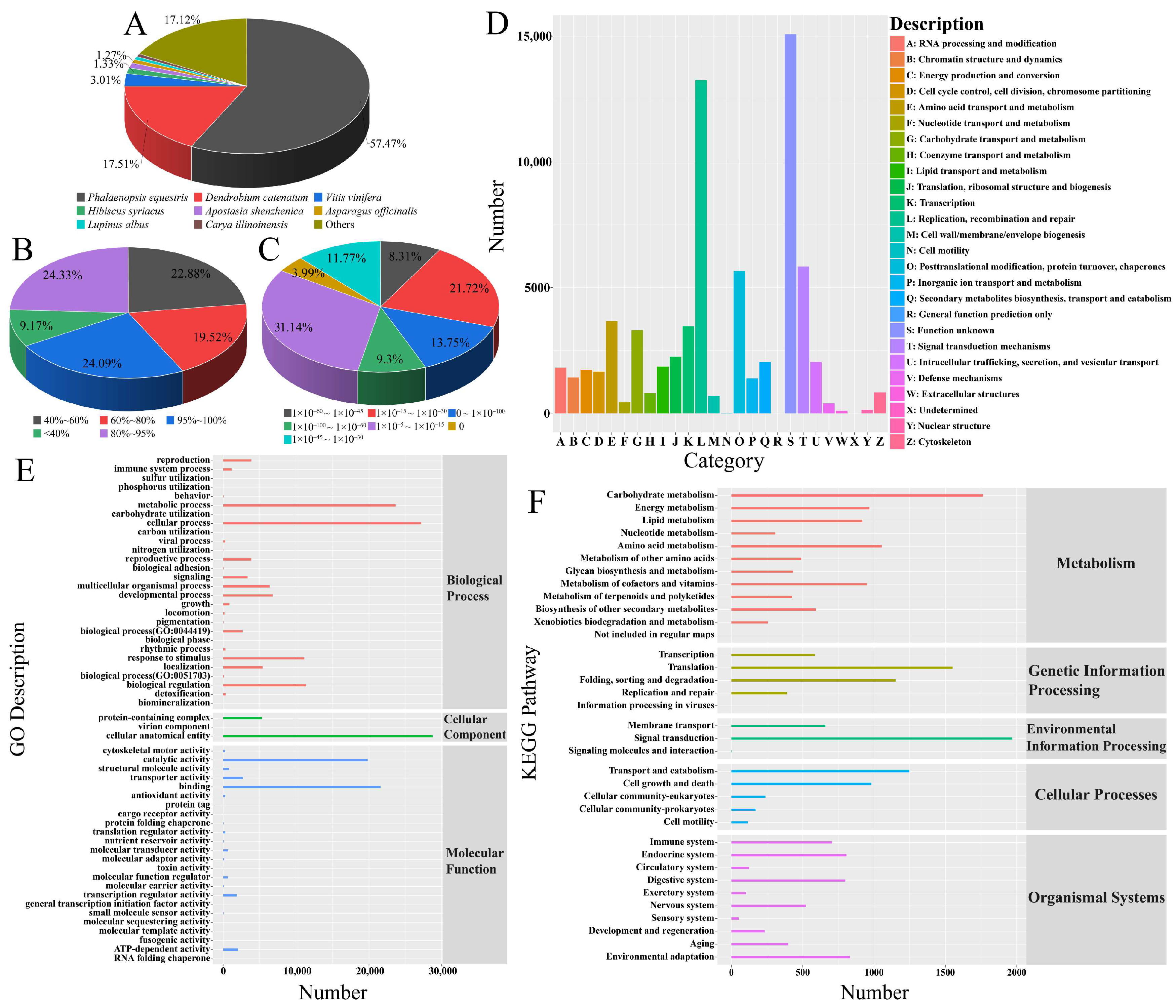
3.5. Unigene Gene Function Annotation
3.6. GO Enrichment Analysis
3.7. KEGG Enrichment Analysis
3.8. Plant–Pathogen Interaction
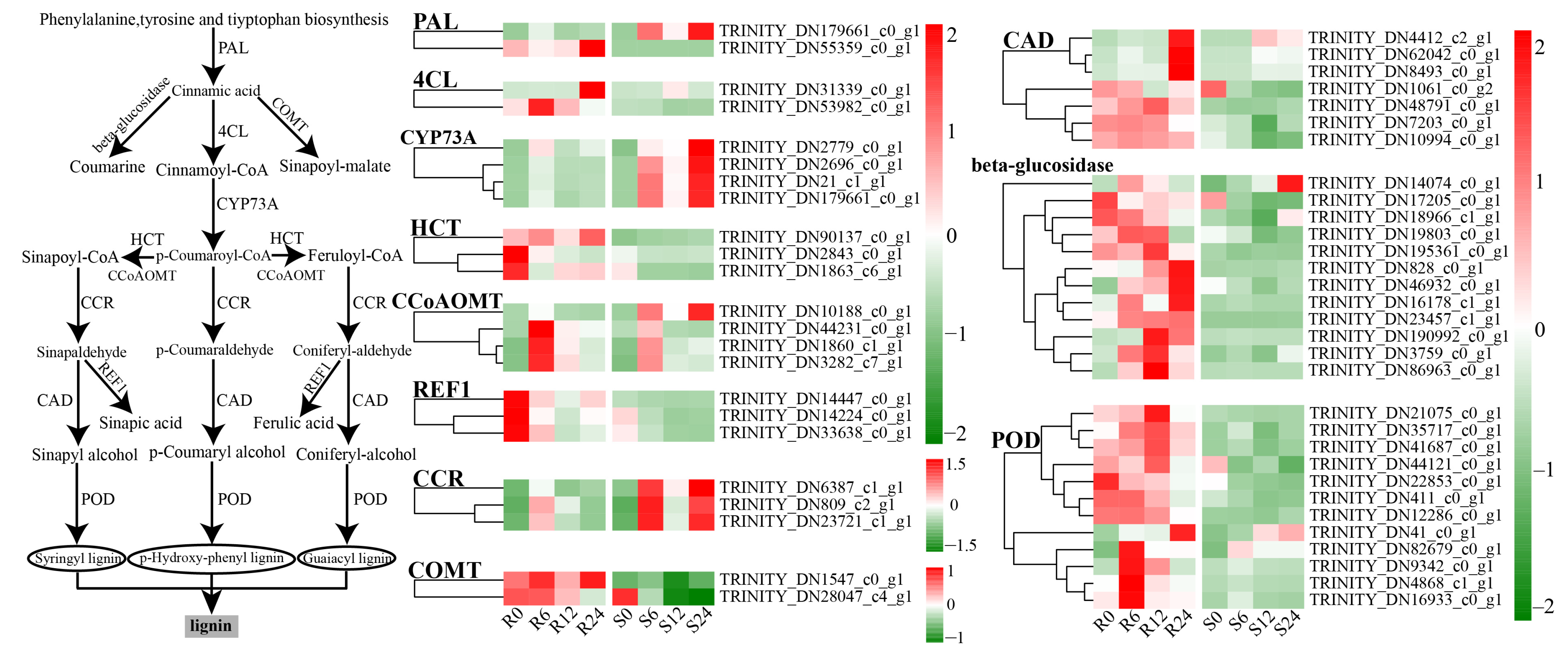
3.9. Differential Expression Analysis of Phenylpropane Metabolism
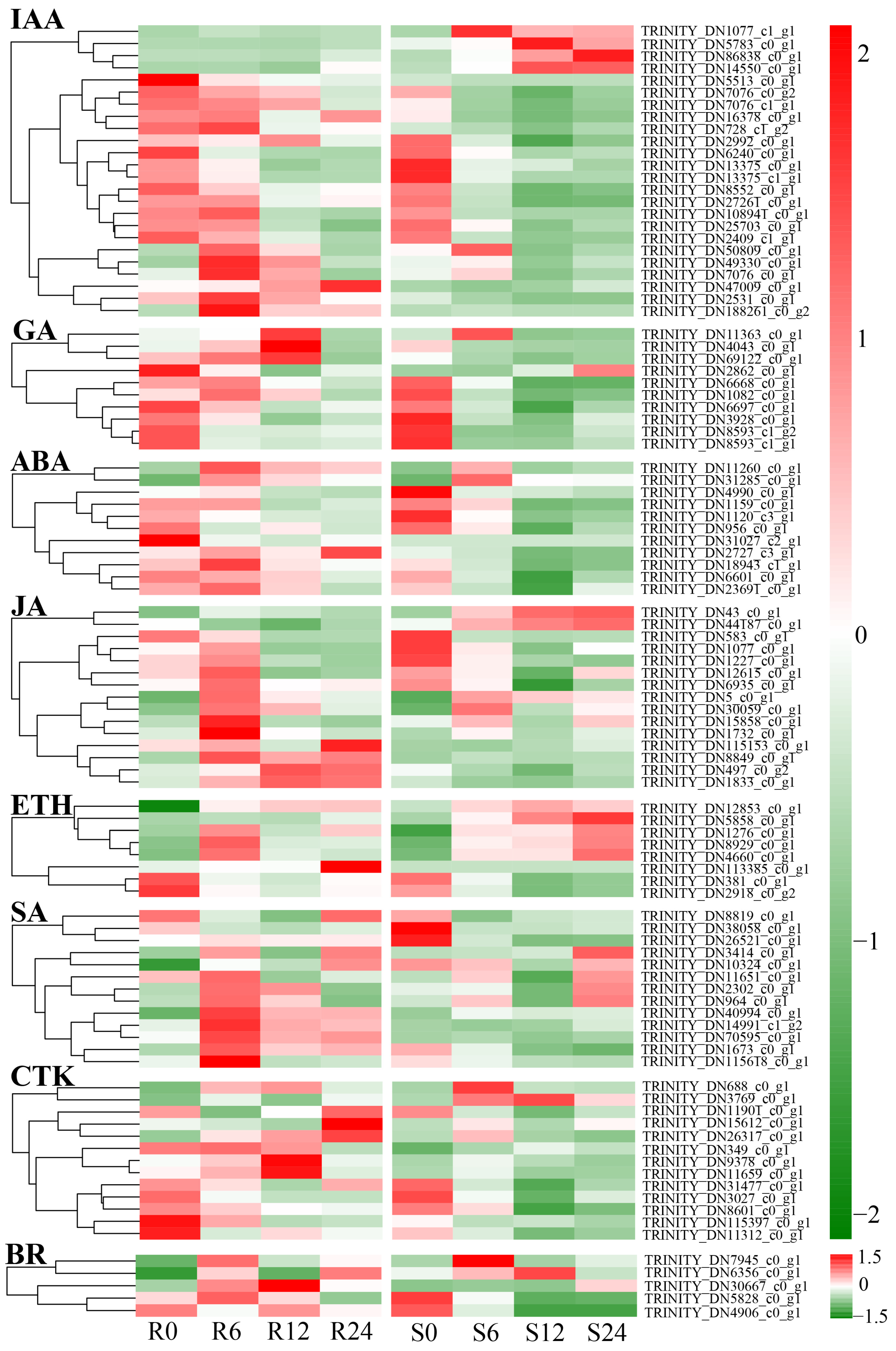
3.10. Differential Expression Analysis of Plant Hormone Signal Transduction Pathways
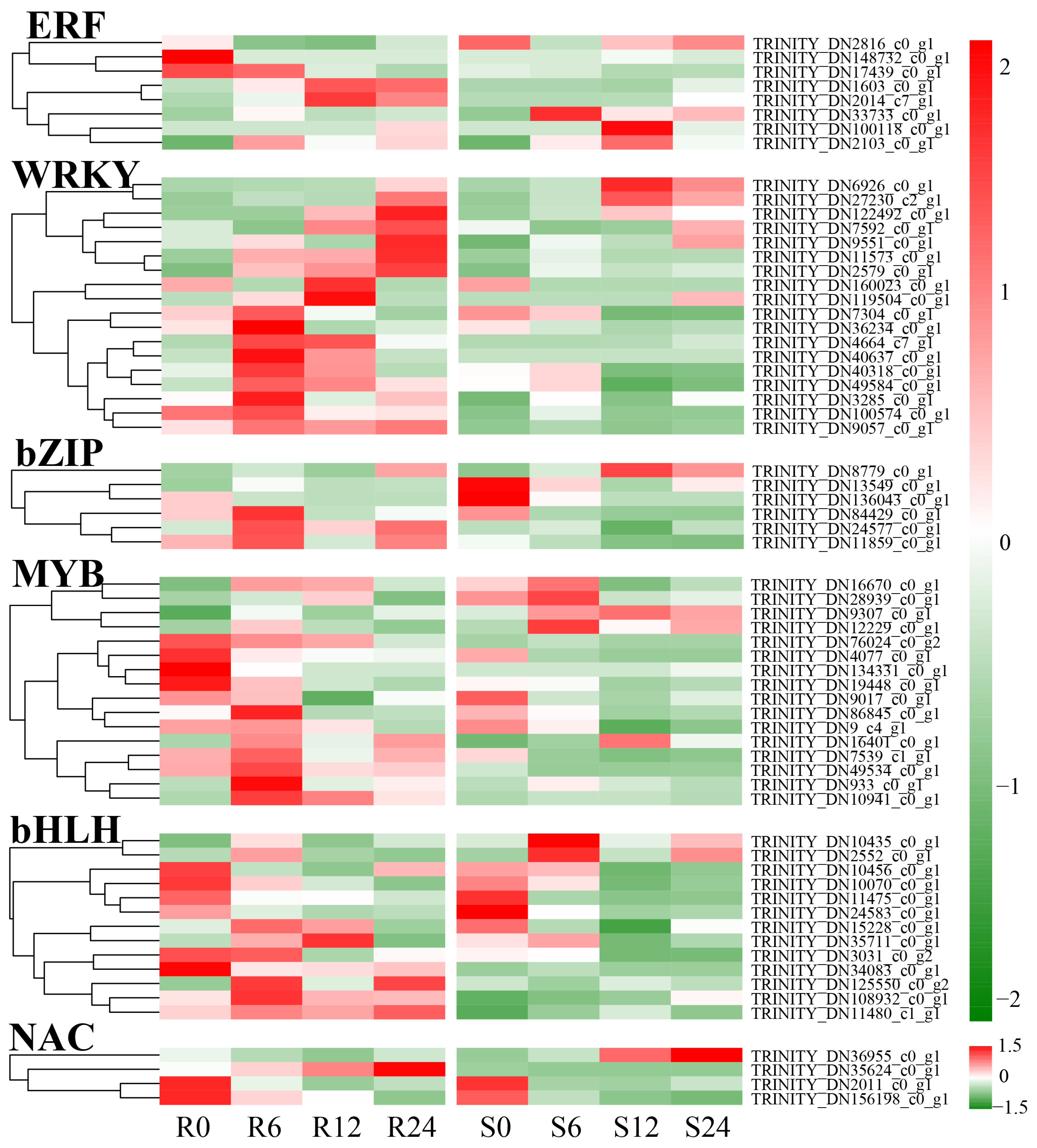
3.11. Transcription Factor Analysis
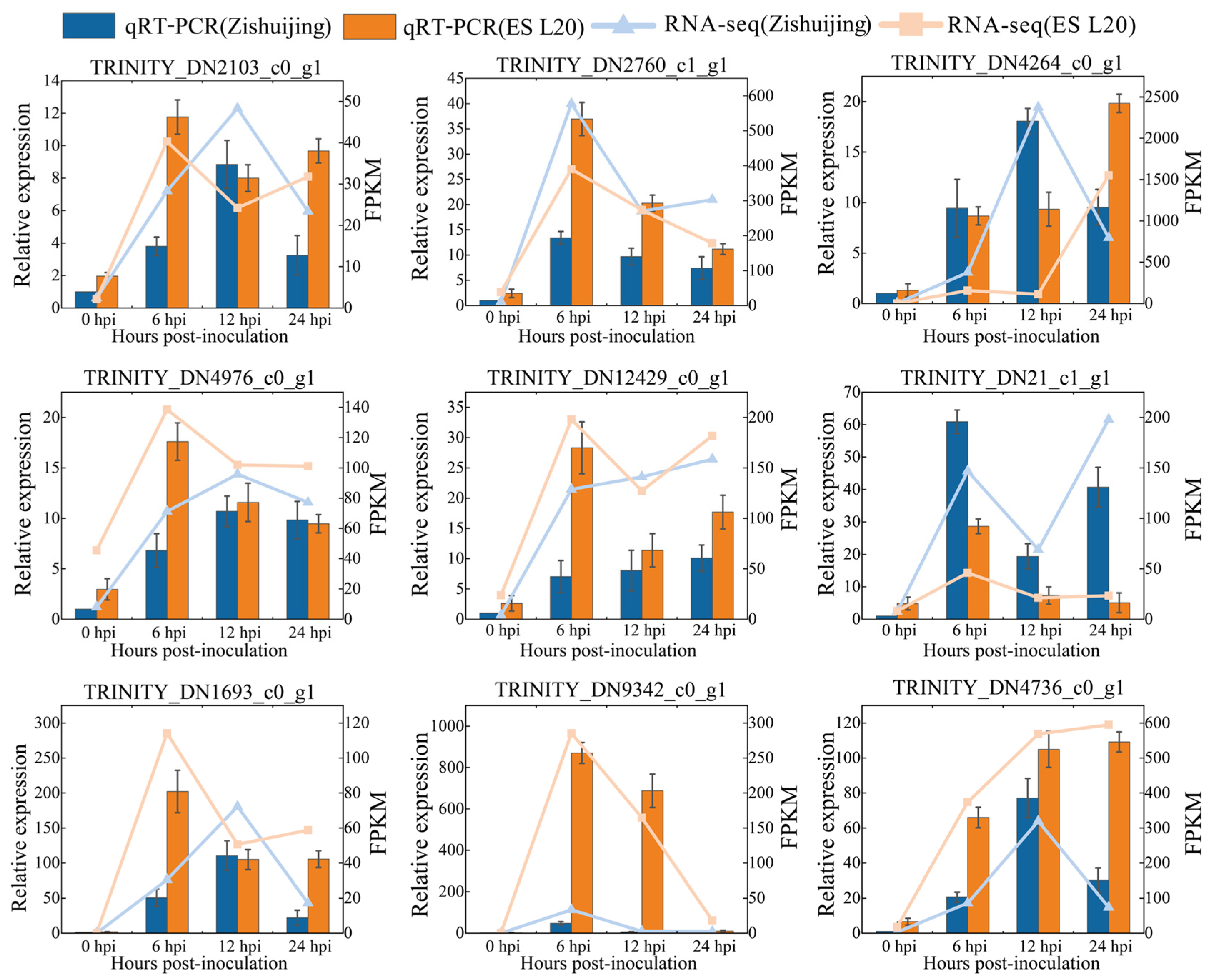
3.12. Transcriptome Results Were Verified by qRT-PCR
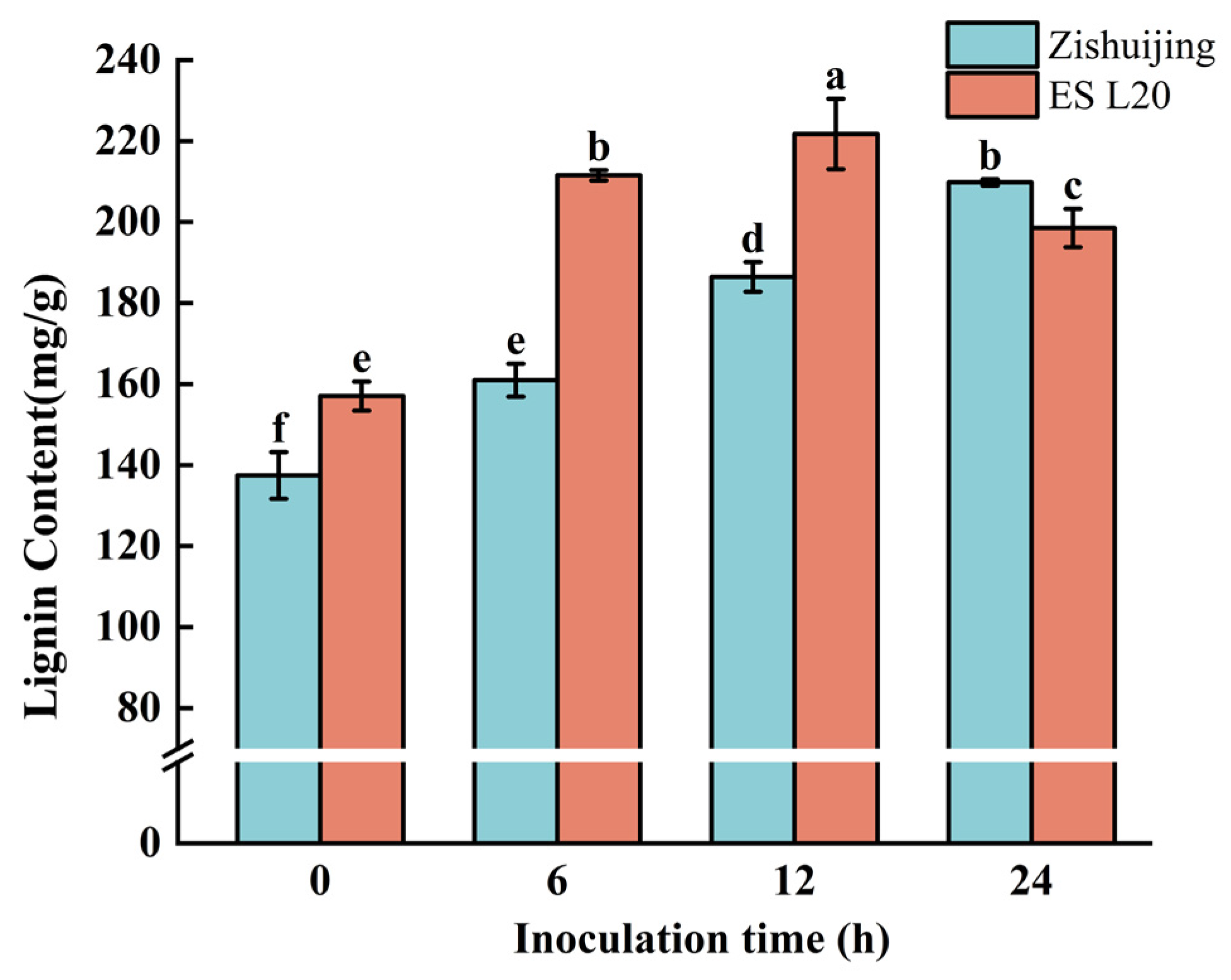
3.13. Determination of Lignin Content at Different Times
4. Discussion
4.1. Gene Expression Patterns and Pathway Enrichment
4.2. Defense-Related Pathways and Gene Functions
4.3. Role of Plant Hormone Signal Transduction Pathways
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Arai, S.; Oro, K.; Koseki, Y.; Nagata, J. The Dynamism of the Phalaenopsis Industry in Taiwan and the International Relay Culture System. E-J. GEO 2011, 6, 16–32. [Google Scholar] [CrossRef]
- Liao, G.J.; Hsieh, T.F.; Chen, H.R. Effect of chemical treatment on prevention of Fusarium yellow of Phalaenopsis seedlings under simulated conditions of shipment. Taiwan Agric. Res. 2012, 61, 124–131. [Google Scholar] [CrossRef]
- Zhou, J.N.; Lin, B.R.; Shen, H.F.; Pu, X.M.; Chen, Z.N.; Feng, J.J. First report of a soft rot of Phalaenopsis aphrodita caused by Dickeya dieffenbachiae in China. Plant Dis. 2012, 96, 760. [Google Scholar] [CrossRef] [PubMed]
- Alič, Š.; Naglič, T.; Tušek-Žnidarič, M.; Ravnikar, M.; Dreo, T. Putative new species of the genus Dickeya as major soft rot pathogens in Phalaenopsis orchid production. Plant Pathol. 2017, 66, 1357–1368. [Google Scholar] [CrossRef]
- Szulta, S.; Kornicka, A. Pectobacterium and Dickeya genus—A review on structural variations of O-polysaccharides and their role in the pathogenic process of plants. Plant Pathol. 2023, 72, 998–1010. [Google Scholar] [CrossRef]
- Lecourieux, D.; Ranjeva, R.; Pugin, A. Calcium in plant defence-signalling pathways. New Phytol. 2006, 171, 249–269. [Google Scholar] [CrossRef]
