
Transcriptome Analysis of Multiple Plant Parts in the Woody Oil Tree Camellia drupifera Loureiro


 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Full-Length Transcriptome Sequencing
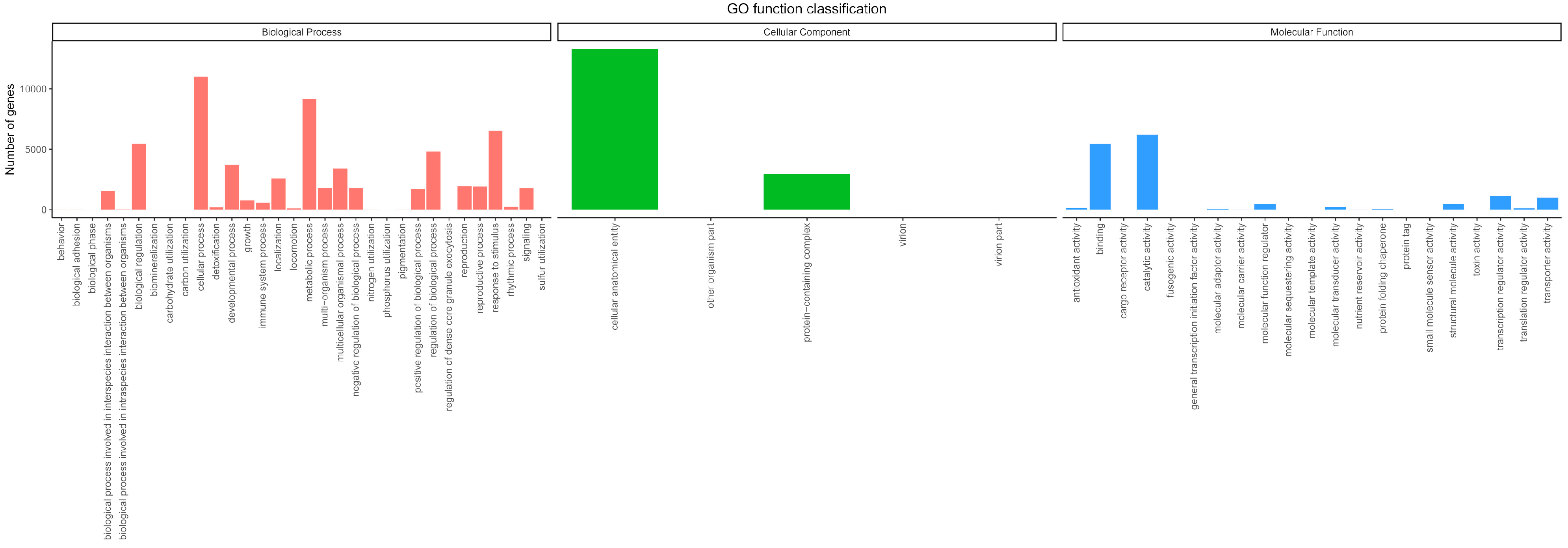
3.2. Functional Classification and Annotation
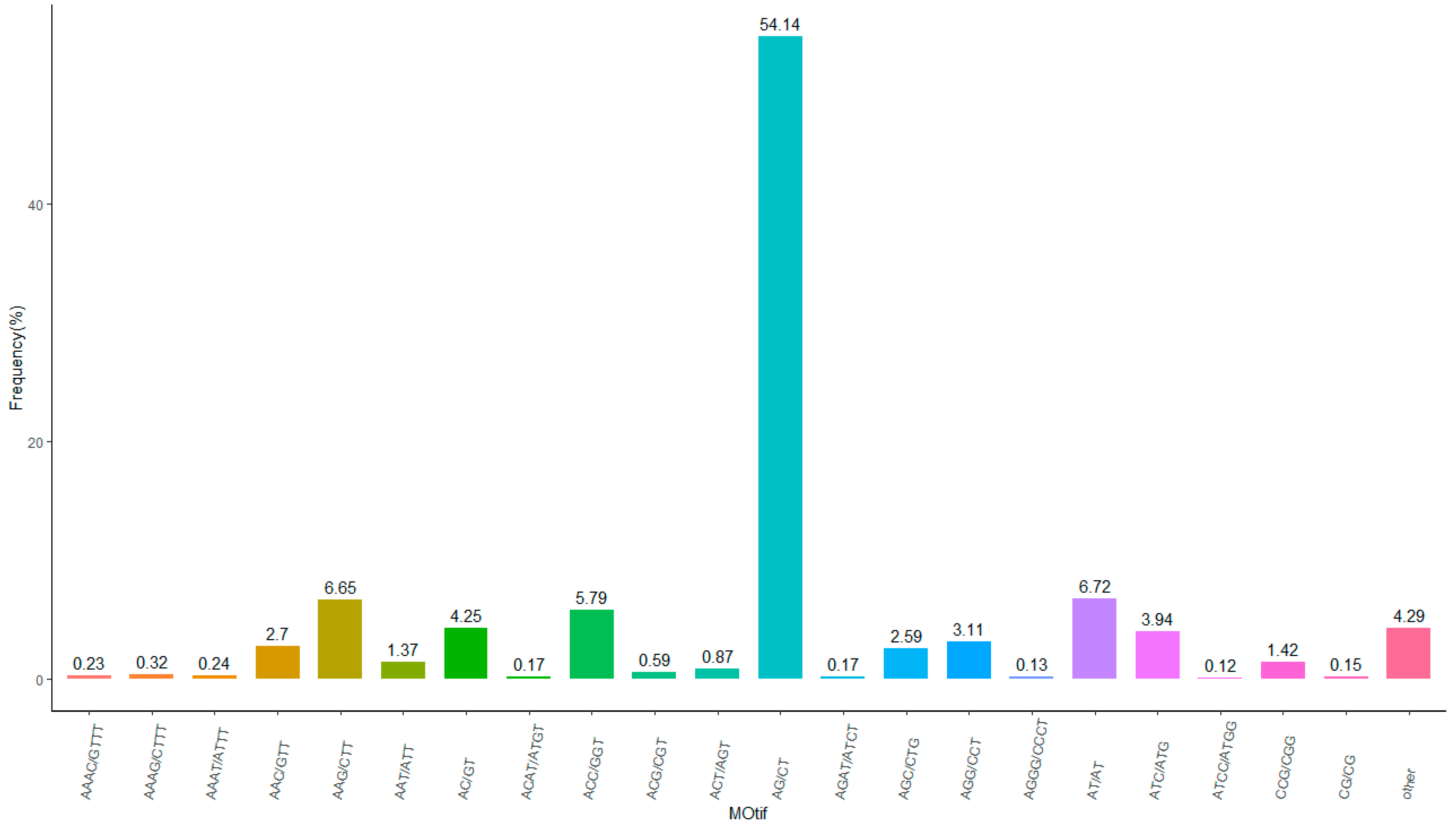
3.3. SSR Analysis
3.4. IncRNA Prediction
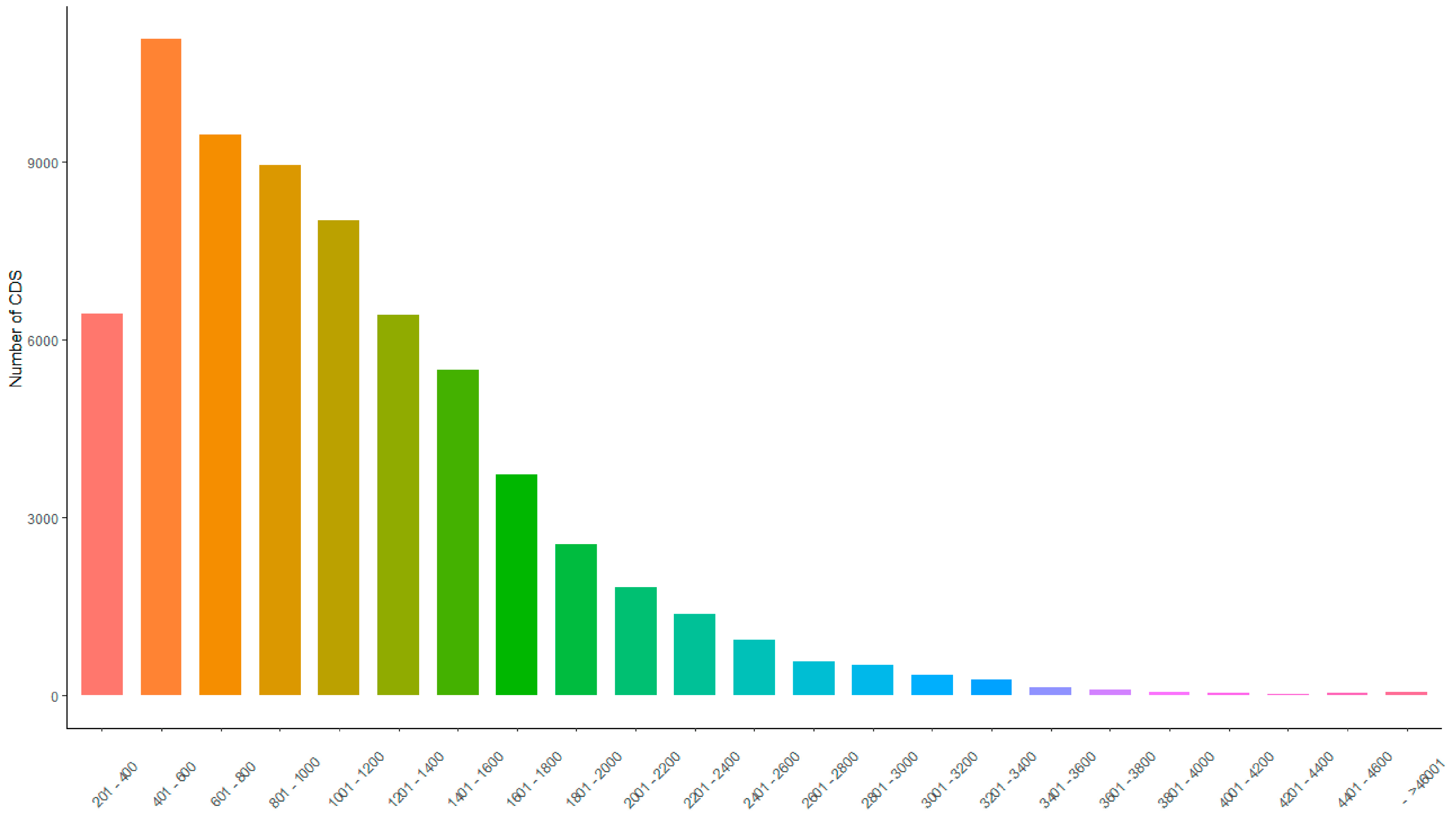
3.5. Protein-Coding Sequence Prediction
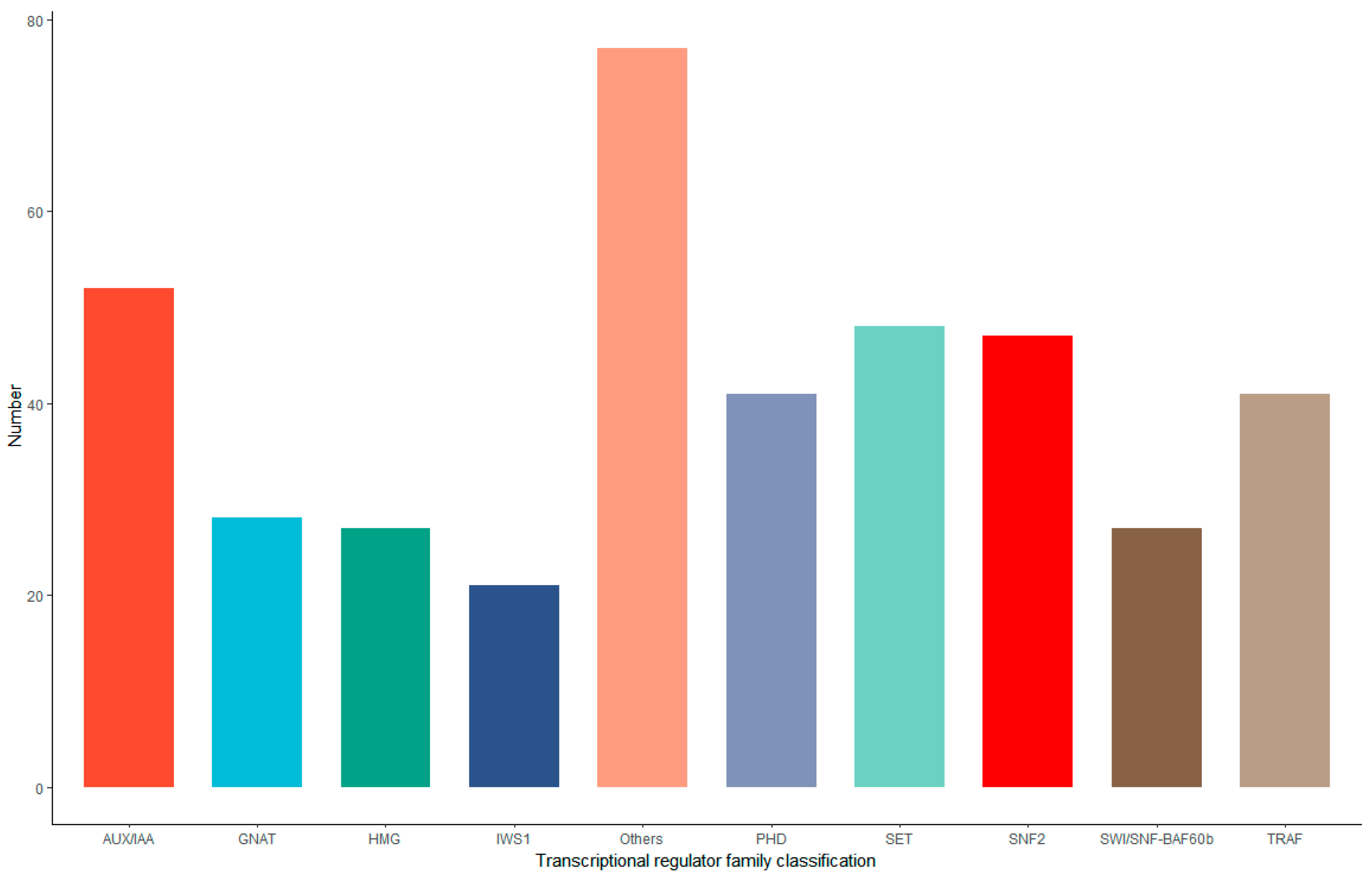
3.6. Transcription Factor and Transcription Regulator Analysis
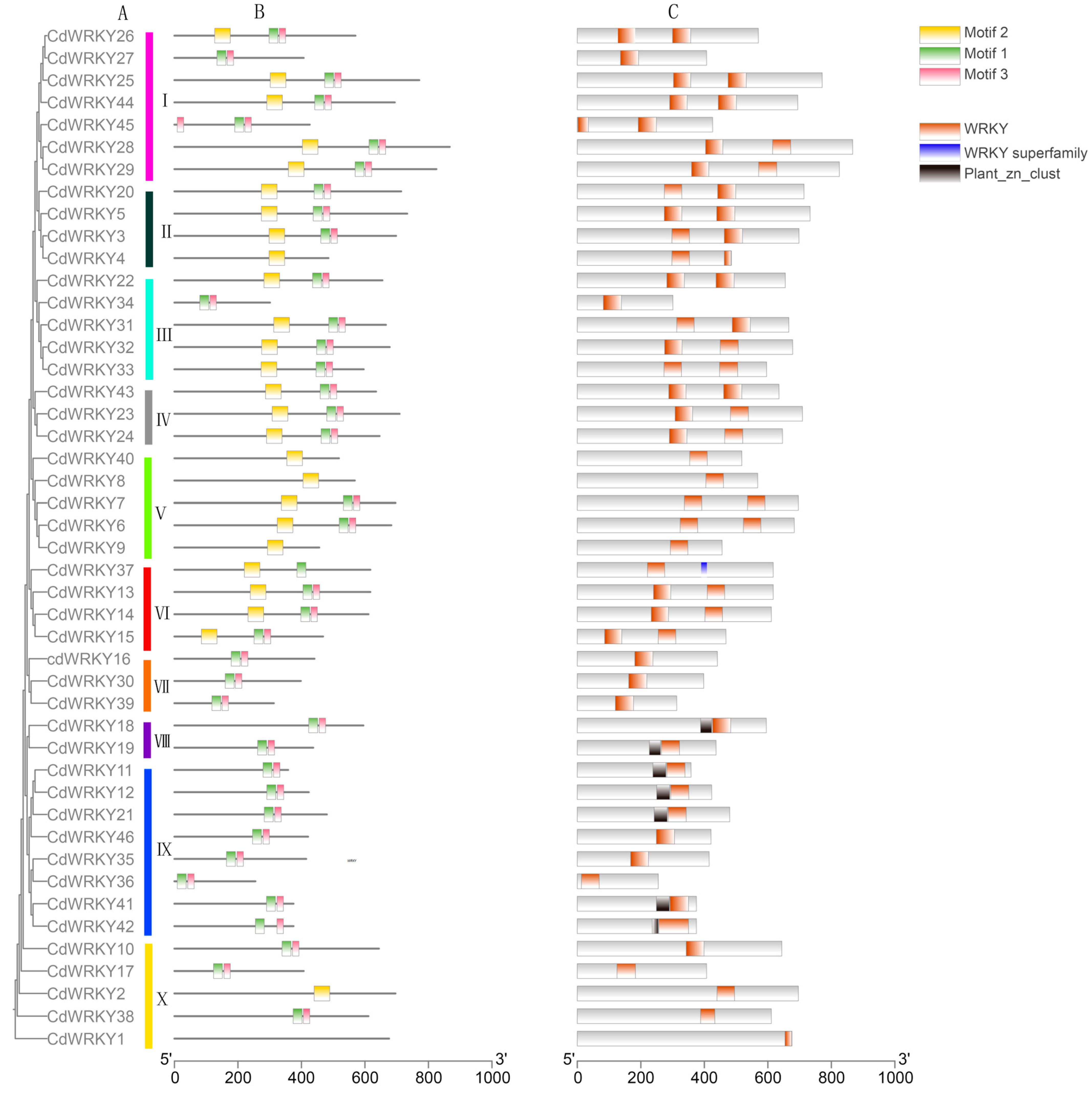
3.7. Identification and Bioinformatics Analysis of WRKY Transcription Factor Families
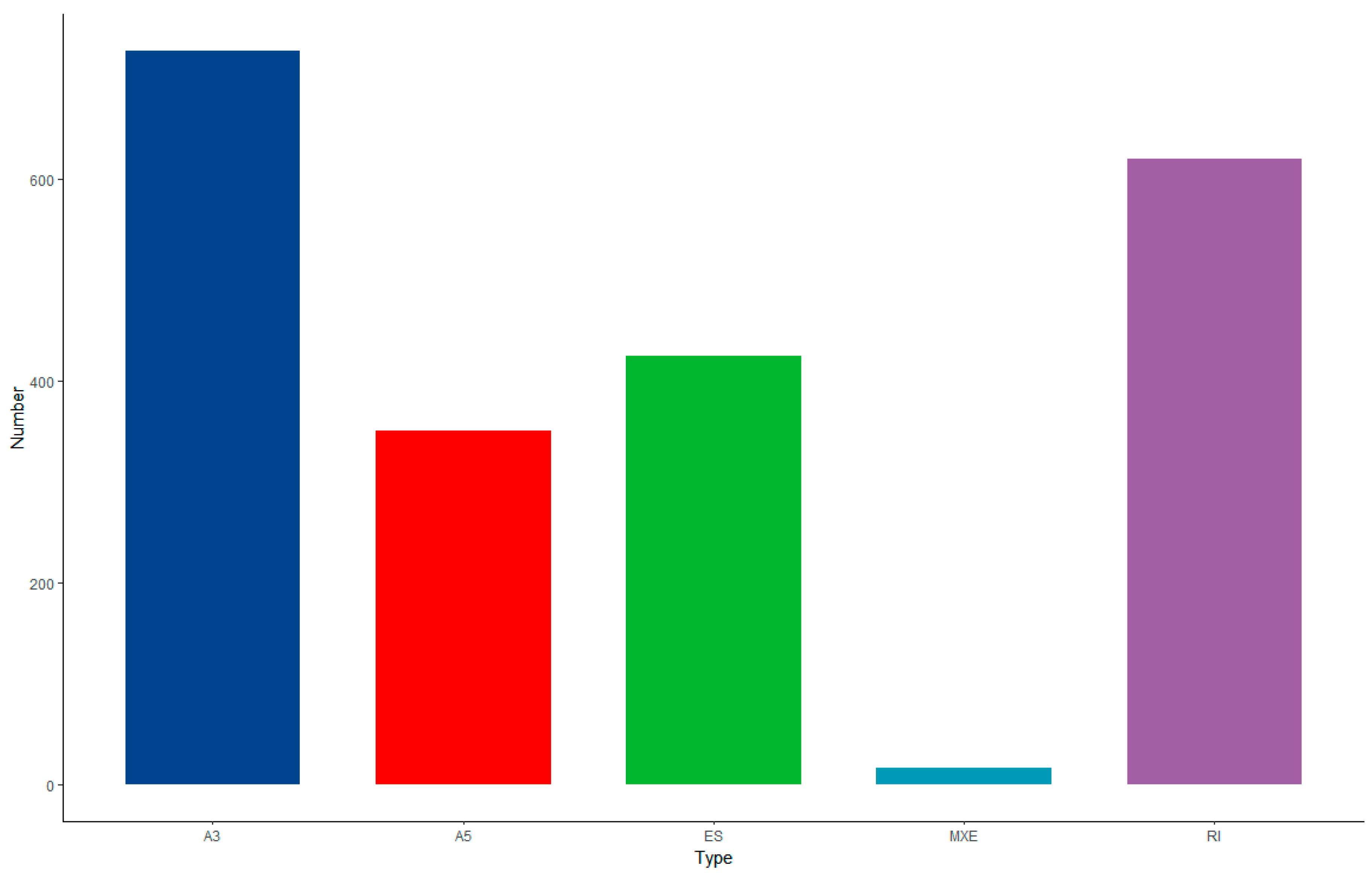
3.8. Alternative Splicing Analysis
3.9. RNA-Seq Characteristics Analysis
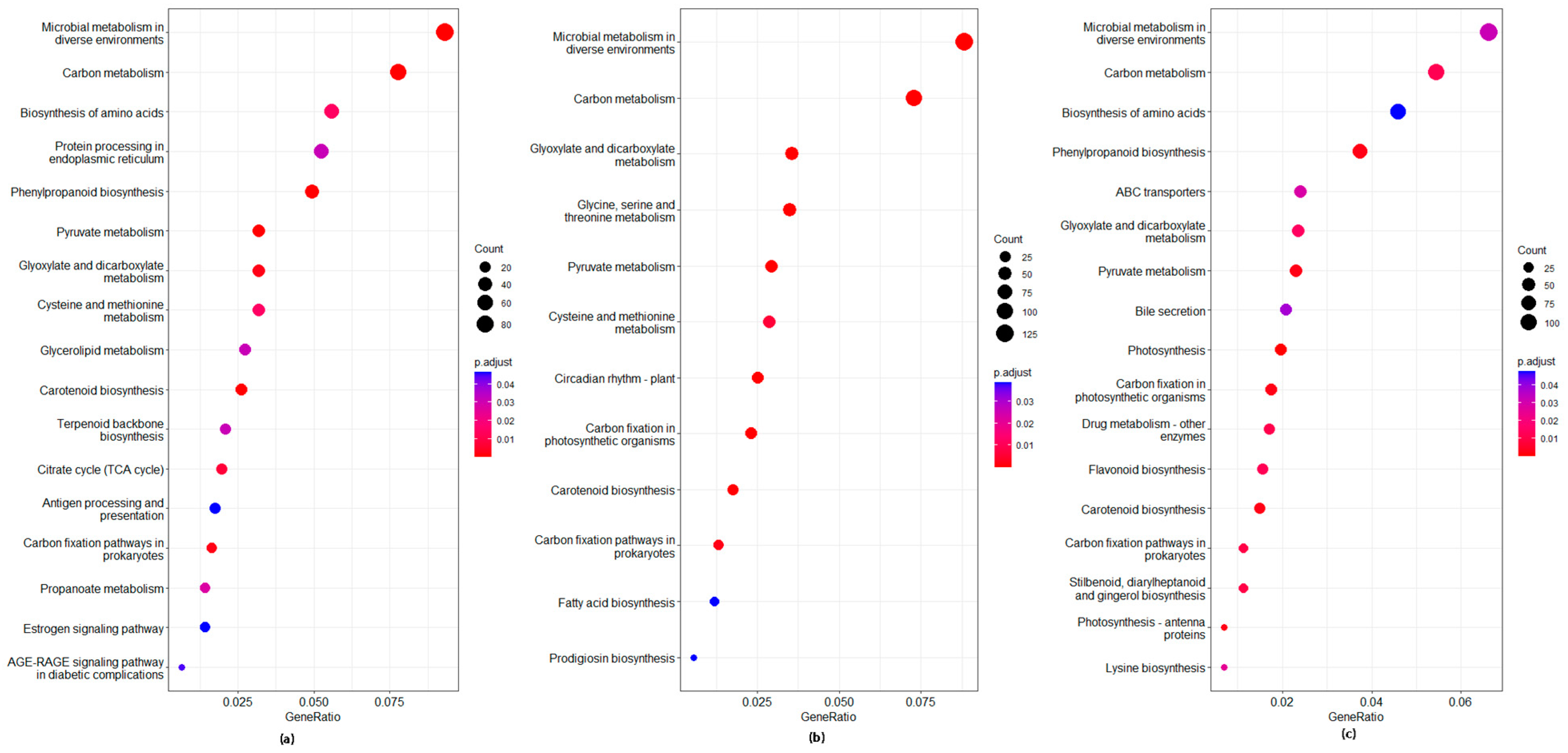
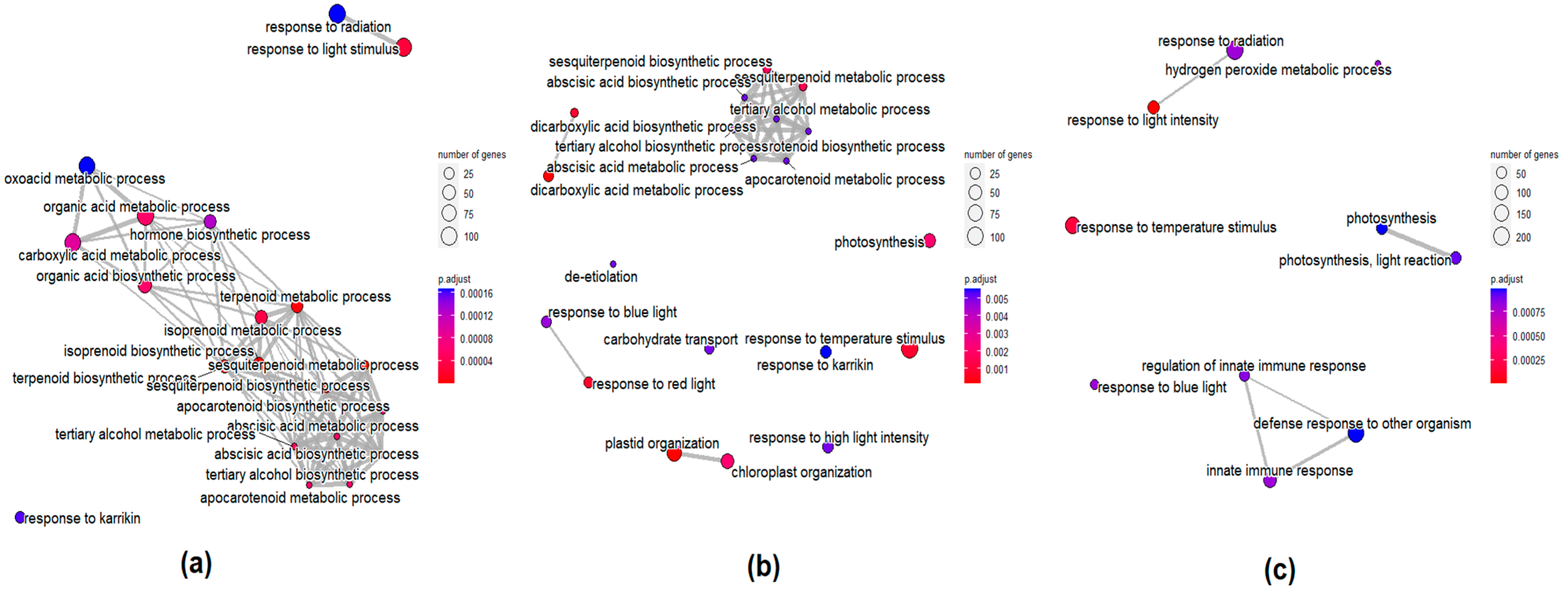
3.10. GO and KEGG Analysis of DEGs
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Y. Physiological Response and Transcriptome of Camellia Oleifera to Drought. Master’s Thesis, Central South University of Forestry and Technology, Changsha, China, 2021. [Google Scholar]
- Chen, S. Research on promotion of Camellia oleifera planting technology. Guangdong Seric. 2022, 56, 66–68. [Google Scholar]
- Tan, X. Advances in the molecular breeding of Camellia oleifera. J. Cen. South Uni. For. Tec. 2023, 43, 1–24. [Google Scholar] [CrossRef]
- Gong, W.; Song, Q.; Ji, K.; Gong, S.; Wang, L.; Chen, L.; Zhang, J.; Yuan, D. Full-length transcriptome from Camellia oleifera seed provides insight into the transcript variants involved in oil biosynthesis. J. Agric. Food Chem. 2020, 68, 14670–14683. [Google Scholar] [CrossRef]
- Ye, Y.; Xing, H.; Chen, X. Anti-inflammatory and analgesic activities of the hydrolyzed sasanquasaponins from the defatted seeds of Camellia oleifera. Arch. Pharm. Res. 2013, 36, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, L.; Mu, Q.; Fang, X.; Zhang, K.; Jia, H.; Li, X.; Bao, Y.; Fang, J. RNA-sequencing reveals biological networks during table grapevine (‘Fujiminori’) fruit development. PLoS ONE 2017, 12, e0170571. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.; Cole, C.; Volden, R.; Vollmers, C. Realizing the potential of full-length transcriptome sequencing. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20190097. [Google Scholar] [CrossRef] [PubMed]
