
Characterizing the Endophytic Microbiome and Microbial Functional Assemblages Associated with Fengtang Plum (Prunus salicina Lindl.) Development and Resistance


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Sample Processing and DNA Extraction
2.3. Library Preparation and 16s/ITS Sequencing
2.4. Data Analysis
3. Results
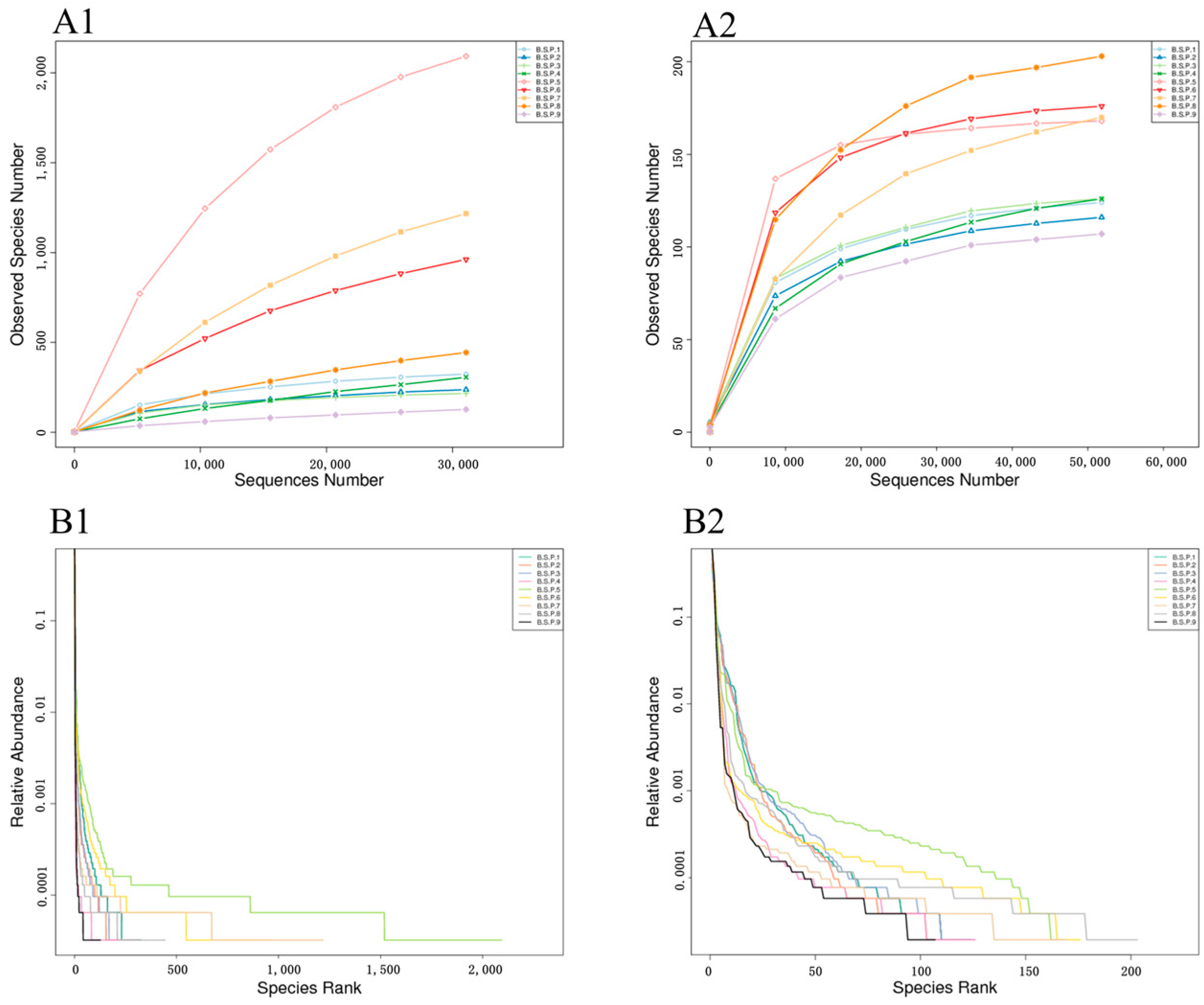
3.1. Diversity of Endophytic Microbial Communities in Fengtang Plum at Different Developmental Stages
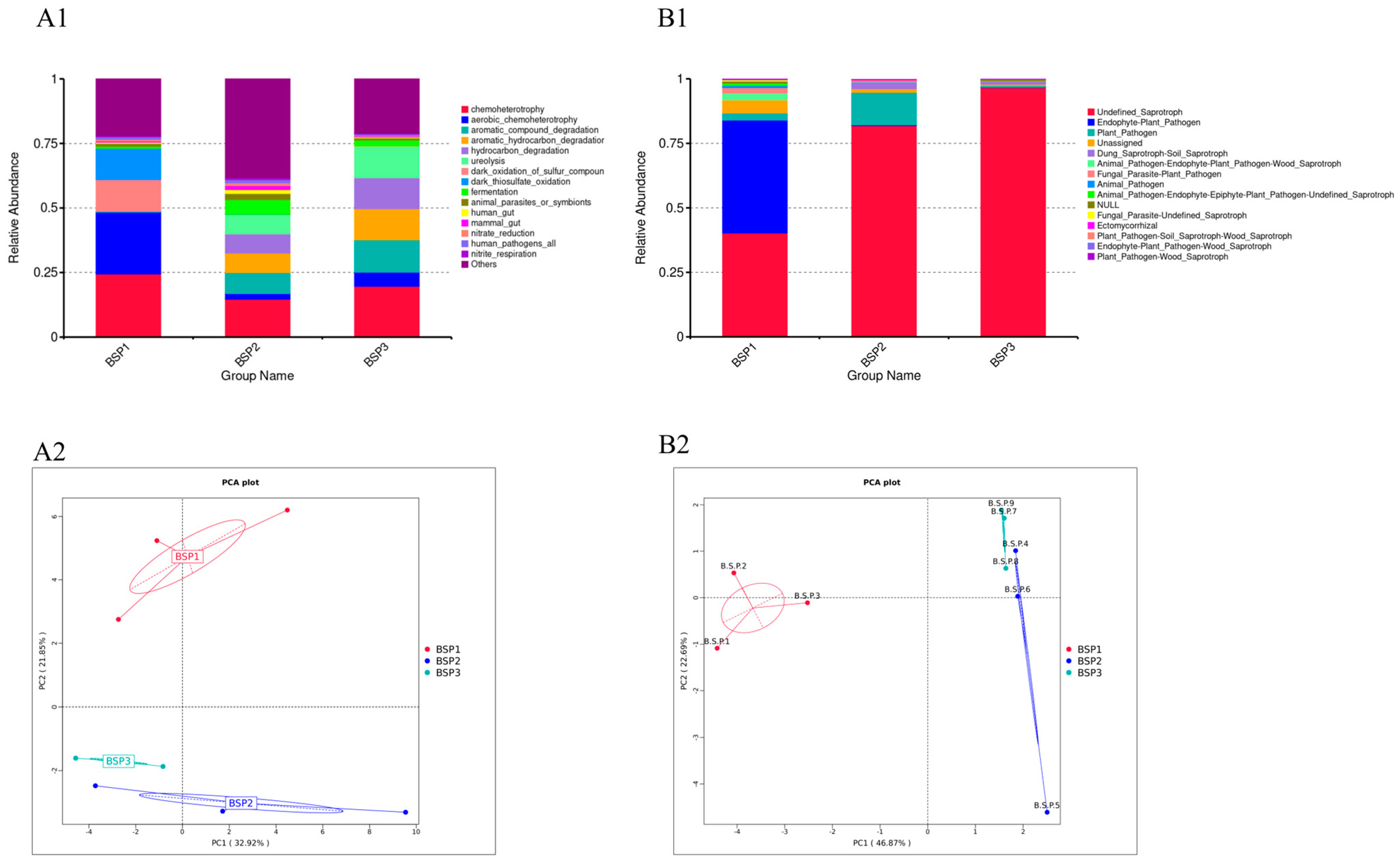
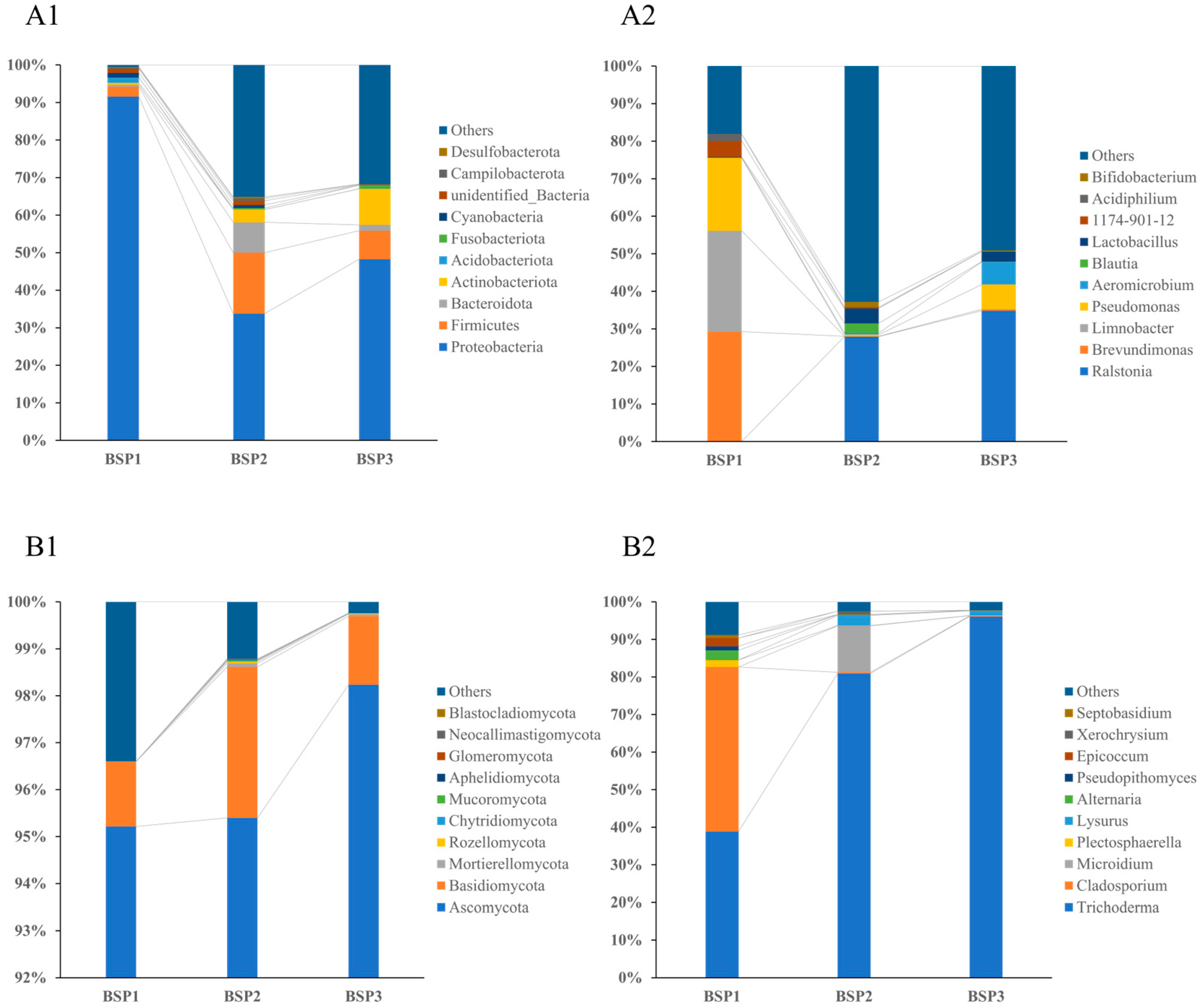
3.2. Composition of Endophytic Microbial Communities in Fengtang Plum at Different Developmental Stages
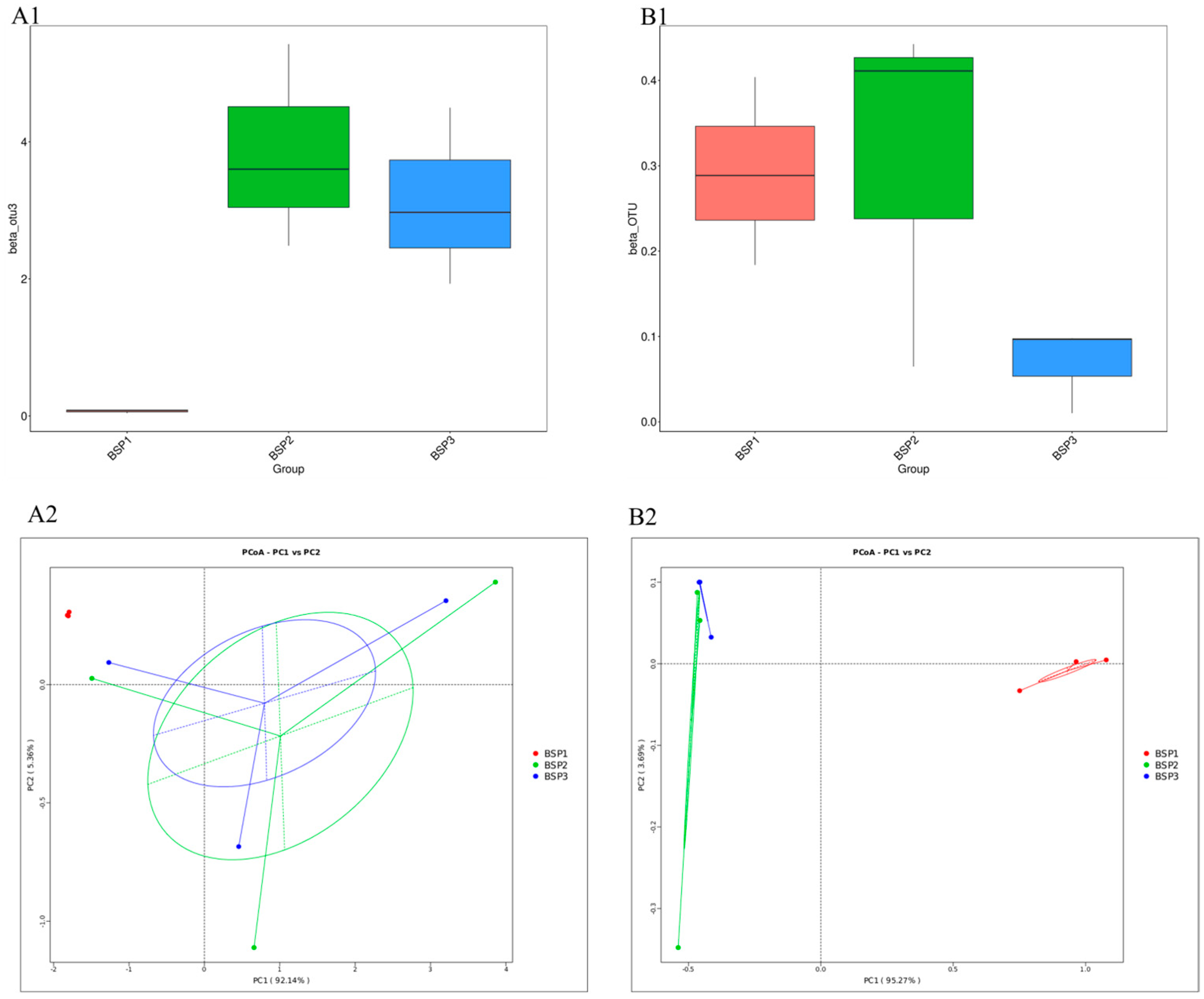
3.3. Structural Differences in Endophytic Microbial Communities in Fengtang Plum at Different Developmental Stages
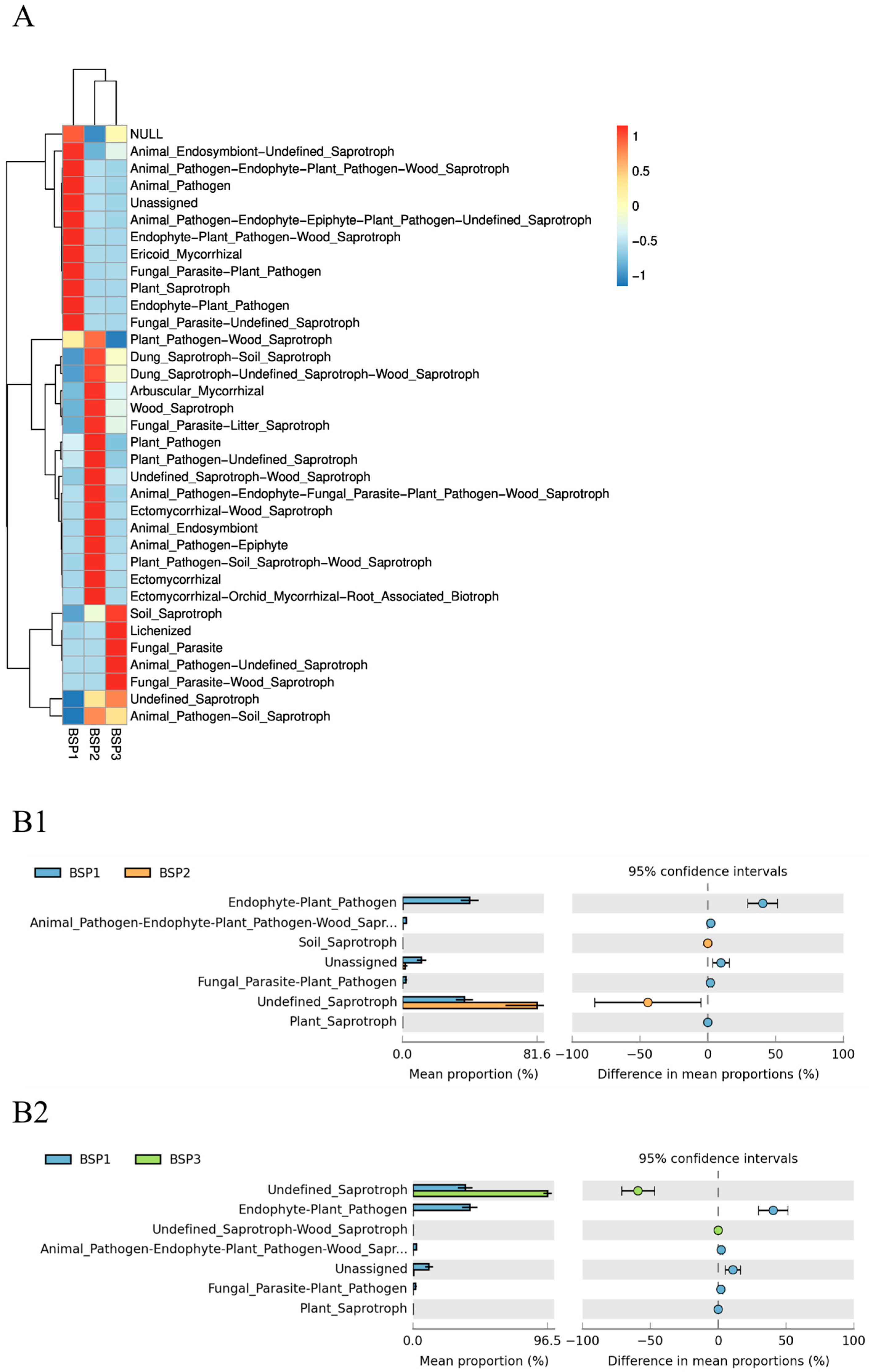
3.4. Functional Prediction of Bacterial Communities in Fengtang Plum at Different Developmental Stages
4. Discussion
4.1. Microbial Diversity and Composition at Different Developmental Stages of Fengtang Plum
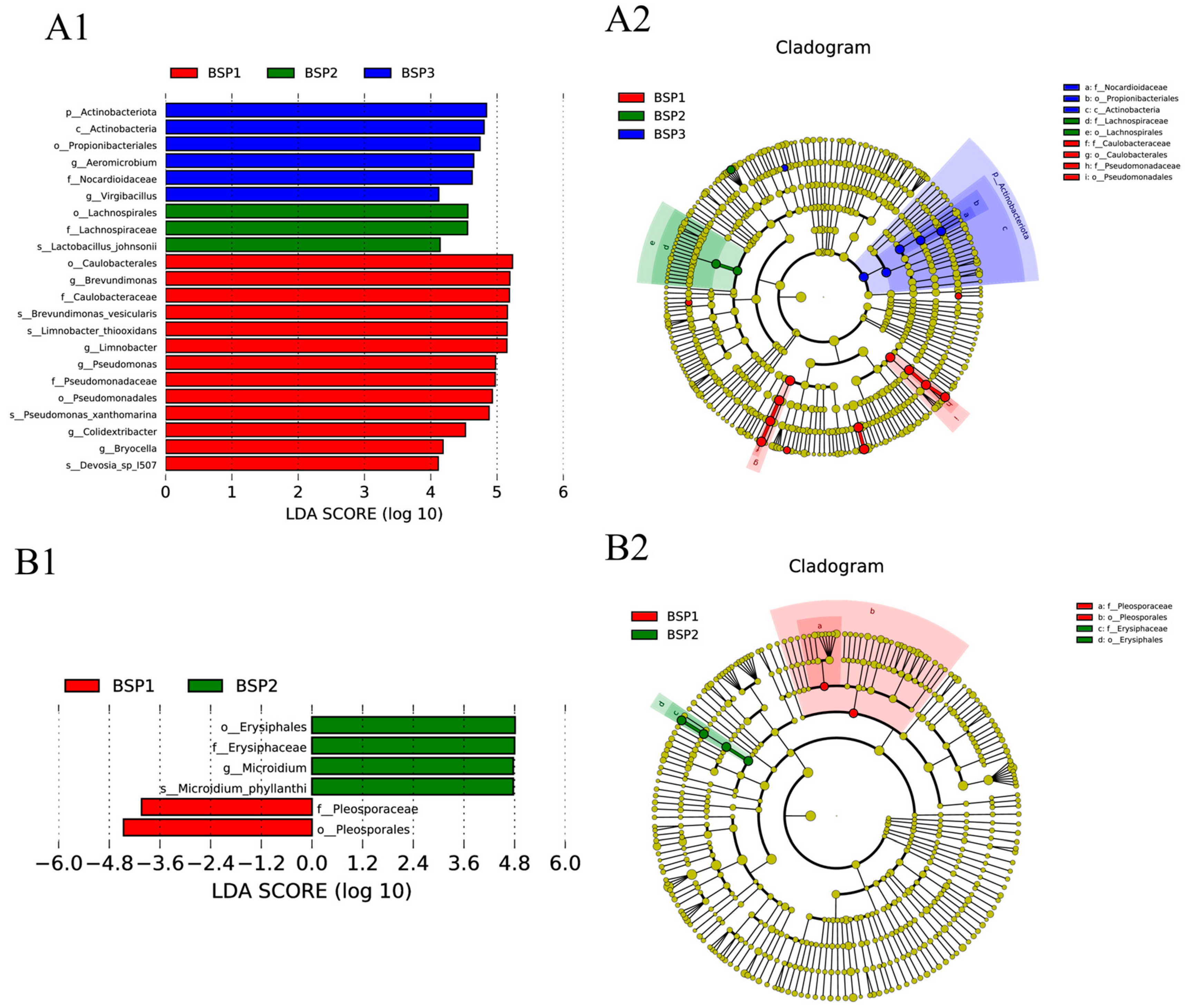
4.2. Corer Microorganisms and Their Potential Functions at Different Developmental Stages of Fengtang Plum
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microbat | Sample Name | RawPE | CleanTags | EffectiveTags | Q20 (%) | Q30 (%) | GC (%) | Effective (%) |
|---|---|---|---|---|---|---|---|---|
| Bacteria | B.S.P.1 | 93,832 | 80,890 | 61,389 | 97.75 | 93.17 | 54.48 | 65.42 |
| B.S.P.2 | 81,761 | 70,979 | 60,899 | 97.78 | 93.21 | 54.66 | 74.48 | |
| B.S.P.3 | 97,798 | 86,483 | 65,095 | 97.77 | 93.05 | 54.57 | 66.56 | |
| B.S.P.4 | 94,152 | 69,602 | 59,957 | 98.64 | 95.32 | 54.90 | 63.68 | |
| B.S.P.5 | 91,524 | 68,248 | 57,710 | 98.10 | 94.15 | 52.74 | 63.05 | |
| B.S.P.6 | 96,374 | 65626 | 59,155 | 98.12 | 94.17 | 52.96 | 61.38 | |
| B.S.P.7 | 109,206 | 72,508 | 66,558 | 98.31 | 94.66 | 53.90 | 60.95 | |
| B.S.P.8 | 79,416 | 56,286 | 49,982 | 98.63 | 95.30 | 54.77 | 62.94 | |
| B.S.P.9 | 90,312 | 65,319 | 59,826 | 98.71 | 95.50 | 55.08 | 66.24 | |
| Fungi | B.S.P.1 | 88,559 | 87,515 | 66,677 | 96.92 | 92.42 | 54.46 | 75.29 |
| B.S.P.2 | 90,317 | 88,996 | 60,189 | 96.99 | 92.64 | 53.82 | 66.64 | |
| B.S.P.3 | 91,497 | 88,988 | 62,810 | 96.29 | 91.26 | 55.37 | 68.65 | |
| B.S.P.4 | 92,168 | 89,345 | 63,028 | 98.45 | 95.00 | 60.07 | 68.38 | |
| B.S.P.5 | 86,001 | 80,175 | 61,329 | 96.88 | 92.02 | 59.72 | 71.31 | |
| B.S.P.6 | 96,512 | 91,365 | 69,558 | 98.28 | 94.68 | 59.77 | 72.07 | |
| B.S.P.7 | 91,810 | 84,993 | 68,839 | 98.41 | 94.89 | 60.02 | 74.98 | |
| B.S.P.8 | 57,553 | 52,789 | 52,551 | 98.58 | 95.34 | 59.67 | 91.31 | |
| B.S.P.9 | 91,940 | 90,400 | 67,129 | 98.51 | 95.14 | 59.94 | 73.01 |
| Classification | Taxonomy | Relative Abundance (%) | Classification | Taxonomy | Relative Abundance (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| BSP1 | BSP2 | BSP3 | BSP1 | BSP2 | BSP3 | ||||
| genus | Ralstonia | 0 ± 0 | 27.9493 ± 24.5603 | 34.8377 ± 11.0352 | genus | Trichoderma | 38.8515 ± 3.7899 | 80.9452 ± 14.2416 | 96.123 ± 2.3117 |
