
Identification and Characterization of Flavonoid Biosynthetic Gene Families in Paeonia Species and Their Roles in Stamen Petalization of Paeonia lactiflora


Abstract
1. Introduction

2. Materials and Methods
2.1. Plant Material Collection, RNA Extraction, and cDNA Synthesis
2.2. Identification of Gene Family Members in the Flavonoid Biosynthesis Pathway
2.3. Chromosome Localization Analysis
2.4. Phylogenetic Analysis
2.5. Prediction of Physicochemical Properties and Subcellular Localization of Proteins
2.6. Protein Domain and Conserved Motif Analysis
2.7. Prediction of Promoter Cis-Acting Elements
2.8. Transcriptional Expression and qRT-PCR Validation of Quercetin Synthesis-Related Genes in P. lactiflora
2.9. Targeted Metabolite Profiling of Quercetin Derivatives
2.10. Structure Preparation and Molecular Docking
3. Results
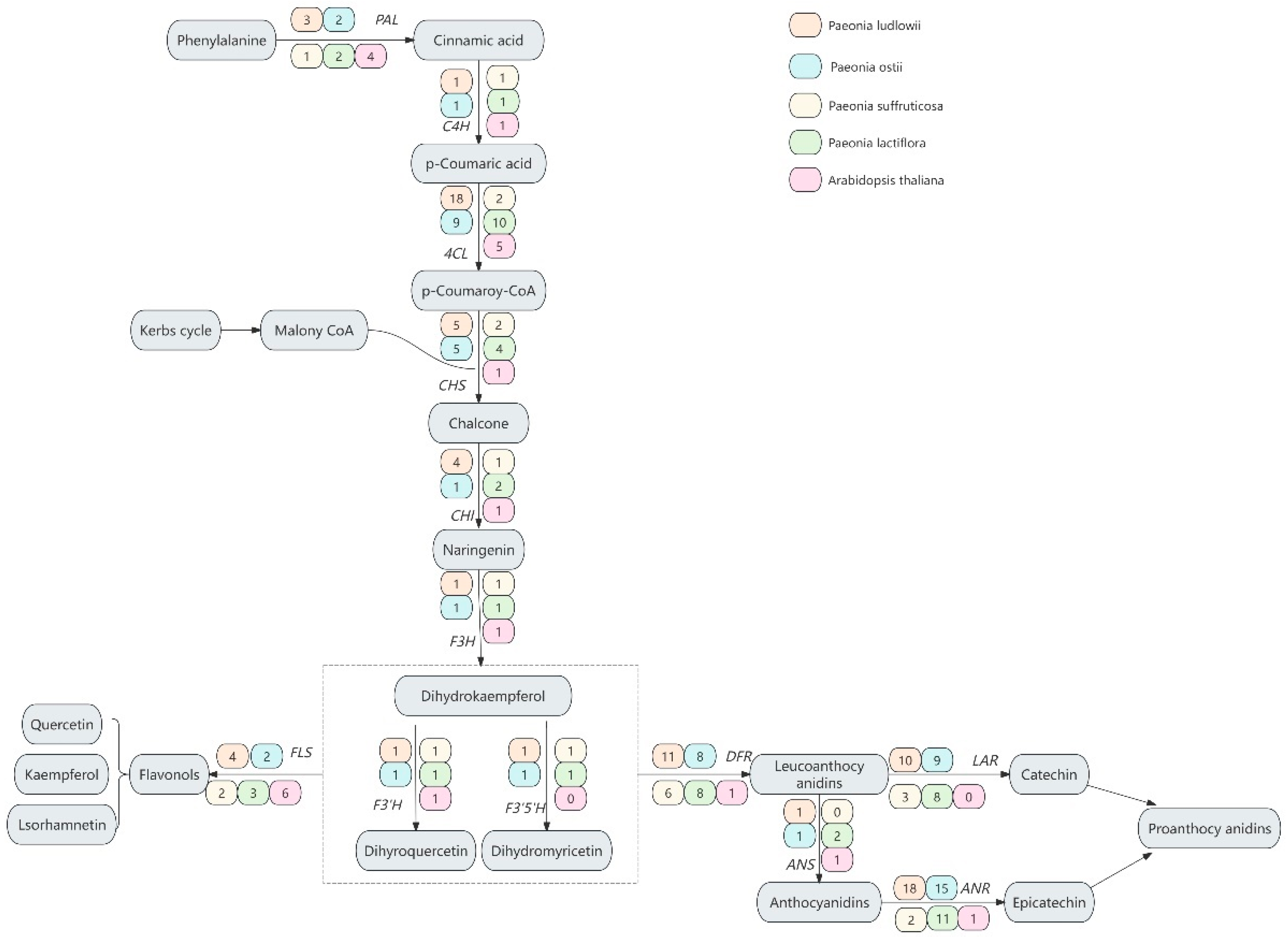
3.1. Identification of 13 Gene Families in the Flavonoid Biosynthesis Pathway of Paeonia
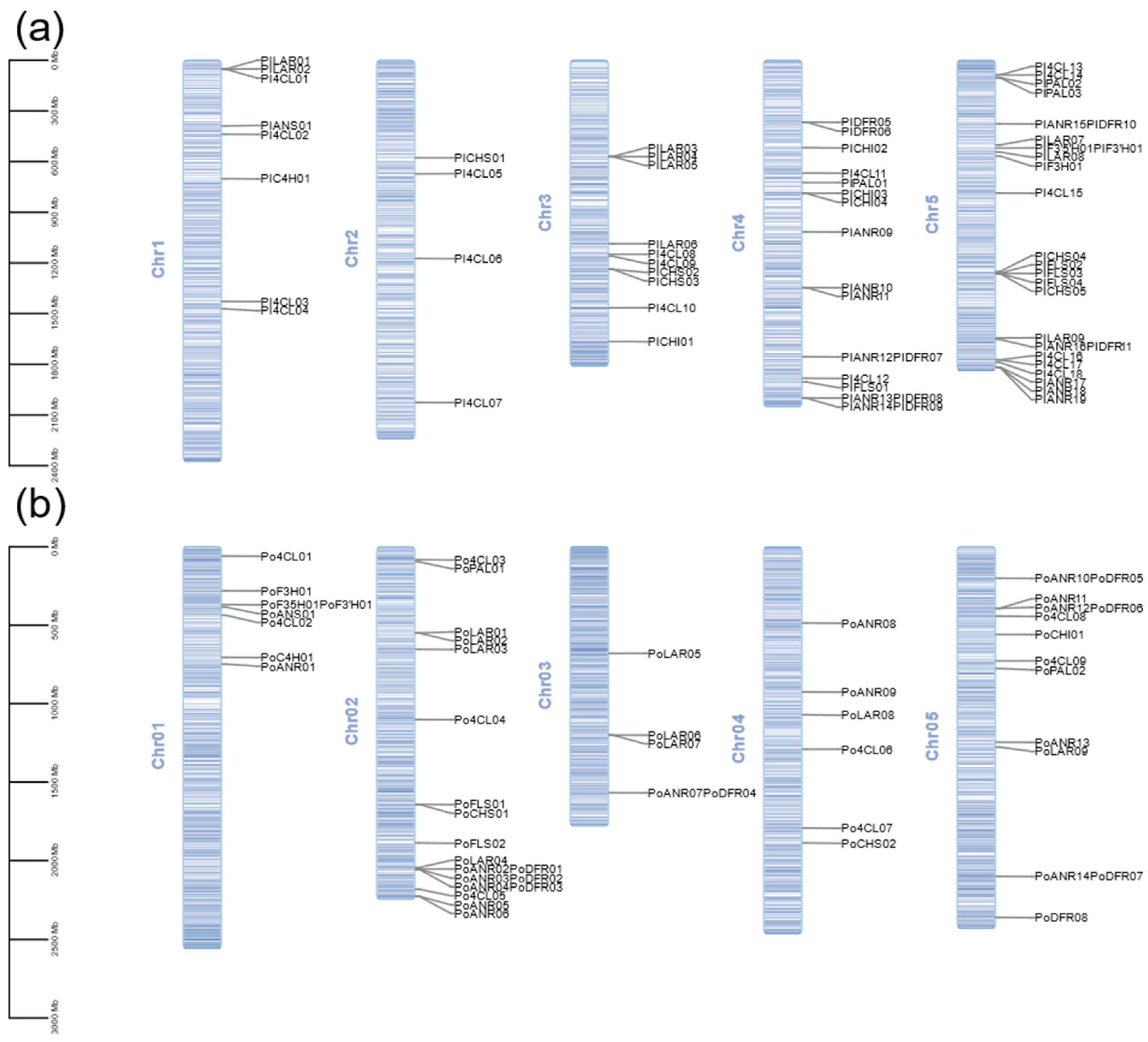
3.2. Chromosome Localization of the Flavonoid Biosynthesis-Related Genes in P. ludlowii and P. ostii
3.3. Physicochemical Properties and Subcellular Localization Prediction of Genes Related to Flavonoid Biosynthesis
3.4. Protein Conserved Domain and Motif Prediction of Genes Related to Flavonoid Biosynthesis
3.5. Prediction of Promoter Cis-Acting Elements of Flavonoid Biosynthesis-Related Genes in P. ostii and P. ludlowii
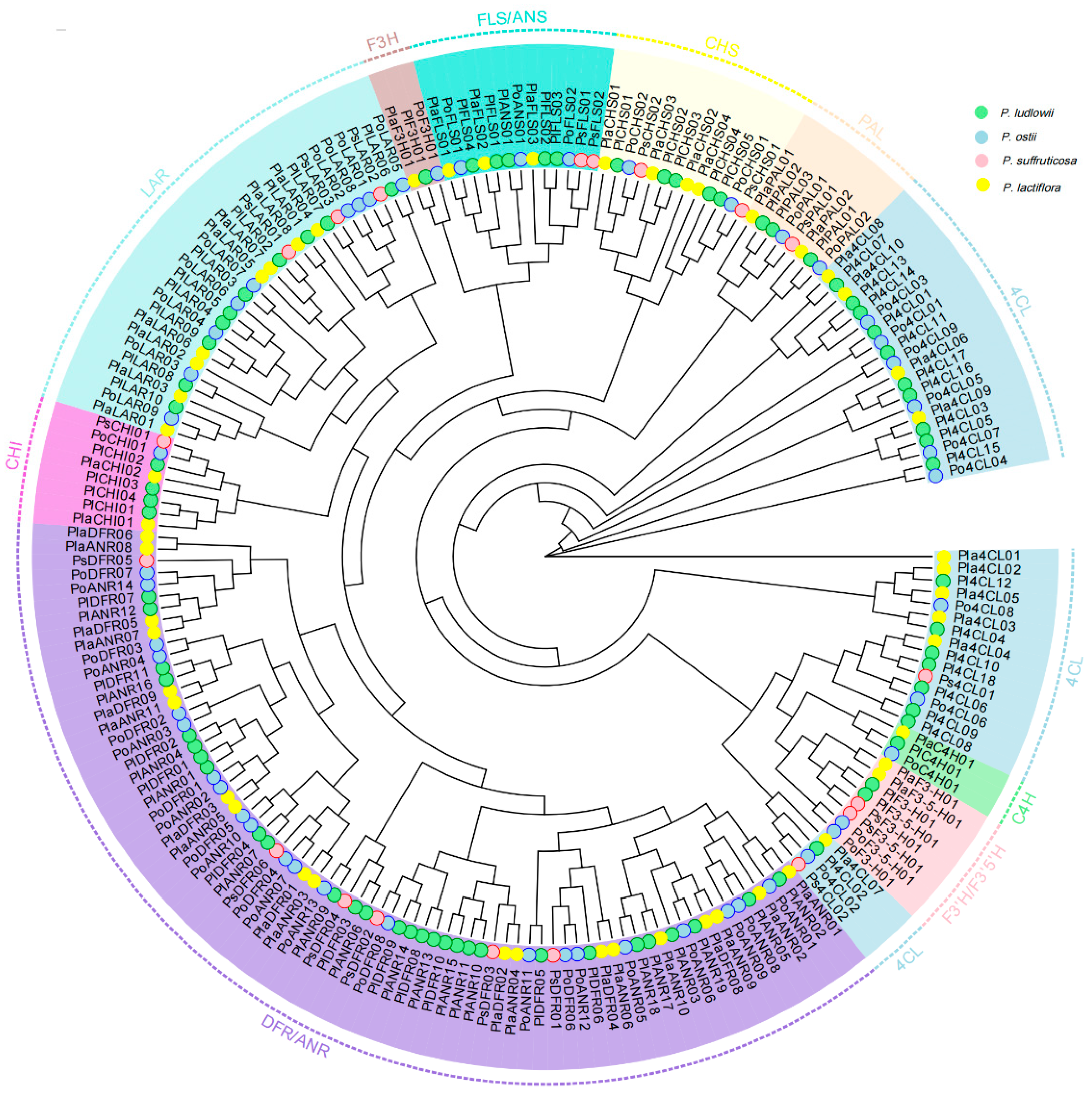
3.6. Phylogenetic Analysis of Gene Families
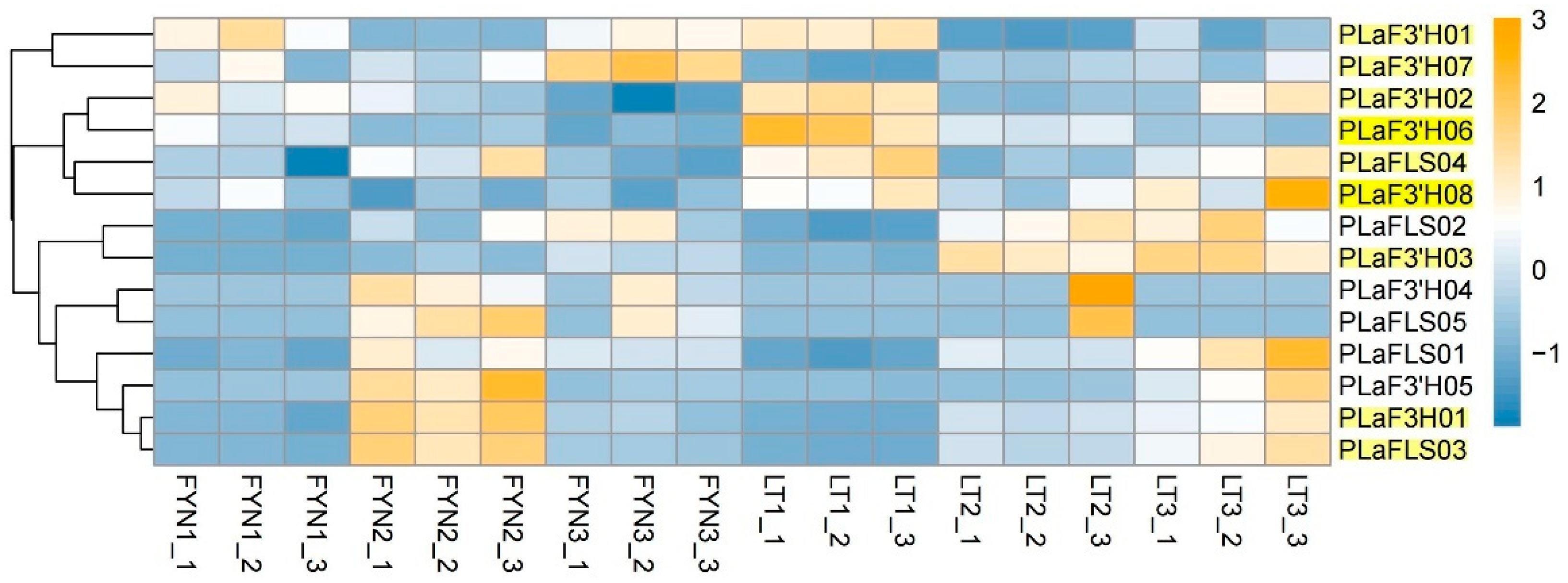
3.7. Expression Pattern Analysis of Quercetin Synthesis Genes in P. lactiflora
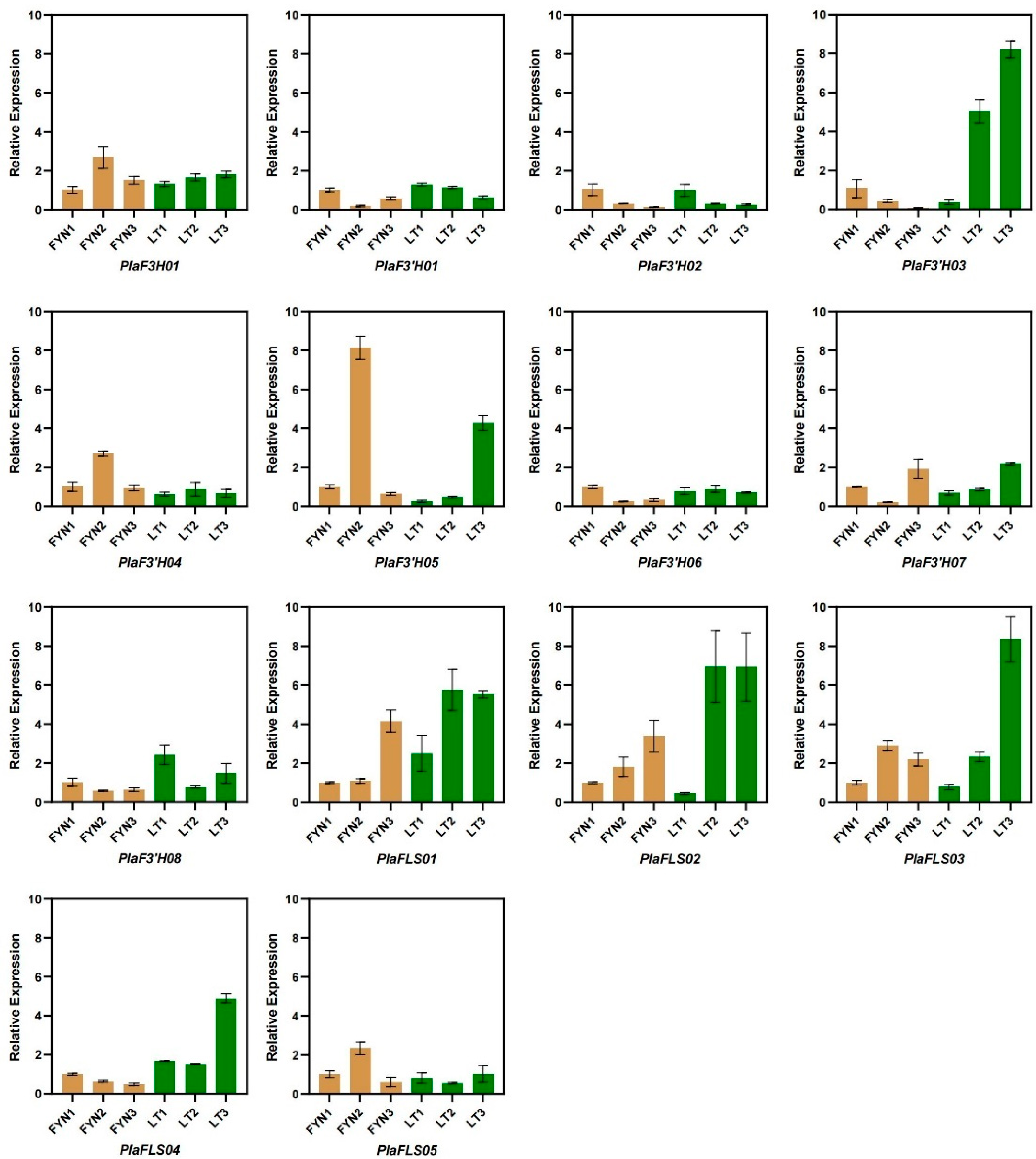
3.8. qPCR Validation of Transcriptome Expression Trends
3.9. Accumulation Patterns of Quercetin Derivatives in P. lactiflora Floral Tissues
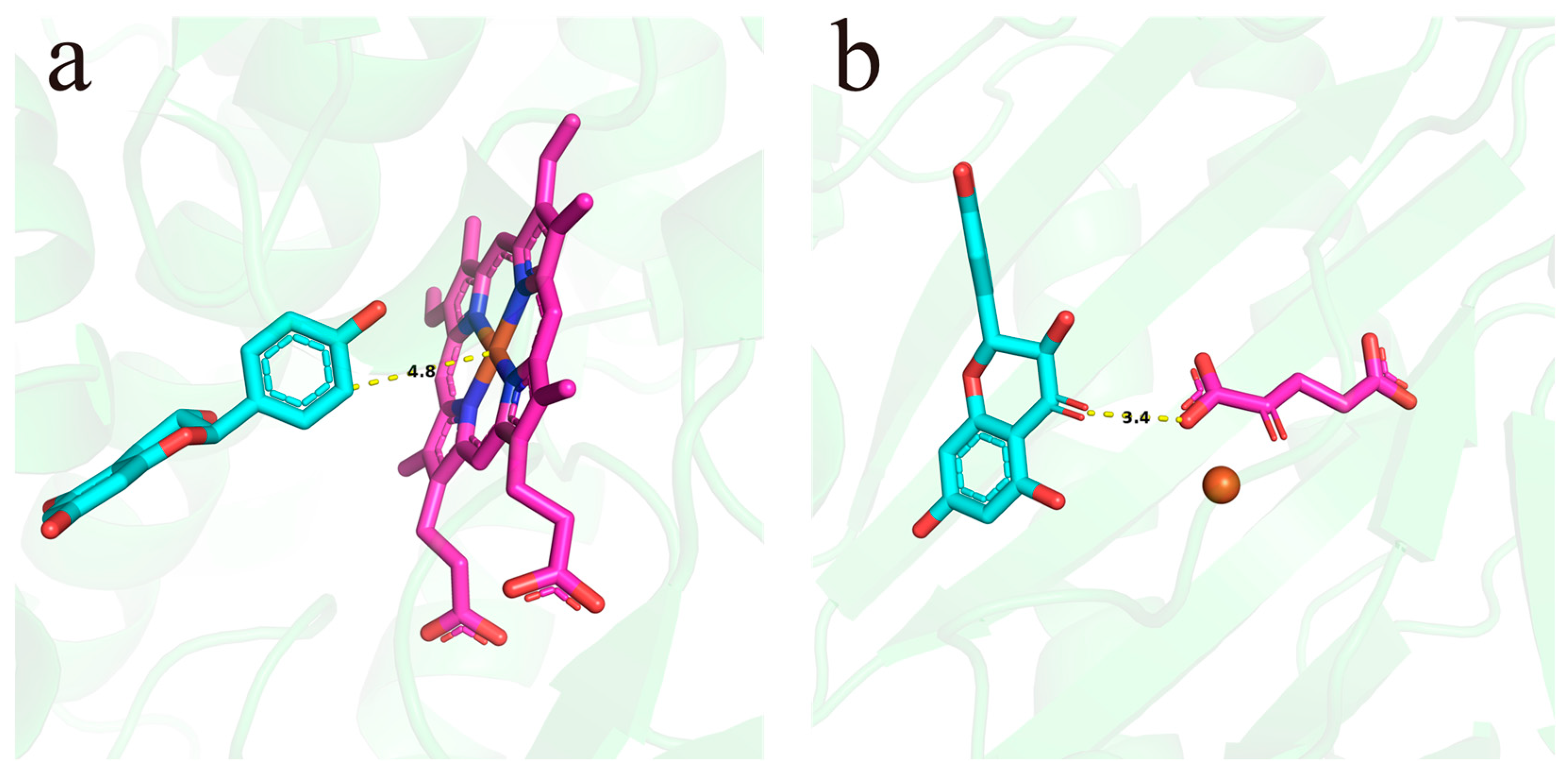
3.10. Screening of Key Enzymes Involved in Quercetin Biosynthesis via Molecular Docking