- Devireddy, A.R.; Zandalinas, S.I.; Fichman, Y.; Mittler, R. Integration of reactive oxygen species and hormone signaling during abiotic stress. Plant J. 2021, 105, 459–476. [Google Scholar] [CrossRef]
- Zhang, M.; Su, J.; Zhang, Y.; Xu, J.; Zhang, S. Conveying endogenous and exogenous signals: MAPK cascades in plant growth and defense. Curr. Opin. Plant Biol. 2018, 45, 1–10. [Google Scholar] [CrossRef]
- Grzyb, M.; Sujkowska-Rybkowska, M.; Mikuła, A. Cell wall remodeling and callose deposition during the embryogenic transition in the tree fern Cyathea delgadii Sternb. Plant Cell Tissue Organ. Cult. 2024, 156, 30. [Google Scholar] [CrossRef]
- Dodds, P.N.; Rathjen, J.P. Plant immunity: Towards an integrated view of plant-pathogen interactions. Nat. Rev. Genet. 2010, 11, 539–548. [Google Scholar] [CrossRef]
- Zipfel, C. Plant pattern-recognition receptors. Trends Immunol. 2014, 35, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, S.; Velásquez, A.C.; Martin, G.B. Assay for pathogen-associated molecular pattern (PAMP)-triggered immunity (PTI) in plants. J. Vis. Exp. 2009, 31, 1442. [Google Scholar] [CrossRef]
- Sanchez-Garrido, J.; Ruano-Gallego, D.; Choudhary, J.S.; Frankel, G. The type III secretion system effector network hypothesis. Trends Microbiol. 2022, 30, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Hurley, B.; Subramaniam, R.; Guttman, D.S.; Desveaux, D. Proteomics of effector-triggered immunity (ETI) in plants. Virulence 2014, 5, 752–760. [Google Scholar] [CrossRef]
- Cui, H.; Tsuda, K.; Parker, J.E. Effector-triggered immunity: From pathogen perception to robust defense. Annu. Rev. Plant Biol. 2015, 66, 487–511. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.G.; Staskawicz, B.J.; Dangl, J.L. The plant immune system: From discovery to deployment. Cell 2024, 187, 2095–2116. [Google Scholar] [CrossRef]
- Dong, X. SA, JA, ethylene, and disease resistance in plants. Curr. Opin. Plant Biol. 1998, 1, 316–323. [Google Scholar] [CrossRef]
- Kimbrel, J.A.; Di, Y.; Cumbie, J.S.; Chang, J.H. RNA-Seq for Plant Pathogenic Bacteria. Genes 2011, 2, 689–705. [Google Scholar] [CrossRef]