| Brevundimonas | 29.2557 ± 2.2169 | 0.0792 ± 0.0396 | 0.3563 ± 0.1814 | Cladosporium | 43.7916 ± 4.3905 | 0.3071 ± 0.1414 | 0.1464 ± 0.0383 | ||
| Limnobacter | 26.8804 ± 2.6311 | 0 ± 0 | 0 ± 0 | Microidium | 0 ± 0 | 12.3632 ± 11.1758 | 0.0664 ± 0.0422 | ||
| Pseudomonas | 19.3983 ± 1.2724 | 0.4355 ± 0.2771 | 6.6112 ± 2.4887 | Plectosphaerella | 1.8495 ± 1.8311 | 0.0606 ± 0.0168 | 0.0394 ± 0.0267 | ||
| Aeromicrobium | 0 ± 0 | 0.4751 ± 0.2743 | 6.0174 ± 1.8619 | Lysurus | 0 ± 0 | 2.7358 ± 1.5099 | 1.247 ± 1.1882 | ||
| Blautia | 0.0792 ± 0.0396 | 2.5337 ± 2.3558 | 0.0792 ± 0.0396 | Alternaria | 2.5997 ± 0.3872 | 0.1903 ± 0.0743 | 0.0942 ± 0.0279 | ||
| Lactobacillus | 0.1584 ± 0.1047 | 3.9984 ± 1.6744 | 2.6524 ± 2.1249 | Pseudopithomyces | 1.0902 ± 1.0004 | 0.0026 ± 0.0026 | 0.0045 ± 0.0023 | ||
| 1174-901-12 | 4.2755 ± 1.5038 | 0.2771 ± 0.2771 | 0 ± 0 | Epicoccum | 2.1785 ± 0.5745 | 0.0484 ± 0.0319 | 0.0548 ± 0.0168 | ||
| Acidiphilium | 1.8211 ± 1.1804 | 0.0396 ± 0.0396 | 0.0396 ± 0.0396 | Xerochrysium | 0 ± 0 | 0.8722 ± 0.8664 | 0 ± 0 | ||
| Bifidobacterium | 0.0396 ± 0.0396 | 1.4252 ± 1.3082 | 0.2771 ± 0.1726 | Septobasidium | 0.8238 ± 0.6154 | 0 ± 0 | 0 ± 0 | ||
| Others | 18.0918 ± 3.2018 | 62.787 ± 22.5108 | 49.1291 ± 14.6311 | Others | 8.8152 ± 0.4412 | 2.4746 ± 0.8314 | 2.2243 ± 1.0105 | ||
| phylum | Proteobacteria | 91.5677 ± 1.3918 | 33.7688 ± 23.119 | 48.2581 ± 15.2248 | phylum | Ascomycota | 95.2163 ± 1.4971 | 95.399 ± 2.5043 | 98.2342 ± 1.2558 |
| Firmicutes | 2.6524 ± 0.7332 | 16.2312 ± 10.4473 | 7.601 ± 4.8694 | Basidiomycota | 1.3813 ± 0.913 | 3.2185 ± 1.7006 | 1.4617 ± 1.2023 | ||
| Bacteroidota | 0.5146 ± 0.3092 | 8.076 ± 4.7362 | 1.5439 ± 0.9889 | Mortierellomycota | 0.0039 ± 0.0039 | 0.074 ± 0.0423 | 0.0437 ± 0.0251 | ||
| Actinobacteriota | 0.4751 ± 0.0686 | 3.4046 ± 1.3267 | 9.5804 ± 0.8949 | Rozellomycota | 0 ± 0 | 0.0367 ± 0.026 | 0.0141 ± 0.0053 | ||
| Acidobacteriota | 1.4252 ± 0.8084 | 0 ± 0 | 0.0396 ± 0.0396 | Chytridiomycota | 0.0006 ± 0.0006 | 0.0193 ± 0.0067 | 0.0064 ± 0.0034 | ||
| Fusobacteriota | 0 ± 0 | 0.3959 ± 0.1726 | 0.9501 ± 0.8314 | Mucoromycota | 0 ± 0 | 0.0154 ± 0.0084 | 0.0006 ± 0.0006 | ||
| Cyanobacteria | 1.2272 ± 0.3382 | 0.8314 ± 0.8314 | 0 ± 0 | Aphelidiomycota | 0 ± 0 | 0.0077 ± 0.0077 | 0 ± 0 | ||
| unidentified_Bacteria | 1.1085 ± 0.1726 | 1.1085 ± 0.5146 | 0.1188 ± 0.0686 | Glomeromycota | 0 ± 0 | 0.0058 ± 0.0058 | 0.0013 ± 0.0013 | ||
| Campilobacterota | 0.2375 ± 0.1814 | 0.673 ± 0.2408 | 0.1584 ± 0.0792 | Neocallimastigomycota | 0 ± 0 | 0.0058 ± 0.0058 | 0 ± 0 | ||
| Desulfobacterota | 0.2375 ± 0.1814 | 0.3563 ± 0.1371 | 0.0396 ± 0.0396 | Blastocladiomycota | 0 ± 0 | 0.0032 ± 0.0032 | 0 ± 0 | ||
| Others | 0.5542 ± 0.0792 | 35.1544 ± 19.578 | 31.7102 ± 15.3388 | Others | 3.3979 ± 0.7014 | 1.2147 ± 0.8178 | 0.2379 ± 0.0348 | ||
References
- Droby, S.; Wisniewski, M. The fruit microbiome: A new frontier for postharvest biocontrol and postharvest biology. Postharvest Biol. Technol. 2018, 140, 107–112. [Google Scholar] [CrossRef]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.-C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar]