- Hestand, M.S.; Ameur, A. The versatility of SMRT sequencing. Genes 2019, 10, 24. [Google Scholar] [CrossRef]
- Deng, A.; Li, J.; Yao, Z.; Afriyie, G.; Chen, Z.; Guo, Y.; Luo, J.; Wang, Z. SMRT sequencing of the full-length transcriptome of the Coelomactra antiquata. Front. Genet. 2021, 12, 741243. [Google Scholar] [CrossRef]
- Yu, H.; Liu, M.; Yin, M.; Shan, T.; Peng, H.; Wang, J.; Chang, X.; Peng, D.; Zha, L.; Gui, S. transcriptome analysis identifies putative genes involved in triterpenoid biosynthesis in Platycodon grandiflorus. Planta 2021, 254, 34. [Google Scholar] [CrossRef]
- Chin, C.-S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio Sequencing and its applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Liu, X.; Mei, W.; Soltis, P.S.; Soltis, D.E.; Barbazuk, W.B. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome. Mol. Ecol. Resour. 2017, 17, 1243–1256. [Google Scholar] [CrossRef]
- Bridges, M.C.; Daulagala, A.C.; Kourtidis, A. LNCcation: lncRNA localization and function. J. Cell Biol. 2021, 220, e202009045. [Google Scholar] [CrossRef]
- Qiu, F.; Wang, X.; Zheng, Y.; Wang, H.; Liu, X.; Su, X. Full-length transcriptome sequencing and different chemotype expression profile analysis of genes related to monoterpenoid biosynthesis in Cinnamomum porrectum. Int. J. Mol. Sci. 2019, 20, 6230. [Google Scholar] [CrossRef]
- Ni, L.; Wang, Z.; Liu, X.; Wu, S.; Hua, J.; Yin, Y.; Li, H.; Gu, C. transcriptome analysis of salt stress in Hibiscus hamabo Sieb. et Zucc based on Pacbio full-length transcriptome sequencing. Int. J. Mol. Sci. 2022, 23, 138. [Google Scholar] [CrossRef]
- Rao, G.; Zhang, J.; Liu, X.; Ying, L. Identification of putative genes for polyphenol biosynthesis in olive fruits and leaves using full-length transcriptome sequencing. Food Chem. 2019, 300, 125246. [Google Scholar]
- Feng, Y.; Zhao, Y.; Zhang, J.; Wang, B.; Yang, C.; Zhou, H.; Qiao, J. Full-length SMRT transcriptome sequencing and microsatellite characterization in Paulownia catalpifolia. Sci. Rep. 2021, 11, 8734. [Google Scholar] [CrossRef]
- Jia, X.; Tang, L.; Mei, X.; Liu, H.; Luo, H.; Deng, Y.; Su, J. Single-molecule long-read sequencing of the full-length transcriptome of Rhododendron lapponicum L. Sci. Rep. 2020, 10, 6755. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Su, Y.; Wang, T. Full-length transcriptome analysis of four different tissues of Cephalotaxus oliveri. Int. J. Mol. Sci. 2021, 22, E787. [Google Scholar] [CrossRef]