- Gao, Y.; He, X.; Wu, B.; Long, Q.; Shao, T.; Wang, Z.; Ding, W. Time-course transcriptome analysis reveals resistance genes of Panax ginseng induced by Cylindrocarpon destructans infection using RNA-seq. PLoS ONE 2016, 11, e0149408. [Google Scholar] [CrossRef]
- Jain, S.; Chittem, K.; Brueggeman, R.; Osorno, J.M.; Richards, J.; Nelson, B.D., Jr. Comparative transcriptome analysis of resistant and susceptible common bean genotypes in response to soybean cyst nematode infection. PLoS ONE 2016, 11, e0159338. [Google Scholar] [CrossRef]
- Yang, D.; Li, S.; Xiao, Y.; Lu, L.; Zheng, Z.; Tang, D.; Cui, H. Transcriptome analysis of rice response to blast fungus identified core genes involved in immunity. Plant Cell Env. 2021, 44, 3103–3121. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.F.; Tsai, T.M.; Chen, Y.R.; Liu, C.P.; Haiso, L.J.; Syue, L.H.; Huang, H.J. Characterization of the early response of the orchid, Phalaenopsis amabilis, to Erwinia chrysanthemi infection using expression profiling. Physiol. Plant 2012, 145, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Draper, H.H.; Squires, E.J.; Mahmoodi, H.; Wu, J.; Agarwal, S.; Hadley, M. A comparative evaluation of thiobarbituric acid methods for the determination of malondialdehyde in biological materials. Free. Radic. Biol. Med. 1993, 15, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kirkham, M.B. Drought-stress-induced changes in activities of superoxide dismutase, catalase, and peroxidase in wheat species. Plant Cell Physiol. 1994, 35, 785–791. [Google Scholar] [CrossRef]
- Dalton, D.A.; Hanus, F.J.; Russell, S.A.; Evans, H.J. Purification, properties, and distribution of ascorbate peroxidase in legume root nodules. Plant Physiol. 1987, 83, 789–794. [Google Scholar] [CrossRef]
- Fu, J.; Huang, B. Involvement of antioxidants and lipid peroxidation in the adaptation of two cool-season grasses to localized drought stress. Environ. Exp. Bot. 2001, 45, 105–114. [Google Scholar] [CrossRef]
- Ruiz, J.M.; Garcia, P.C.; Rivero, R.M.; Romero, L. Response of phenolic metabolism to the application of carbendazim plus boron in tobacco. Physiol. Plant. 1999, 106, 151–157. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Jiang, Z.; Ma, B.; Erinle, K.O.; Cao, B.; Liu, X.; Ye, S.; Zhang, Y. Enzymatic antioxidant defense in resistant plant: Pennisetum americanum (L.) K. Schum during long-term atrazine exposure. Pestic. Biochem. Physiol. 2016, 133, 59–66. [Google Scholar] [CrossRef]