- Ou, T.; Xu, W.-f.; Wang, F.; Strobel, G.; Zhou, Z.-y.; Xiang, Z.-h.; Liu, J.; Xie, J. A microbiome study reveals seasonal variation in endophytic bacteria among different mulberry cultivars. Comput. Struct. Biotechnol. J. 2019, 17, 1091–1100. [Google Scholar] [CrossRef]
- Wu, Y.; Qu, M.; Pu, X.; Lin, J.; Shu, B. Distinct microbial communities among different tissues of citrus tree Citrus reticulata cv. Chachiensis. Sci. Rep. 2020, 10, 6068. [Google Scholar] [CrossRef] [PubMed]
- Goforth, M.; Cooper, M.A.; Oliver, A.S.; Pinzon, J.; Skots, M.; Obergh, V.; Suslow, T.V.; Flores, G.E.; Huynh, S.; Parker, C.T. Bacterial community shifts of commercial apples, oranges, and peaches at different harvest points across multiple growing seasons. PLoS ONE 2024, 19, e0297453. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.; Qureshi, K.A.; Jaremko, M.; White, J.; Singh, S.K.; Sharma, V.K.; Singh, K.K.; Santoyo, G.; Puopolo, G.; Kumar, A. Deciphering the role of endophytic microbiome in postharvest diseases management of fruits: Opportunity areas in commercial up-scale production. Front. Plant Sci. 2022, 13, 1026575. [Google Scholar] [CrossRef]
- Droby, S.; Zhimo, V.Y.; Wisniewski, M.; Freilich, S. The pathobiome concept applied to postharvest pathology and its implication on biocontrol strategies. Postharvest Biol. Technol. 2022, 189, 111911. [Google Scholar] [CrossRef]
- Zhang, H.; Serwah Boateng, N.A.; Ngolong Ngea, G.L.; Shi, Y.; Lin, H.; Yang, Q.; Wang, K.; Zhang, X.; Zhao, L.; Droby, S. Unravelling the fruit microbiome: The key for developing effective biological control strategies for postharvest diseases. Compr. Rev. Food Sci. Food Saf. 2021, 20, 4906–4930. [Google Scholar] [CrossRef]
- Wang, B.; Wang, X.; Wang, Z.; Zhu, K.; Wu, W. Comparative metagenomic analysis reveals rhizosphere microbial community composition and functions help protect grapevines against salt stress. Front. Microbiol. 2023, 14, 1102547. [Google Scholar] [CrossRef]
- Kusstatscher, P.; Cernava, T.; Abdelfattah, A.; Gokul, J.; Korsten, L.; Berg, G. Microbiome approaches provide the key to biologically control postharvest pathogens and storability of fruits and vegetables. FEMS Microbiol. Ecol. 2020, 96, fiaa119. [Google Scholar] [CrossRef]
- Zhimo, V.Y.; Kumar, A.; Biasi, A.; Salim, S.; Feygenberg, O.; Toamy, M.A.; Abdelfattaah, A.; Medina, S.; Freilich, S.; Wisniewski, M.; et al. Compositional shifts in the strawberry fruit microbiome in response to near-harvest application of Metschnikowia fructicola, a yeast biocontrol agent. Postharvest Biol. Technol. 2021, 175, 111469. [Google Scholar] [CrossRef]
- Kumar, A.; Zhimo, Y.; Biasi, A.; Salim, S.; Feygenberg, O.; Wisniewski, M.; Droby, S. Endophytic microbiome in the carposphere and its importance in fruit physiology and pathology. In Postharvest Pathology; Plant Pathology in the 21st Century; Springer: Berlin/Heidelberg, Germany, 2021; Chapter 5; pp. 73–88. [Google Scholar]