- Qu, X.; Zhou, J.; Masabni, J.; Yuan, J. Phosphorus relieves aluminum toxicity in oil tea seedlings by regulating the metabolic profiling in the roots. Plant Physiol. Biochem. 2020, 152, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhong, Q.; Tian, F.; Zhou, X.; Tan, X.; Luo, Z. Transcriptome analysis reveals putative induction of floral initiation by old leaves in tea-oil tree (Camellia oleifera ’Changlin53′). Int. J. Mol. Sci. 2022, 23, 13021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Cui, Z.; Guo, M.; Xi, R. Characteristics of the soil microbial community in the forestland of Camellia oleifera. PeerJ 2020, 8, e9117. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liang, D.; Song, Z.; Tan, Y.; Guo, X.; Wang, D. Genetic diversity analysis and core germplasm collection construction of Camellia oleifera based on fruit phenotype and SSR data. Genes 2022, 13, 2351. [Google Scholar] [CrossRef]
- Long, W.; Huang, G.; Yao, X.; Lv, L.; Yu, C.; Wang, K. Untargeted metabolism approach reveals difference of varieties of bud and relation among characteristics of grafting seedlings in Camellia oleifera. Front. Plant Sci. 2022, 13, 1024353. [Google Scholar] [CrossRef] [PubMed]
- Hao, B.-Q.; Liao, H.-Z.; Xia, Y.-Y.; Wang, D.-X.; Ye, H. BSR and full-length transcriptome approaches identified candidate genes for high seed ratio in Camellia vietnamensis. Curr. Issues Mol. Biol. 2022, 45, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Miao, B.-B.; Dong, W.; Gu, Y.-X.; Han, Z.-F.; Luo, X.; Ke, C.-H.; You, W.-W. OmicsSuite: A customized and pipelined suite for analysis and visualization of multi-omics big data. Hortic. Res. 2023, 10, uhad195. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding rnas and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.C.; Maddox, J.D.; Rodríguez, H.N.; Castro, C.G.; Imán-Correa, S.A.; Cobos, M.; Paredes, J.D.; Marapara, J.L.; Braga, J.; Adrianzén, P.M. Dataset of de novo assembly and functional annotation of the transcriptome during germination and initial growth of seedlings of Myrciaria dubia “Camu-Camu”. Data Brief 2020, 31, 105834. [Google Scholar] [CrossRef] [PubMed]