- Liu, N.; Lin, Z.; Guan, L.; Gaughan, G.; Lin, G. Antioxidant enzymes regulate reactive oxygen species during pod elongation in Pisum sativum and Brassica chinensis. PLoS ONE 2014, 9, e87588. [Google Scholar] [CrossRef]
- Shahidi, P.; Bahramnejad, B.; Vafaee, Y.; Dastan, D.; Heidari, P. Isolation and characterization of Phenylalanine ammonia lyase (PAL) genes in Ferula pseudalliacea: Insights into the phenylpropanoid pathway. Genes 2024, 15, 771. [Google Scholar] [CrossRef]
- Duszyn, M.; Świeżawska, B.; Szmidt-Jaworska, A.; Jaworski, K. Cyclic nucleotide gated channels (CNGCs) in plant signalling-Current knowledge and perspectives. J. Plant Physiol. 2019, 241, 153035. [Google Scholar] [CrossRef]
- Boudsocq, M.; Sheen, J. CDPKs in immune and stress signaling. Trends Plant Sci. 2013, 18, 30–40. [Google Scholar] [CrossRef]
- Yang, D.; Chen, T.; Wu, Y.; Tang, H.; Yu, J.; Dai, X.; Tan, X. Genome-wide analysis of the peanut CaM/CML gene family reveals that the AhCML69 gene is associated with resistance to Ralstonia solanacearum. BMC Genom. 2024, 25, 200. [Google Scholar] [CrossRef]
- Soltabayeva, A.; Dauletova, N.; Serik, S.; Sandybek, M.; Omondi, J.O.; Kurmanbayeva, A.; Srivastava, S. Receptor-like kinases (LRR-RLKs) in response of plants to biotic and abiotic stresses. Plants 2022, 11, 2660. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Xian, L.; Zhuang, H.; Macho, A.P. SGT1 is not required for plant LRR-RLK-mediated immunity. Mol. Plant Pathol. 2021, 22, 145–150. [Google Scholar] [CrossRef]
- Hsiao, Y.C.; Yamada, M. The roles of peptide hormones and their receptors during plant root development. Genes 2020, 12, 22. [Google Scholar] [CrossRef]
- Wu, G.; Wang, W. Recent advances in understanding the role of two mitogen-activated protein kinase cascades in plant immunity. J. Exp. Bot. 2024, 75, 2256–2265. [Google Scholar] [CrossRef]