- Lukša, J.; Vepštaitė-Monstavičė, I.; Yurchenko, V.; Serva, S.; Servienė, E. High content analysis of sea buckthorn, black chokeberry, red and white currants microbiota–a pilot study. Food Res. Int. 2018, 111, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Shen, J.; Zhang, L.; Zou, X.; Jin, L. Metabolomic and transcriptomic integration reveals the mechanism of aroma formation as strawberries naturally turn colors while ripening. Food Chem. 2024, 460, 140765. [Google Scholar] [CrossRef] [PubMed]
- Todhanakasem, T.; Van Tai, N.; Pornpukdeewattana, S.; Charoenrat, T.; Young, B.M.; Wattanachaisaereekul, S. The relationship between microbial communities in coffee fermentation and aroma with metabolite attributes of finished products. Foods 2024, 13, 2332. [Google Scholar] [CrossRef] [PubMed]
- Escobar Rodriguez, C.; Novak, J.; Buchholz, F.; Uetz, P.; Bragagna, L.; Gumze, M.; Antonielli, L.; Mitter, B. The bacterial microbiome of the tomato fruit is highly dependent on the cultivation approach and correlates with flavor chemistry. Front. Plant Sci. 2021, 12, 775722. [Google Scholar] [CrossRef] [PubMed]
- Topp, B.L.; Russell, D.M.; Neumüller, M.; Dalbó, M.A.; Liu, W. Plum. In Fruit Breeding; Springer: Berlin/Heidelberg, Germany, 2012; pp. 571–621. [Google Scholar]
- Zhang, Y.; Li, Y.Q.; Xiao, Y.; Wu, J.L.; Cheng, L. Breeding and cultivation techniques of the new plum variety “fengtangli”. S. China Fruits 2018, 47, 146–148. [Google Scholar] [CrossRef]
- Wang, H.L.; Xie, P.; Ma, Y.H.; Zhao, K.; Wang, H.; Zhao, X.; Zheng, Q. Differences in sugar and acid components and quality evaluation of “fengtangli” plum fruits from different production areas in Guizhou. Guizhou Agric. Sci. 2024, 52, 84–93. [Google Scholar]
- Rungjindamai, N.; Jeffries, P.; Xu, X.-M. Epidemiology and management of brown rot on stone fruit caused by Monilinia laxa. Eur. J. Plant Pathol. 2014, 140, 1–17. [Google Scholar] [CrossRef]
- De Miccolis Angelini, R.M.; Landi, L.; Raguseo, C.; Pollastro, S.; Faretra, F.; Romanazzi, G. Tracking of diversity and evolution in the brown rot fungi Monilinia fructicola, Monilinia fructigena, and Monilinia laxa. Front. Microbiol. 2022, 13, 854852. [Google Scholar] [CrossRef]
- Janisiewicz, W.J.; Jurick II, W.M.; Vico, I.; Peter, K.A.; Buyer, J.S. Culturable bacteria from plum fruit surfaces and their potential for controlling brown rot after harvest. Postharvest Biol. Technol. 2013, 76, 145–151. [Google Scholar] [CrossRef]
- Janakiev, T.; Dimkić, I.; Bojić, S.; Fira, D.; Stanković, S.; Berić, T. Bacterial communities of plum phyllosphere and characterization of indigenous antagonistic Bacillus thuringiensis R3/3 isolate. J. Appl. Microbiol. 2020, 128, 528–543. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Gao, L.; Zhang, Z.; Ming, Z.; Gao, F.; Ma, S.; Zou, M. Diversity analysis of microorganisms on the surface of four summer fruit varieties in Baotou, inner Mongolia, China. PeerJ 2024, 12, e18752. [Google Scholar] [CrossRef] [PubMed]