- Foissac, S.; Sammeth, M. ASTALAVISTA: Dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucleic Acids Res. 2007, 35, W297–W299. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.-Y.; Chen, X.; Gao, G.; Zhang, H.; Zhu, Q.-H.; Liu, X.-C.; Zhong, Y.-F.; Gu, X.; He, K.; Luo, J. PlantTFDB: A comprehensive plant transcription factor database. Nucleic Acids Res. 2008, 36, D966–D969. [Google Scholar] [CrossRef] [PubMed]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER Web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Diao, P.; Chen, C.; Zhang, Y.; Meng, Q.; Lv, W.; Ma, N. The role of NAC transcription factor in plant cold response. Plant Signal. Behav. 2020, 15, 1785668. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.; Duan, Y.; Wang, C.; Xu, M.; He, C.; Su, L.; Zheng, Y. Identification and bioinformatics analysis of Bhlh transcription factor family in Clerodendrum japonicum. Mol. Plant Breed. 2023, 1–17. [Google Scholar]
- Dai, Z.; Sheridan, J.M.; Gearing, L.J.; Moore, D.L.; Su, S.; Wormald, S.; Wilcox, S.; O’Connor, L.; Dickins, R.A.; Blewitt, M.E.; et al. EdgeR: A versatile tool for the analysis of shRNA-Seq and CRISPR-Cas9 genetic screens. F1000Res 2014, 3, 95. [Google Scholar] [CrossRef]
- Zhang, Z.; Xu, Y.; Chen, Y.; Li, Z.; Wang, X.; Chen, L.; Peng, S.; Ma, L.; Wang, R.; Li, M.; et al. Transcriptome sequencing and analysis of SSR characteristics of Camellia oleifera. J. South. For. Uni. 2018, 38, 63–68. [Google Scholar]
- Zhang, C.; Ren, H.; Yao, X.; Wang, K.; Chang, J. Full-length transcriptome analysis of pecan (Carya illinoinensis) kernels. G3-Genes Genom. Genet. 2021, 11, jkab182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Z.; Hu, A.; Wu, H.; Zhu, J.; Wang, F.; Cao, P.; Yang, X.; Zhang, H. Full-length transcriptome analysis of the halophyte Nitraria sibirica Pall. Genes 2022, 13, 661. [Google Scholar] [CrossRef] [PubMed]
- Lin, P. The genome of oil-Camellia and population genomics analysis provide insights into seed oil domestication. Genom. Biol. 2022, 23, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Liao, B.; Hang, R.; Luo, H.; Tu, P.; Wang, Y.; Dai, W.; Lu, Y.; Li, Y. Populations construction and genetic evaluation of hybrid F1 generation of Camellia gauchowensis Chang II. Non-Wood For. Res. 2023, 41, 91–105. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, B.; Wen, D.; Liu, R.; Yao, X.; Chen, Z.; Mu, R.; Pei, H.; Liu, M.; Song, B.; et al. Chromosome-scale genome assembly of Camellia sinensis combined with multi-omics provides insights into its responses to infestation with green leafhoppers. Front. Plant Sci. 2022, 13, 1004387. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Kumar, V.; Olson, A.; Ware, D. Reviving the transcriptome studies: An insight into the emergence of single-molecule transcriptome sequencing. Front. Genet. 2019, 10, 384. [Google Scholar] [CrossRef] [PubMed]