- Ray, S.K.; Macoy, D.M.; Kim, W.Y.; Lee, S.Y.; Kim, M.G. Role of RIN4 in regulating PAMP-triggered immunity and effector-triggered immunity: Current status and future perspectives. Mol. Cells 2019, 42, 503–511. [Google Scholar] [CrossRef]
- Hou, S.; Yang, Y.; Wu, D.; Zhang, C. Plant immunity: Evolutionary insights from PBS1, Pto, and RIN4. Plant Signal. Behav. 2011, 6, 794–799. [Google Scholar] [CrossRef]
- Ahn, Y.J.; Kim, H.; Choi, S.; Mazo-Molina, C.; Prokchorchik, M.; Zhang, N.; Sohn, K.H. Ptr1 and ZAR1 immune receptors confer overlapping and distinct bacterial pathogen effector specificities. New Phytol. 2023, 239, 1935–1953. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Wang, D.; Xie, H.; Zhao, Z.; Chen, Y.; Zhang, D.; Sun, Y. Genome-wide identification and expression analysis of the KCS gene family in soybean (Glycine max) reveal their potential roles in response to abiotic stress. Front. Plant Sci. 2023, 14, 1291731. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Pan, X.; Wan, Y.; Li, Z.; Xie, Z.; Yang, P. A 3-Ketoacyl-CoA Synthase 10 (KCS10) homologue from alfalfa enhances drought tolerance by regulating cuticular wax biosynthesis. J. Agric. Food Chem. 2023, 71, 14493–14504. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Feng, K.; Xie, M.; Barros, J.; Tschaplinski, T.J.; Tuskan, G.A.; Chen, J.G. Phylogenetic occurrence of the phenylpropanoid pathway and lignin biosynthesis in plants. Front. Plant Sci. 2021, 12, 704697. [Google Scholar] [CrossRef] [PubMed]
- Ge, W.; Xin, J.; Tian, R. Phenylpropanoid pathway in plants and its role in response to heavy metal stress: A review. Chin. J. Biotechnol. 2023, 39, 425–445. [Google Scholar] [CrossRef]
- Sharma, A.; Shahzad, B.; Rehman, A.; Bhardwaj, R.; Landi, M.; Zheng, B. Response of phenylpropanoid pathway and the role of polyphenols in plants under abiotic stress. Molecules 2019, 24, 2452. [Google Scholar] [CrossRef]
- Day, A.; Neutelings, G.; Nolin, F.; Grec, S.; Habrant, A.; Crônier, D.; Hawkins, S. Caffeoyl coenzyme A O-methyltransferase down-regulation is associated with modifications in lignin and cell-wall architecture in flax secondary xylem. Plant Physiol. Biochem. 2009, 47, 9–19. [Google Scholar] [CrossRef]