- Bram, B.; Michiel, O.D.B.; Sofie, T.; Sascha, T.; Nele, W.; Wout, B.; Jaco, V. Performance of 16s rdna primer pairs in the study of rhizosphere and endosphere bacterial microbiomes in metabarcoding studies. Front. Microbiol. 2016, 7, 650. [Google Scholar]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Fungal Barcoding Consortium; Bolchacova, E.; Voigt, K.; et al. Nuclear ribosomal internal transcribed spacer (its) region as a universal DNA barcode marker for fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef]
- Degnan, P.H.; Howard, O. Illumina-based analysis of microbial community diversity. ISME J. 2012, 6, 183–194. [Google Scholar] [CrossRef]
- Caporaso, G.J.; Lauber, L.C.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the illumina hiseq and miseq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. Flash: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. Vsearch: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal database project: Data and tools for high throughput rRNA analysis. Nat. Rev. Microbiol. 2014, 42, D633–D642. [Google Scholar] [CrossRef]
- Abarenkov, K.; Nilsson, R.H.; Larsson, K.-H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E.; Pennanen, T.; et al. The unite database for molecular identification of fungi–recent updates and future perspectives. New Phytol. 2010, 186, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Peng, C.; Cao, H.; Song, J.; Gong, B.; Li, L.; Wang, L.; He, Y.; Liang, M.; Lin, J. Microbial functional assemblages predicted by the FAPROTAX analysis are impacted by physicochemical properties, but C, N and S cycling genes are not in mangrove soil in the Beibu Gulf, China. Ecol. Indic. 2022, 139, 108887. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Piombo, E.; Abdelfattah, A.; Danino, Y.; Salim, S.; Feygenberg, O.; Spadaro, D.; Wisniewski, M.; Droby, S. Characterizing the fungal microbiome in date (Phoenix dactylifera) fruit pulp and peel from early development to harvest. Microorganisms 2020, 8, 641. [Google Scholar] [CrossRef]
- Lukša, J.; Vepštaitė-Monstavičė, I.; Apšegaitė, V.; Blažytė-Čereškienė, L.; Stanevičienė, R.; Strazdaitė-Žielienė, Ž.; Ravoitytė, B.; Aleknavičius, D.; Būda, V.; Mozūraitis, R.; et al. Fungal microbiota of sea buckthorn berries at two ripening stages and volatile profiling of potential biocontrol yeasts. Microorganisms 2020, 8, 456. [Google Scholar] [CrossRef]
- Bill, M.; Chidamba, L.; Gokul, J.K.; Korsten, L. Mango endophyte and epiphyte microbiome composition during fruit development and post-harvest stages. Horticulturae 2021, 7, 495. [Google Scholar] [CrossRef]
- Balbino Miguel, P.S.; Delvaux, J.C.; de Oliveira, M.N.V.; Monteiro, L.C.P.; de Souza Freitas, F.; Costa, M.D.; Tótola, M.R.; de Moraes, C.A.; Borges, A.C. Diversity of endophytic bacteria in the fruits of Coffea canephora. Afr. J. Microbiol. Res. 2013, 7, 586–594. [Google Scholar] [CrossRef]
- Wang, Y.-R.; Zeng, S.-X.; Leng, J.-S.; Huang, B.-Y.; Chen, H.; Wang, Y.; Liu, J. Metagenomic analysis of the epiphytic and endophytic microbiota of plum fruits at different storage temperatures. Res. Sq. 2024. [Google Scholar] [CrossRef]