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shen, N.; Wang, T.; Gan, Q.; Liu, S.; Wang, L.; Jin, B. Plant Flavonoids: Classification, Distribution, Biosynthesis, and Antioxidant Activity. Food Chem. 2022, 383, 132531. [Google Scholar] [CrossRef]
- Kurepa, J.; Shull, T.E.; Smalle, J.A. Friends in Arms: Flavonoids and the Auxin/Cytokinin Balance in Terrestrialization. Plants 2023, 12, 517. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Feng, Y.; Yu, S.; Fan, Z.; Li, X.; Li, J.; Yin, H. The Flavonoid Biosynthesis Network in Plants. Int. J. Mol. Sci. 2021, 22, 12824. [Google Scholar] [CrossRef] [PubMed]
- Daryanavard, H.; Postiglione, A.E.; Mühlemann, J.K.; Muday, G.K. Flavonols Modulate Plant Development, Signaling, and Stress Responses. Curr. Opin. Plant Biol. 2023, 72, 102350. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Arif, Y.; Bajguz, A.; Hayat, S. The Role of Quercetin in Plants. Plant Physiol. Biochem. 2021, 166, 10–19. [Google Scholar] [CrossRef]
- Ylstra, B.; Touraev, A.; Moreno, R.M.B.; Stöger, E.; Van Tunen, A.J.; Vicente, O.; Mol, J.N.M.; Heberle-Bors, E. Flavonols Stimulate Development, Germination, and Tube Growth of Tobacco Pollen. Plant Physiol. 1992, 100, 902–907. [Google Scholar] [CrossRef]
- Van Eldik, G.J.; Reijnen, W.H.; Ruiter, R.K.; Van Herpen, M.M.; Schrauwen, J.A.; Wullems, G.J. Regulation of Flavonol Biosynthesis during Anther and Pistil Development, and during Pollen Tube Growth in Solanum tuberosum. Plant J. 1997, 11, 105–113. [Google Scholar] [CrossRef]
- Teale, W.D.; Pasternak, T.; Dal Bosco, C.; Dovzhenko, A.; Kratzat, K.; Bildl, W.; Schwörer, M.; Falk, T.; Ruperti, B.; Schaefer, J.V.; et al. Flavonol-mediated Stabilization of PIN Efflux Complexes Regulates Polar Auxin Transport. EMBO J. 2021, 40, e104416. [Google Scholar] [CrossRef]