- Ji, P.; Lin, M.; Chen, M.; Kashif, M.H.; Fan, Y.; Ali, T.; Liu, Z. Caffeoyl-coenzyme A O-methyltransferase mediates regulation of carbon flux fluctuations during phenylpropenes and lignin biosynthesis in the vegetative organ roots of Asarum sieboldii Miq. Plant Physiol. Biochem. 2023, 201, 107855. [Google Scholar] [CrossRef]
- Zhong, R.; Morrison, W.H., III; Himmelsbach, D.S.; Poole, F.L.; Ye, Z.H. Essential role of caffeoyl coenzyme AO-methyltransferase in lignin biosynthesis in woody poplar plants. Plant Physiol. 2000, 124, 563–578. [Google Scholar] [CrossRef]
- Do, C.T.; Pollet, B.; Thévenin, J.; Sibout, R.; Denoue, D.; Barrière, Y.; Jouanin, L. Both caffeoyl Coenzyme A 3-O-methyltransferase 1 and caffeic acid O-methyltransferase 1 are involved in redundant functions for lignin, flavonoids and sinapoyl malate biosynthesis in Arabidopsis. Planta 2007, 226, 1117–1129. [Google Scholar] [CrossRef]
- Li, Y.; Wang, R.; Pei, Y.; Yu, W.; Wu, W.; Li, D.; Hu, Z. Phylogeny and functional characterization of the cinnamyl alcohol dehydrogenase gene family in Phryma leptostachya. Int. J. Biol. Macromol. 2022, 217, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Muro-Villanueva, F.; Kim, H.; Ralph, J.; Chapple, C. H-lignin can be deposited independently of cinnamyl alcohol dehydrogenase C and D in Arabidopsis. Plant Physiol. 2022, 189, 2015–2028. [Google Scholar] [CrossRef] [PubMed]
- Opassiri, R.; Maneesan, J.; Akiyama, T.; Pomthong, B.; Jin, S.; Kimura, A.; Cairns, J.R.K. Rice Os4BGlu12 is a wound-induced β-glucosidase that hydrolyzes cell wall-β-glucan-derived oligosaccharides and glycosides. Plant Sci. 2010, 179, 273–280. [Google Scholar] [CrossRef]
- Du, W.; Yang, J.; Li, Q.; Jiang, W.; Pang, Y. Medicago truncatula β-glucosidase 17 contributes to drought and salt tolerance through antioxidant flavonoid accumulation. Plant Cell Env. 2024, 47, 3076–3089. [Google Scholar] [CrossRef] [PubMed]
- De Araujo Ribeiro, G.C.; De Assis, S.A. β-glucosidases from Saccharomyces cerevisiae: Production, protein precipitation, characterization, and application in the enzymatic hydrolysis of delignified sugarcane bagasse. Prep. Biochem. Biotechnol. 2024, 54, 317–327. [Google Scholar] [CrossRef]