- Minio, A.; Massonnet, M.; Figueroa-Balderas, R.; Vondras, A.M.; Blanco-Ulate, B.; Cantu, D. Iso-seq allows genome-independent transcriptome profiling of grape berry development. G3-Genes Genom. Genet. 2019, 9, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yan, H.; Wang, S.; Chen, Y.; Shang, J. Camellia oleifera “Hengchong 89” transcriptomes and gene expression of photosynthesis and lipid pathway. Non-Wood For. Res. 2022, 40, 31–39. [Google Scholar] [CrossRef]
- Ha, Y.J.; Sa, K.J.; Lee, J.K. Identifying SSR markers associated with seed characteristics in perilla (Perilla frutescens L.). Physiol. Mol. Biol. Plants 2021, 27, 93–105. [Google Scholar] [CrossRef]
- Jia, X.; Deng, Y.; Sun, X.; Liang, L.; Su, J. De novo assembly of the transcriptome of Neottopteris nidus using Illumina paired-end sequencing and development of EST-SSR markers. Mol. Breed. 2016, 36, 94. [Google Scholar] [CrossRef]
- Ma, D.; Fang, J.; Ding, Q.; Wei, L.; Li, Y.; Zhang, L.; Zhang, X. A survey of transcriptome complexity using full-length isoform sequencing in the tea plant Camellia sinensis. Mol. Genet. Genom. 2022, 297, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Ji, X.; Zhang, Z.; Li, H.; Zhang, H.; Song, W. Bioinformatics analysis of Capsicum C3H transcription factor family. Mol. Plant Breed. 2020, 18, 1784–1791. [Google Scholar] [CrossRef]
- Bao, Y.; Nie, T.; Wang, D.; Chen, Q. Anthocyanin regulatory networks in Solanum tuberosum L. leaves elucidated via integrated metabolomics, transcriptomics, and StAN1 overexpression. BMC Plant Biol. 2022, 22, 228. [Google Scholar] [CrossRef] [PubMed]
- Szakonyi, D.; Duque, P. Alternative splicing as a regulator of early plant development. Front. Plant Sci. 2018, 9, 1174. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Xiang, Y.; Xiong, Y.; Lin, Z.; Xue, Y.; Mao, M.; Sun, L.; Zhou, Y.; Li, X.; Huang, Z. SMRT sequencing analysis reveals the full-length transcripts and alternative splicing patterns in Ananas comosus var. bracteatus. PeerJ 2019, 7, e7062. [Google Scholar] [CrossRef] [PubMed]