- Lewis, D.R.; Ramirez, M.V.; Miller, N.D.; Vallabhaneni, P.; Ray, W.K.; Helm, R.F.; Winkel, B.S.J.; Muday, G.K. Auxin and Ethylene Induce Flavonol Accumulation through Distinct Transcriptional Networks. Plant Physiol. 2011, 156, 144–164. [Google Scholar] [CrossRef]
- Buer, C.S.; Muday, G.K. The Transparent testa4 Mutation Prevents Flavonoid Synthesis and Alters Auxin Transport and the Response of Arabidopsis Roots to Gravity and Light. Plant Cell 2004, 16, 1191–1205. [Google Scholar] [CrossRef]
- Bouchard, R.; Bailly, A.; Blakeslee, J.J.; Oehring, S.C.; Vincenzetti, V.; Lee, O.R.; Paponov, I.; Palme, K.; Mancuso, S.; Murphy, A.S.; et al. Immunophilin-like TWISTED DWARF1 Modulates Auxin Efflux Activities of Arabidopsis P-Glycoproteins*. J. Biol. Chem. 2006, 281, 30603–30612. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.R.; Miller, N.D.; Splitt, B.L.; Wu, G.; Spalding, E.P. Separating the Roles of Acropetal and Basipetal Auxin Transport on Gravitropism with Mutations in Two Arabidopsis Multidrug Resistance-Like ABC Transporter Genes. Plant Cell 2007, 19, 1838–1850. [Google Scholar] [CrossRef] [PubMed]
- Bailly, A.; Sovero, V.; Vincenzetti, V.; Santelia, D.; Bartnik, D.; Koenig, B.W.; Mancuso, S.; Martinoia, E.; Geisler, M. Modulation of P-Glycoproteins by Auxin Transport Inhibitors Is Mediated by Interaction with Immunophilins*. J. Biol. Chem. 2008, 283, 21817–21826. [Google Scholar] [CrossRef]
- Kumar, V.; Suman, U.; Rubal; Yadav, S.K. Flavonoid Secondary Metabolite: Biosynthesis and Role in Growth and Development in Plants. In Recent Trends and Techniques in Plant Metabolic Engineering; Yadav, S.K., Kumar, V., Singh, S.P., Eds.; Springer: Singapore, 2018; pp. 19–45. ISBN 978-981-13-2251-8. [Google Scholar]
- Hong, D.-Y. Peonies of the World: Taxonomy and Phytogeography; Royal Botanic Gardens Kew: London, UK, 2010; Volume 1, ISBN 1-84246-392-6. [Google Scholar]