- Abdelfattah, A.; Freilich, S.; Bartuv, R.; Zhimo, V.Y.; Kumar, A.; Biasi, A.; Salim, S.; Feygenberg, O.; Burchard, E.; Dardick, C.; et al. Global analysis of the apple fruit microbiome: Are all apples the same? Environ. Microbiol. 2021, 23, 6038–6055. [Google Scholar] [CrossRef]
- Aydin, D.; Coskun, O.F. Comparison of edta-enhanced phytoextraction strategies with Nasturtium officinale (Watercress) on an artificially arsenic contaminated water. Pak. J. Bot 2013, 45, 1423–1429. [Google Scholar]
- Wassermann, B.; Müller, H.; Berg, G. An apple a day: Which bacteria do we eat with organic and conventional apples? Front. Microbiol. 2019, 10, 475179. [Google Scholar] [CrossRef] [PubMed]
- Abdelfattah, A.; Wisniewski, M.; Droby, S.; Schena, L. Spatial and compositional variation in the fungal communities of organic and conventionally grown apple fruit at the consumer point-of-purchase. Hortic. Res. 2016, 3, 16047. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.L.; Hermosa, R.; Lorito, M.; Monte, E. Trichoderma: A multipurpose, plant-beneficial microorganism for eco-sustainable agriculture. Nat. Rev. Microbiol. 2023, 21, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Qiao, C.; Penton, C.R.; Xiong, W.; Liu, C.; Wang, R.; Liu, Z.; Xu, X.; Li, R.; Shen, Q. Reshaping the rhizosphere microbiome by bio-organic amendment to enhance crop yield in a maize-cabbage rotation system. Appl. Soil Ecol. 2019, 142, 136–146. [Google Scholar] [CrossRef]
- Su, J.; Wang, Y.; Bai, M.; Peng, T.; Li, H.; Xu, H.-J.; Guo, G.; Bai, H.; Rong, N.; Sahu, S.K.; et al. Soil conditions and the plant microbiome boost the accumulation of monoterpenes in the fruit of Citrus reticulata ‘Chachi’. Microbiome 2023, 11, 61. [Google Scholar] [CrossRef]
- Lugtenberg, B.; Kamilova, F. Plant-growth-promoting rhizobacteria. Annu. Rev. Microbiol. 2009, 63, 541–556. [Google Scholar] [CrossRef]
- Verma, M.; Mishra, J.; Arora, N.K. Plant growth-promoting rhizobacteria: Diversity and applications. In Environmental Biotechnology: For Sustainable Future; Springer: Berlin/Heidelberg, Germany, 2018; pp. 129–173. [Google Scholar]
- Oleńska, E.; Małek, W.; Wójcik, M.; Swiecicka, I.; Thijs, S.; Vangronsveld, J. Beneficial features of plant growth-promoting rhizobacteria for improving plant growth and health in challenging conditions: A methodical review. Sci. Total Environ. 2020, 743, 140682. [Google Scholar] [CrossRef] [PubMed]
- Hakim, S.; Naqqash, T.; Nawaz, M.S.; Laraib, I.; Siddique, M.J.; Zia, R.; Mirza, M.S.; Imran, A. Rhizosphere engineering with plant growth-promoting microorganisms for agriculture and ecological sustainability. Front. Sustain. Food Syst. 2021, 5, 617157. [Google Scholar] [CrossRef]
- Zeng, Q.; Ding, X.; Wang, J.; Han, X.; Iqbal, H.M.; Bilal, M. Insight into soil nitrogen and phosphorus availability and agricultural sustainability by plant growth-promoting rhizobacteria. Environ. Sci. Pollut. Res. 2022, 29, 45089–45106. [Google Scholar] [CrossRef]