- Qiao, D.; Yang, C.; Chen, J.; Guo, Y.; Li, S. Comprehensive identification of the full-length transcripts and alternative splicing related to the secondary metabolism pathways in the tea plant (Camellia sinensis). Sci. Rep. 2019, 9, 2709. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Cao, S.-F.; Shi, L.-Y.; Chen, W.; Yin, X.-R.; Yang, Z.-F. Abscisic acid biosynthesis, metabolism and signaling in ripening fruit. Front. Plant Sci. 2023, 14, 1279031. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yuan, B.; Leng, P. The role of ABA in triggering ethylene biosynthesis and ripening of tomato fruit. J. Exp. Bot. 2009, 60, 1579–1588. [Google Scholar] [CrossRef]
- Shen, Y.; Duan, W.; Hu, J.; Cui, N.; Cao, Z.; Shu, Q. Relationships between peel anatomy structure of Camellia oleifera and resistance to Colletotrichum gloeosporioides. Plant Prot. 2015, 41, 98–102. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions, and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Number | N50 Length (bp) | Mean Length (bp) |
|---|---|---|---|
| Subreads | 48,000,902 | 1587 | 1332 |
| CCS | 733,008 | 1904 | 1640 |
| FLNC | 649,567 | 1691 | 1431 |
| Cluster | 72,253 | 1735 | 1476 |
| Corrected consensus | 71,615 | 1727 | 1470 |
| Repeat Number | A | C | G | T | Total |
|---|---|---|---|---|---|
| 5–15 | 2776 | 216 | 132 | 7393 | 10,517 |
| 16–25 | 821 | 212 | 81 | 2433 | 3547 |
| 26–35 | 68 | 7 | 1 | 54 | 130 |
| 36–45 | 15 | 1 | 0 | 1 | 17 |
| 46–55 | 5 | 0 | 0 | 0 | 5 |
| 56–65 | 1 | 0 | 0 | 0 | 1 |
| Total | 3686 | 436 | 214 | 9882 | 14,218 |
| Gene Name | Gene ID | Number of Amino Acids (aa) | Molecular Weight (Da) | pI | Aliphatic Index | GRAVY |
|---|---|---|---|---|---|---|
| CdWRKY1 | transcript/12715-0F | 676 | 77,558.85 | 9.14 | 79.56 | −0.267 |
| CdWRKY2 | transcript/12715-1F | 696 | 80,332.99 | 9.70 | 75.33 | −0.444 |
| CdWRKY3 | transcript/13333-0F | 698 | 78,409.60 | 8.82 | 53.34 | −0.816 |
| CdWRKY4 | transcript/32049-0F | 485 | 53,980.17 | 8.59 | 51.05 | −0.979 |
| CdWRKY5 | transcript/10090-1F | 733 | 81,630.75 | 8.61 | 56.23 | −0.599 |
| CdWRKY6 | transcript/13640-0F | 683 | 76,683.25 | 8.74 | 66.19 | −0.629 |
| CdWRKY7 | transcript/12770-1F | 696 | 78,325.12 | 8.82 | 67.20 | −0.655 |
| CdWRKY8 | transcript/22628-2F | 568 | 63,432.05 | 6.76 | 64.21 | −0.644 |
| CdWRKY9 | transcript/35057-2F | 456 | 50,736.29 | 7.71 | 59.43 | −0.786 |
| CdWRKY10 | transcript/16490-0F | 644 | 71,338.50 | 7.82 | 74.24 | −0.400 |