- Li, P.; Shen, J.; Wang, Z.; Liu, S.; Liu, Q.; Li, Y.; He, C.; Xiao, P. Genus Paeonia: A Comprehensive Review on Traditional Uses, Phytochemistry, Pharmacological Activities, Clinical Application, and Toxicology. J. Ethnopharmacol. 2021, 269, 113708. [Google Scholar] [CrossRef]
- Zuo, D.-D.; Guo, D.-L.; Hou, X.-G. Development Mechanism of Flower Type and Color in Tree Peony: A Review. Crop Breed. Genet. Genom. 2024, 6, e240009. [Google Scholar] [CrossRef]
- Kamenetsky-Goldstein, R.; Yu, X. Cut Peony Industry: The First 30 Years of Research and New Horizons. Hortic. Res. 2022, 9, uhac079. [Google Scholar] [CrossRef]
- Fan, Y.; Zheng, Y.; Teixeira Da Silva, J.A.; Yu, X. Comparative Transcriptomics and WGCNA Reveal Candidate Genes Involved in Petaloid Stamens in Paeonia lactiflora. J. Hortic. Sci. Biotechnol. 2021, 96, 588–603. [Google Scholar] [CrossRef]
- Lee, S.; Park, N.-I.; Park, Y.; Heo, K.; Kwon, Y.; Kim, E.S.; Son, Y.K.; Lee, K.J.; Choi, S.Y.; Choi, B.-S.; et al. Distribution of Flavonoids in Paeonia suffruticosa and Analyses of the Genes Involved in Flavonoid Biosynthesis in Plants. Plant Gene 2025, 41, 100490. [Google Scholar] [CrossRef]
- Zhang, T.; Yuan, Y.; Zhan, Y.; Cao, X.; Liu, C.; Zhang, Y.; Gai, S. Metabolomics Analysis Reveals Embden Meyerhof Parnas Pathway Activation and Flavonoids Accumulation during Dormancy Transition in Tree Peony. BMC Plant Biol. 2020, 20, 484. [Google Scholar] [CrossRef]
- Wang, X.; Tang, Y.H.; Luan, Y.T.; Zhang, H.C.; Zhao, D.Q.; Tao, J. Flavonoids Composition and Transcriptome Analysis in Herbaceous Peony (Paeonia lactiflora) of Double-Colored Flowers. Russ. J. Plant Physiol. 2022, 69, 66. [Google Scholar] [CrossRef]
- Gu, Z.; Zhu, J.; Hao, Q.; Yuan, Y.-W.; Duan, Y.-W.; Men, S.; Wang, Q.; Hou, Q.; Liu, Z.-A.; Shu, Q.; et al. A Novel R2R3-MYB Transcription Factor Contributes to Petal Blotch Formation by Regulating Organ-Specific Expression of PsCHS in Tree Peony (Paeonia suffruticosa). Plant Cell Physiol. 