- Hazman, M.; Sühnel, M.; Schäfer, S.; Zumsteg, J.; Lesot, A.; Beltran, F.; Heitz, T. Characterization of Jasmonoyl-Isoleucine (JA-Ile) hormonal catabolic pathways in rice upon wounding and salt stress. Rice 2019, 12, 45. [Google Scholar] [CrossRef]
- Sheard, L.; Tan, X.; Mao, H.; Withers, J.; Ben-Nissan, G.; Hinds, T.R.; Zheng, N. Jasmonate perception by inositol-phosphate-potentiated COI1-JAZ co-receptor. Nature 2010, 468, 400–405. [Google Scholar] [CrossRef]
- Melotto, M.; Mecey, C.; Niu, Y.; Chung, H.S.; Katsir, L.; Yao, J.; He, S.Y. A critical role of two positively charged amino acids in the Jas motif of Arabidopsis JAZ proteins in mediating coronatine- and jasmonoyl isoleucine-dependent interactions with the COI1 F-box protein. Plant J. 2008, 55, 979–988. [Google Scholar] [CrossRef]
- Mittler, R. Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci. 2002, 7, 405–410. [Google Scholar] [CrossRef]
- Dixon, R.A.; Paiva, N.L. Stress-induced phenylpropanoid metabolism. Plant Cell 1995, 7, 1085–1097. [Google Scholar] [CrossRef]
- Cheval, C.; Aldon, D.; Galaud, J.P.; Ranty, B. Calcium/calmodulin-mediated regulation of plant immunity. Biochim. Biophys. Acta 2013, 1833, 1766–1771. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, C.; Robatzek, S.; Navarro, L.; Oakeley, E.J.; Jones, J.D.; Felix, G.; Boller, T. Bacterial disease resistance in Arabidopsis through flagellin perception. Nature 2004, 440, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.C.; Chou, M.Y.; Chou, S.J.; Li, Y.R.; Peng, H.P.; Shih, M.C. Submergence confers immunity mediated by the WRKY22 transcription factor in Arabidopsis. Plant Cell 2013, 25, 2699–2713. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, X.; Yang, B.; Xu, S.; Wei, X.; Zhao, P.; Jiang, Y.Q. WRKY55 transcription factor positively regulates leaf senescence and the defense response by modulating the transcription of genes implicated in the biosynthesis of reactive oxygen species and salicylic acid in Arabidopsis. Development 2020, 147, dev189647. [Google Scholar] [CrossRef]