| CdWRKY11 | transcript/47559-1F | 358 | 39,265.84 | 9.31 | 81.70 | −0.284 |
| CdWRKY12 | transcript/40146-1F | 423 | 46,659.14 | 9.03 | 82.96 | −0.253 |
| CdWRKY13 | transcript/18070-2F | 617 | 68,544.64 | 5.53 | 67.94 | −0.643 |
| CdWRKY14 | transcript/19438-0F | 611 | 67,895.13 | 5.70 | 67.18 | −0.622 |
| CdWRKY15 | transcript/33657-1F | 468 | 52,261.53 | 9.02 | 69.15 | −0.518 |
| CdWRKY16 | transcript/36057-2F | 441 | 50,114.73 | 9.01 | 70.48 | −0.441 |
| CdWRKY17 | transcript/39571-1F | 407 | 46,644.36 | 5.54 | 71.13 | −0.514 |
| CdWRKY18 | transcript/21604-1F | 595 | 67,947.45 | 9.62 | 74.07 | −0.346 |
| CdWRKY19 | transcript/38354-2F | 437 | 50,029.91 | 9.88 | 70.50 | −0.676 |
| CdWRKY20 | transcript/12244-1F | 714 | 80,637.60 | 8.61 | 56.27 | −0.680 |
| CdWRKY21 | transcript/31845-2F | 480 | 53,644.37 | 9.56 | 82.83 | −0.360 |
| CdWRKY22 | transcript/15476-1F | 655 | 74,111.36 | 9.25 | 73.98 | −0.516 |
| CdWRKY23 | transcript/11987-0F | 709 | 79,137.66 | 6.27 | 76.8 | −0.562 |
| CdWRKY24 | transcript/14844-1F | 646 | 71,250.54 | 6.26 | 73.16 | −0.646 |
| CdWRKY25 | transcript/9489-0F | 771 | 85,095.23 | 7.43 | 71.91 | −0.522 |
| CdWRKY26 | transcript/22886-1F | 570 | 62,944.15 | 7.70 | 68.82 | −0.617 |
| CdWRKY27 | transcript/37754-2F | 407 | 45,222.69 | 8.25 | 76.54 | −0.551 |
| CdWRKY28 | transcript/6076-1F | 867 | 95,656.72 | 6.84 | 61.75 | −0.647 |
| CdWRKY29 | transcript/7374-1F | 825 | 90,366.89 | 6.84 | 60.88 | −0.629 |
| CdWRKY30 | transcript/41541-0F | 398 | 44,614.59 | 5.83 | 63.47 | −0.615 |
| CdWRKY31 | transcript/14886-0F | 666 | 73,648.13 | 7.01 | 59.64 | −0.713 |
| CdWRKY32 | transcript/13119-0F | 678 | 75,034.93 | 8.47 | 62.04 | −0.695 |
| CdWRKY33 | transcript/18293-2F | 596 | 65,716.90 | 8.80 | 54.90 | −0.842 |
| CdWRKY34 | transcript/53940-0F | 301 | 33,767.11 | 8.74 | 75.45 | −0.598 |
| CdWRKY35 | transcript/39534-1F | 415 | 46,453.20 | 7.99 | 55.64 | −0.560 |
| CdWRKY36 | transcript/59382-2F | 255 | 28,797.70 | 6.98 | 58.82 | −0.403 |
| CdWRKY37 | transcript/17933-1F | 617 | 68,401.17 | 6.08 | 61.94 | −0.701 |
| CdWRKY38 | transcript/17933-2F | 611 | 69,898.00 | 10.45 | 103.32 | −0.118 |
| CdWRKY39 | transcript/52505-0F | 313 | 35,571.78 | 9.17 | 75.91 | −0.422 |
| CdWRKY40 | transcript/27301-2F | 518 | 57,145.69 | 8.66 | 74.92 | −0.423 |
| CdWRKY41 | transcript/43765-2F | 375 | 41,119.73 | 9.66 | 69.7 | −0.503 |