2019, 60, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Duan, J.; Wang, Q.; Zhang, M.; Zhi, H.; Bai, Z.; Zhang, Y.; Luo, J. The Paeonia Qiui R2R3-MYB Transcription Factor PqMYBF1 Positively Regulates Flavonol Accumulation. Plants 2023, 12, 1427. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Zheng, Y.; Chen, L.; Xu, L.; Teixeira Da Silva, J.A.; Wu, B.; Yu, X. Transcriptomic and Proteomic Analyses Reveal Changes in the Metabolic Pathways of Paeonia lactiflora Petaloid Stamens. Sci. Hortic. 2023, 312, 111859. [Google Scholar] [CrossRef]
- Liu, X.; Chen, Y.; Zhang, J.; He, Y.; Ya, H.; Gao, K.; Yang, H.; Xie, W.; Li, L. Widely Targeted Metabolomics Reveals Stamen Petaloid Tissue of Paeonia lactiflora Pall. Being a Potential Pharmacological Resource. PLoS ONE 2022, 17, e0274013. [Google Scholar] [CrossRef]
- Zhou, S.; Ma, C.; Gao, S.; Hou, D.; Song, P.; Guo, Z.; Zhang, Y.; Shi, G. Cloning and Expression of Floral Organ Identity Genes in Paeonia Ostii ‘Fengdan’. Pak. J. Bot. 2022, 54, 2115–2123. [Google Scholar] [CrossRef]
- Lv, S.; Cheng, S.; Wang, Z.; Li, S.; Jin, X.; Lan, L.; Yang, B.; Yu, K.; Ni, X.; Li, N.; et al. Draft Genome of the Famous Ornamental Plant Paeonia suffruticosa. Ecol. Evol. 2020, 10, 4518–4530. [Google Scholar] [CrossRef]
- Xiao, P.-X.; Li, Y.; Lu, J.; Zuo, H.; Pingcuo, G.; Ying, H.; Zhao, F.; Xu, Q.; Zeng, X.; Jiao, W.-B. High-Quality Assembly and Methylome of a Tibetan Wild Tree Peony Genome (Paeonia ludlowii) Reveal the Evolution of Giant Genome Architecture. Hortic. Res. 2023, 10, uhad241. [Google Scholar] [CrossRef]
- Zheng, Y.; Fan, Y.; Chen, Q.; Sun, M.; Chen, X.; Zhu, W.; Wang, Y.; Teixeira Da Silva, J.A.; Yu, X. Exploring miRNA-Target Modules in Paeonia lactiflora Stamen Petalization through Integrated miRNAome, Transcriptome, and Degradome Analysis. Ind. Crops Prod. 2024, 215, 118662. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Yuan, J.; Jiang, S.; Jian, J.; Liu, M.; Yue, Z.; Xu, J.; Li, J.; Xu, C.; Lin, L.; Jing, Y.; et al. Genomic Basis of the Giga-Chromosomes and Giga-Genome of Tree Peony Paeonia Ostii. Nat. Commun. 2022, 13, 7328. [Google Scholar] [CrossRef]