- Xamxidin, M.; Huang, X.-P.; Yang, X.-W.; Wang, T.; Chen, C.; Wu, M. Limnobacter parvuscolonica sp. Nov., thiosulfate-oxidizing bacterium isolated from lake water. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Chen, Y.; Feng, X.; He, Y.; Wang, F. Genome analysis of a Limnobacter sp. Identified in an anaerobic methane-consuming cell consortium. Front. Mar. Sci. 2016, 3, 257. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, J.; Sun, P.; Chen, C.; Shen, J. Community structure of phyllosphere bacteria in different cultivars of fingered citron (Citrus medica ‘fingered’) and their correlations with fragrance. Front. Plant Sci. 2022, 13, 936252. [Google Scholar] [CrossRef] [PubMed]
- Sangiorgio, D.; Cellini, A.; Spinelli, F.; Pastore, C.; Farneti, B.; Savioli, S.; Rodriguez-Estrada, M.T.; Donati, I. Contribution of fruit microbiome to raspberry volatile organic compounds emission. Postharvest Biol. Technol. 2022, 183, 111742. [Google Scholar] [CrossRef]
- Agarwal, H.; Dowarah, B.; Baruah, P.M.; Bordoloi, K.S.; Krishnatreya, D.B.; Agarwala, N. Endophytes from Gnetum gnemon l. Can protect seedlings against the infection of phytopathogenic bacterium Ralstonia solanacearum as well as promote plant growth in tomato. Microbiol. Res. 2020, 238, 126503. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, T.R.; Jacquiod, S.; Nour, E.H.; Sørensen, S.J.; Smalla, K. Biocontrol of bacterial wilt disease through complex interaction between tomato plant, antagonists, the indigenous rhizosphere microbiota, and Ralstonia solanacearum. Front. Microbiol. 2020, 10, 2835. [Google Scholar] [CrossRef]
- Mori, T.; Fujiyoshi, T.; Inada, T.; Matsusaki, H.; Ogawa, K.; Matsuzoe, N. Phenotypic conversion of Ralstonia solanacearum in susceptible and resistant Solanum plants. Environ. Control. Biol. 2011, 49, 165–176. [Google Scholar] [CrossRef]
- Mori, T.; Inada, T.; Ogawa, K.; Matsusaki, H.; Matsuzoe, N. Phenotypic conversion of Ralstonia solanacearum in water extract of Solanum toxicarium. J. Plant Pathol. 2012, 94, 535–542. [Google Scholar]
- Nakahara, H.; Mori, T.; Sadakari, N.; Matsusaki, H.; Matsuzoe, N. Selection of effective non-pathogenic ralstonia solanacearum as biocontrol agents against bacterial wilt in eggplant. J. Plant Dis. Prot. 2016, 123, 119–124. [Google Scholar] [CrossRef]
- Konappa, N.; Krishnamurthy, S.; Siddaiah, C.N.; Ramachandrappa, N.S.; Chowdappa, S. Evaluation of biological efficacy of Trichoderma asperellum against tomato bacterial wilt caused by Ralstonia solanacearum. Egypt. J. Biol. Pest Control. 2018, 28, 63. [Google Scholar] [CrossRef]
- Mohamed, B.F.; Sallam, N.M.; Alamri, S.A.; Abo-Elyousr, K.A.; Mostafa, Y.S.; Hashem, M. Approving the biocontrol method of potato wilt caused by Ralstonia solanacearum (Smith) using enterobacter cloacae PS14 and Trichoderma asperellum T34. Egypt. J. Biol. Pest Control. 2020, 30, 61. [Google Scholar] [CrossRef]

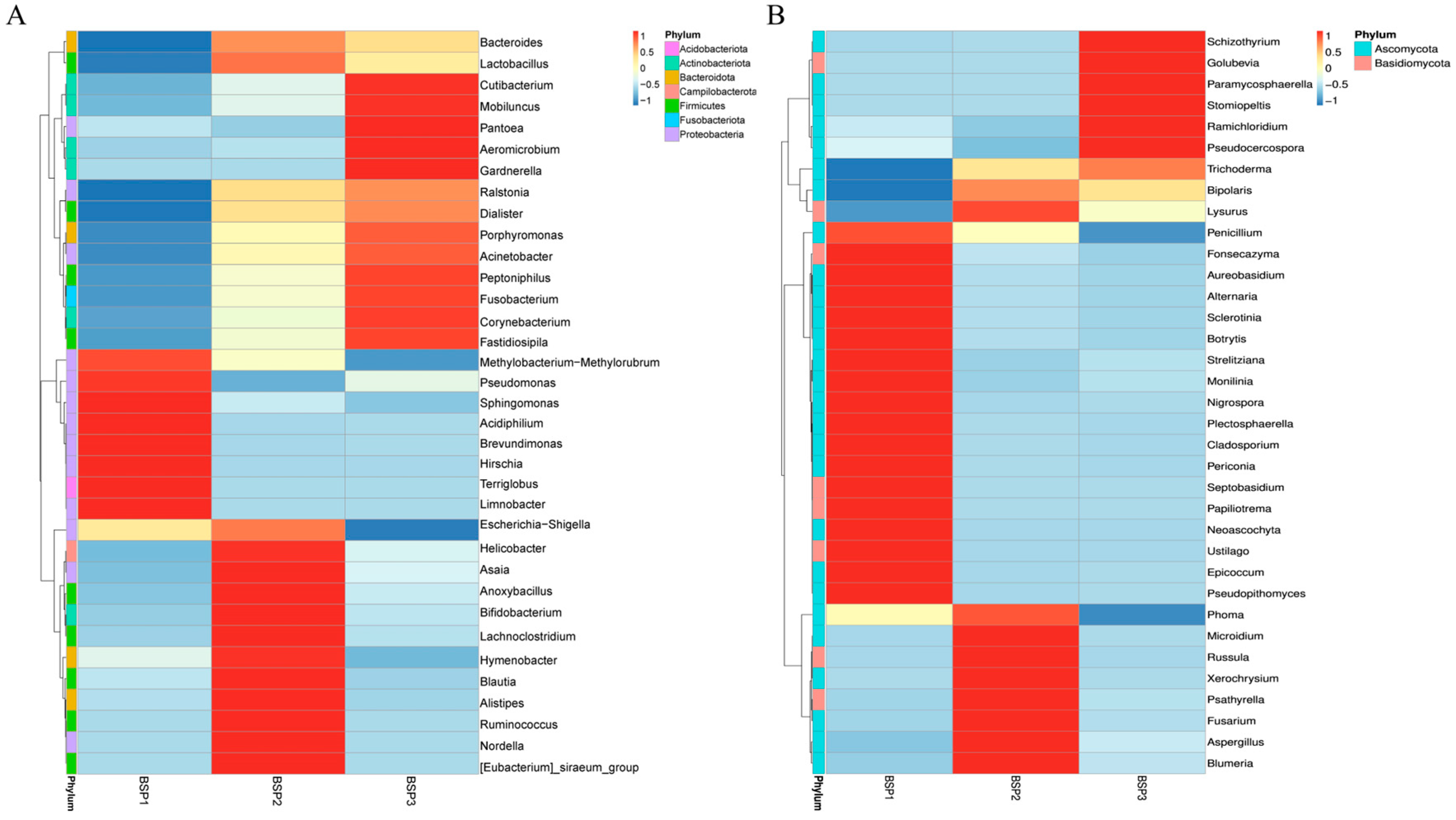

| Bacteria Species | BSP1 vs. BSP2 | BSP1 vs. BSP3 | BSP2 vs. BSP3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Average Abundance (%) | Contribution (%) | Average Abundance (%) | Contribution (%) | Average Abundance (%) | Contribution (%) | ||||
| BSP1 | BSP2 | BSP1 | BSP3 | BSP2 | BSP3 | ||||