| CdWRKY42 | transcript/42177-0F | 375 | 42,509.98 | 9.46 | 87.87 | −0.107 |
| CdWRKY43 | transcript/16548-1F | 635 | 71,526.47 | 9.16 | 75.06 | −0.601 |
| CdWRKY44 | transcript/13308-2F | 694 | 76,182.39 | 8.34 | 69.48 | −0.596 |
| CdWRKY45 | transcript/38850-1F | 426 | 46,782.05 | 7.43 | 55.19 | −0.821 |
| CdWRKY46 | transcript/38829-1F | 421 | 46,790.26 | 9.78 | 70.17 | −0.570 |
| FASTNAME | BF_Total Bases (G) | BF_Q20 Bases (%) | BF_Q30 Bases (%) | BF_GC Content (%) | AF_total Bases (G) | AF_Q20 Bases (%) | AF_Q30 Bases (%) | AF_GC Content (%) |
|---|---|---|---|---|---|---|---|---|
| A | 5.61 | 97.64 | 94.00 | 45.26 | 5.46 | 98.25 | 94.76 | 45.25 |
| B | 6.90 | 96.83 | 92.85 | 52.54 | 6.53 | 98.11 | 94.38 | 51.90 |
| C | 6.55 | 97.66 | 93.93 | 45.07 | 6.40 | 98.20 | 94.60 | 45.08 |
| D | 8.19 | 97.13 | 93.12 | 46.28 | 7.86 | 98.04 | 94.25 | 46.03 |
| E1 | 7.02 | 96.62 | 92.24 | 6.67 | 6.67 | 97.78 | 93.64 | 46.34 |
| E2 | 9.03 | 97.39 | 97.39 | 46.16 | 8.68 | 98.26 | 94.84 | 45.89 |
| F1 | 8.98 | 97.23 | 93.36 | 46.62 | 8.69 | 98.04 | 94.36 | 46.57 |
| F2 | 6.22 | 96.74 | 92.20 | 47.08 | 6.00 | 97.56 | 93.20 | 46.99 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, H.; Liao, B.; Deng, J.; Liu, B.; Shen, Y.; Xiong, W.; He, S.; Zou, P.; Chen, F.; Srihawech, T.; et al. Transcriptome Analysis of Multiple Plant Parts in the Woody Oil Tree Camellia drupifera Loureiro. Horticulturae 2024, 10, 914. https://doi.org/10.3390/horticulturae10090914
Shen H, Liao B, Deng J, Liu B, Shen Y, Xiong W, He S, Zou P, Chen F, Srihawech T, et al. Transcriptome Analysis of Multiple Plant Parts in the Woody Oil Tree Camellia drupifera Loureiro. Horticulturae. 2024; 10(9):914. https://doi.org/10.3390/horticulturae10090914
Chicago/Turabian StyleShen, Hongjian, Boyong Liao, Jinqing Deng, Biting Liu, Yang Shen, Wanyu Xiong, Shan He, Peishan Zou, Fang Chen, Thitaree Srihawech, and et al. 2024. "Transcriptome Analysis of Multiple Plant Parts in the Woody Oil Tree Camellia drupifera Loureiro" Horticulturae 10, no. 9: 914. https://doi.org/10.3390/horticulturae10090914
APA StyleShen, H., Liao, B., Deng, J., Liu, B., Shen, Y., Xiong, W., He, S., Zou, P., Chen, F., Srihawech, T., Lee, S. Y., & Li, Y. (2024). Transcriptome Analysis of Multiple Plant Parts in the Woody Oil Tree Camellia drupifera Loureiro. Horticulturae, 10(9), 914. https://doi.org/10.3390/horticulturae10090914