- Du, L.; Lu, C.; Wang, Z.; Zou, L.; Xiong, Y.; Zhang, Q. GFAnno: Integrated Method for Plant Flavonoid Biosynthesis Pathway Gene Annotation. Beverage Plant Res. 2024, 4, e008. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “One for All, All for One” Bioinformatics Platform for Biological Big-Data Mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a Database of Plant Cis-Acting Regulatory Elements and a Portal to Tools for in Silico Analysis of Promoter Sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Li, P.; Piao, Y.; Shon, H.S.; Ryu, K.H. Comparing the Normalization Methods for the Differential Analysis of Illumina High-Throughput RNA-Seq Data. BMC Bioinform. 2015, 16, 347. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing Real-Time PCR Data by the Comparative CT Method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Sanner, M.F. Python: A Programming Language for Software Integration and Development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
- Biłas, R.; Szafran, K.; Hnatuszko-Konka, K.; Kononowicz, A.K. Cis-Regulatory Elements Used to Control Gene Expression in Plants. Plant Cell Tissue Organ Cult. 2016, 127, 269–287. [Google Scholar] [CrossRef]
- Kerwin, R.E.; Hart, J.E.; Fiesel, P.D.; Lou, Y.-R.; Fan, P.; Jones, A.D.; Last, R.L. Tomato Root Specialized Metabolites Evolved through Gene Duplication and Regulatory Divergence within a Biosynthetic Gene Cluster. Sci. Adv. 2024, 10, eadn3991. [Google Scholar] [CrossRef]
- Shirai, K.; Matsuda, F.; Nakabayashi, R.; Okamoto, M.; Tanaka, M.; Fujimoto, A.; Shimizu, M.; Shinozaki, K.; Seki, M.; Saito, K.; et al. A Highly Specific Genome-Wide Association Study Integrated with Transcriptome Data Reveals the Contribution of Copy Number Variations to Specialized Metabolites in Arabidopsis thaliana Accessions. Mol. Biol. Evol. 2017, 34, 3111–3122. [Google Scholar] [CrossRef]