| Ralstonia | 0.00 | 27.91 | 22.45 | 0.00 | 34.77 | 20.34 | 27.91 | 34.77 | 32.28 |
| Brevundimonas | 30.74 | 0.00 | 16.88 | 30.74 | 0.20 | 17.86 | 0.00 | 0.20 | 28.61 |
| Limnobacter | 28.15 | 0.00 | 15.46 | 28.15 | 0.00 | 17.82 | 0.00 | 0.00 | 5.38 |
| Pseudomonas | 20.06 | 0.56 | 15.33 | 20.06 | 6.38 | 16.46 | 0.56 | 6.38 | 5.23 |
| Aeromicrobium | 0.00 | 0.44 | 10.71 | 0.00 | 6.10 | 8.00 | 0.44 | 6.10 | 2.88 |
| Blautia | 0.20 | 2.59 | 2.17 | 0.20 | 0.04 | 3.57 | 2.59 | 0.04 | 2.36 |
| Lactobacillus | 0.16 | 4.11 | 1.34 | 0.16 | 2.87 | 1.59 | 4.11 | 2.87 | 1.31 |
| Acidiphilium | 1.79 | 0.00 | 0.99 | 1.79 | 0.04 | 1.03 | 0.00 | 0.04 | 1.12 |
| Bifidobacterium | 0.04 | 1.48 | 0.94 | 0.04 | 0.16 | 1.00 | 1.48 | 0.16 | 1.10 |
| Escherichia-Shigella | 0.84 | 1.12 | 0.80 | 0.84 | 0.28 | 0.77 | 1.12 | 0.28 | 0.88 |
| Fungi Species | BSP1 vs. BSP2 | BSP1 vs. BSP3 | BSP2 vs. BSP3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Average Abundance (%) | Contribution (%) | Average Abundance (%) | Contribution (%) | Average Abundance (%) | Contribution (%) | ||||
| BSP1 | BSP2 | BSP1 | BSP3 | BSP2 | BSP3 | ||||
| Trichoderma | 38.85 | 80.95 | 22.45 | 38.85 | 96.13 | 20.34 | 80.95 | 96.13 | 32.28 |
| Cladosporium | 43.80 | 0.31 | 16.88 | 43.80 | 0.15 | 17.86 | 0.31 | 0.15 | 28.61 |
| Microidium | 0.00 | 12.36 | 15.46 | 0.00 | 0.07 | 17.82 | 12.36 | 0.07 | 5.38 |
| Plectosphaerella | 1.85 | 0.06 | 15.33 | 1.85 | 0.04 | 16.46 | 0.06 | 0.04 | 5.23 |
| Lysurus | 0.00 | 2.73 | 10.71 | 0.00 | 1.24 | 8.00 | 2.73 | 1.24 | 2.88 |
| Alternaria | 2.60 | 0.19 | 2.17 | 2.60 | 0.09 | 3.57 | 0.19 | 0.09 | 2.36 |
| Pseudopithomyces | 1.09 | 0.00 | 1.34 | 1.09 | 0.00 | 1.59 | 0.00 | 0.00 | 1.31 |
| Epicoccum | 2.18 | 0.05 | 0.99 | 2.18 | 0.05 | 1.03 | 0.05 | 0.05 | 1.12 |
| Xerochrysium | 0.00 | 0.87 | 0.94 | 0.00 | 0.00 | 1.00 | 0.87 | 0.00 | 1.10 |
| Septobasidium | 0.83 | 0.00 | 0.80 | 0.83 | 0.00 | 0.77 | 0.00 | 0.00 | 0.88 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, J.; Shi, Y.; Li, H.; Wang, R. Characterizing the Endophytic Microbiome and Microbial Functional Assemblages Associated with Fengtang Plum (Prunus salicina Lindl.) Development and Resistance. Horticulturae 2025, 11, 483. https://doi.org/10.3390/horticulturae11050483
Lei J, Shi Y, Li H, Wang R. Characterizing the Endophytic Microbiome and Microbial Functional Assemblages Associated with Fengtang Plum (Prunus salicina Lindl.) Development and Resistance. Horticulturae. 2025; 11(5):483. https://doi.org/10.3390/horticulturae11050483
Chicago/Turabian StyleLei, Jiqing, Yinna Shi, Hong Li, and Rui Wang. 2025. "Characterizing the Endophytic Microbiome and Microbial Functional Assemblages Associated with Fengtang Plum (Prunus salicina Lindl.) Development and Resistance" Horticulturae 11, no. 5: 483. https://doi.org/10.3390/horticulturae11050483
APA StyleLei, J., Shi, Y., Li, H., & Wang, R. (2025). Characterizing the Endophytic Microbiome and Microbial Functional Assemblages Associated with Fengtang Plum (Prunus salicina Lindl.) Development and Resistance. Horticulturae, 11(5), 483. https://doi.org/10.3390/horticulturae11050483