- Wang, Z.; Wei, X.; Wang, Y.; Sun, M.; Zhao, P.; Wang, Q.; Jiang, Y.Q. WRKY29 transcription factor regulates ethylene biosynthesis and response in Arabidopsis. Plant Physiol. Biochem. 2023, 194, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Ai, G.; Yang, D.L.; Dou, D. The warfare for plant highway: Vascular plant–microbe interaction pinpoints lignin. Stress. Biol. 2022, 2, 24. [Google Scholar] [CrossRef]
- Mou, Z.; Zhao, Y.; Ye, F.; Shi, Y.; Kennelly, E.J.; Chen, S.; Zhao, D. Identification, biological activities and biosynthetic pathway of Dendrobium alkaloids. Front. Pharmacol. 2021, 12, 605994. [Google Scholar] [CrossRef]
- Pieterse, C.M.; Van der Does, D.; Zamioudis, C.; Leon-Reyes, A.; Van Wees, S.C. Hormonal modulation of plant immunity. Annu. Rev. Cell Dev. Biol. 2012, 28, 489–521. [Google Scholar] [CrossRef]
- Takahashi, H.; Kanayama, Y.; Zheng, M.S.; Kusano, T.; Hase, S.; Ikegami, M.; Shah, J. Antagonistic interactions between the SA and JA signaling pathways in Arabidopsis modulate expression of defense genes and gene-for-gene resistance to cucumber mosaic virus. Plant Cell Physiol. 2004, 45, 803–809. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xin, J.; Tang, J.; Mao, Y.; Ren, D.; Luo, P.; Cui, Y. Comparative Transcriptome Profiling Reveals Differential Defense Responses of Resistant and Susceptible Phalaenopsis to Dickeya fangzhongdai. Horticulturae 2025, 11, 534. https://doi.org/10.3390/horticulturae11050534
Xin J, Tang J, Mao Y, Ren D, Luo P, Cui Y. Comparative Transcriptome Profiling Reveals Differential Defense Responses of Resistant and Susceptible Phalaenopsis to Dickeya fangzhongdai. Horticulturae. 2025; 11(5):534. https://doi.org/10.3390/horticulturae11050534
Chicago/Turabian StyleXin, Jingjing, Jiyong Tang, Ying Mao, Dongdong Ren, Ping Luo, and Yongyi Cui. 2025. "Comparative Transcriptome Profiling Reveals Differential Defense Responses of Resistant and Susceptible Phalaenopsis to Dickeya fangzhongdai" Horticulturae 11, no. 5: 534. https://doi.org/10.3390/horticulturae11050534
APA StyleXin, J., Tang, J., Mao, Y., Ren, D., Luo, P., & Cui, Y. (2025). Comparative Transcriptome Profiling Reveals Differential Defense Responses of Resistant and Susceptible Phalaenopsis to Dickeya fangzhongdai. Horticulturae, 11(5), 534. https://doi.org/10.3390/horticulturae11050534