- Chang, J.; Marczuk-Rojas, J.P.; Waterman, C.; Garcia-Llanos, A.; Chen, S.; Ma, X.; Hulse-Kemp, A.; Deynze, A.V.; de Peer, Y.V.; Carretero-Paulet, L. Chromosome-Scale Assembly of the Moringa Oleifera Lam. Genome Uncovers Polyploid History and Evolution of Secondary Metabolism Pathways through Tandem Duplication. Plant Genome 2022, 15, e20238. [Google Scholar] [CrossRef]
- Liu, S.; Meng, Z.; Zhang, H.; Chu, Y.; Qiu, Y.; Jin, B.; Wang, L. Identification and Characterization of Thirteen Gene Families Involved in Flavonoid Biosynthesis in Ginkgo biloba. Ind. Crops Prod. 2022, 188, 115576. [Google Scholar] [CrossRef]
- Dubos, C.; Le Gourrierec, J.; Baudry, A.; Huep, G.; Lanet, E.; Debeaujon, I.; Routaboul, J.; Alboresi, A.; Weisshaar, B.; Lepiniec, L. MYBL2 Is a New Regulator of Flavonoid Biosynthesis in Arabidopsis thaliana. Plant J. 2008, 55, 940–953. [Google Scholar] [CrossRef]
- Li, B.; Fan, R.; Guo, S.; Wang, P.; Zhu, X.; Fan, Y.; Chen, Y.; He, K.; Kumar, A.; Shi, J. The Arabidopsis MYB Transcription Factor, MYB111 Modulates Salt Responses by Regulating Flavonoid Biosynthesis. Environ. Exp. Bot. 2019, 166, 103807. [Google Scholar] [CrossRef]
- Yue, L.; Zhong, M.; Kang, D.; Qin, H.; Li, H.; Chai, X.; Kang, Y.; Yang, X. Genome-Wide Identification and Characterization of Flavonol Synthase (FLS) Gene Family in Brassica Vegetables and Their Roles in Response to Biotic and Abiotic Stresses. Sci. Hortic. 2024, 331, 113168. [Google Scholar] [CrossRef]
- Peer, W.A.; Murphy, A.S. Flavonoids and Auxin Transport: Modulators or Regulators? Trends Plant Sci. 2007, 12, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Kumari, G.; Nigam, V.K.; Pandey, D.M. The Molecular Docking and Molecular Dynamics Study of Flavonol Synthase and Flavonoid 3′-Monooxygenase Enzymes Involved for the Enrichment of Kaempferol. J. Biomol. Struct. Dyn. 2023, 41, 2478–2491. [Google Scholar] [CrossRef] [PubMed]
- Stracke, R.; De Vos, R.C.H.; Bartelniewoehner, L.; Ishihara, H.; Sagasser, M.; Martens, S.; Weisshaar, B. Metabolomic and Genetic Analyses of Flavonol Synthesis in Arabidopsis thaliana Support the in Vivo Involvement of Leucoanthocyanidin Dioxygenase. Planta 2009, 229, 427–445. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Lowest Binding Free Energy |
|---|---|
| PlaF3’H07 | −4.7 kcal/mol |
| PlaF3’H01 | −2.1 kcal/mol |
| PlaF3’H03 | −2.4 kcal/mol |
| PlaF3’H06 | −0.3 kcal/mol |
| PlaF3’H08 | −kcal/mol |
| PlaF3’H02 | −2.3 kcal/mol |
| PlaF3’H04 | — |
| PlaFLS01 | −3.2 kcal/mol |
| PlaFLS02 | −1.7 kcal/mol |
| PlaFLS03 | — |
| PlaFLS04 | −2.7 kcal/mol |
| PlaFLS05 | −4.6 kcal/mol |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Y.; Fan, Y.; Ji, X.; Wu, X. Identification and Characterization of Flavonoid Biosynthetic Gene Families in Paeonia Species and Their Roles in Stamen Petalization of Paeonia lactiflora. Horticulturae 2025, 11, 463. https://doi.org/10.3390/horticulturae11050463
Zheng Y, Fan Y, Ji X, Wu X. Identification and Characterization of Flavonoid Biosynthetic Gene Families in Paeonia Species and Their Roles in Stamen Petalization of Paeonia lactiflora. Horticulturae. 2025; 11(5):463. https://doi.org/10.3390/horticulturae11050463
Chicago/Turabian StyleZheng, Yanyi, Yongming Fan, Xiang Ji, and Xiaopei Wu. 2025. "Identification and Characterization of Flavonoid Biosynthetic Gene Families in Paeonia Species and Their Roles in Stamen Petalization of Paeonia lactiflora" Horticulturae 11, no. 5: 463. https://doi.org/10.3390/horticulturae11050463
APA StyleZheng, Y., Fan, Y., Ji, X., & Wu, X. (2025). Identification and Characterization of Flavonoid Biosynthetic Gene Families in Paeonia Species and Their Roles in Stamen Petalization of Paeonia lactiflora. Horticulturae, 11(5), 463. https://doi.org/10.3390/horticulturae11050463