

Chromosome-Scale Genome Assembly Provides Insights into Fresh Pine Wood Decay Strategies of the Wolfiporia hoelen

Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Culture Conditions
2.2. Genome Features Estimation from K-mer Method
2.3. Sequencing Libraries
2.4. Genome Assembly
2.5. Genome Annotation
2.6. Growth Test of Edible Mushroom-Forming Fungi on Pine Wood Sawdust
2.7. Comparative Genomics Analysis
3. Results
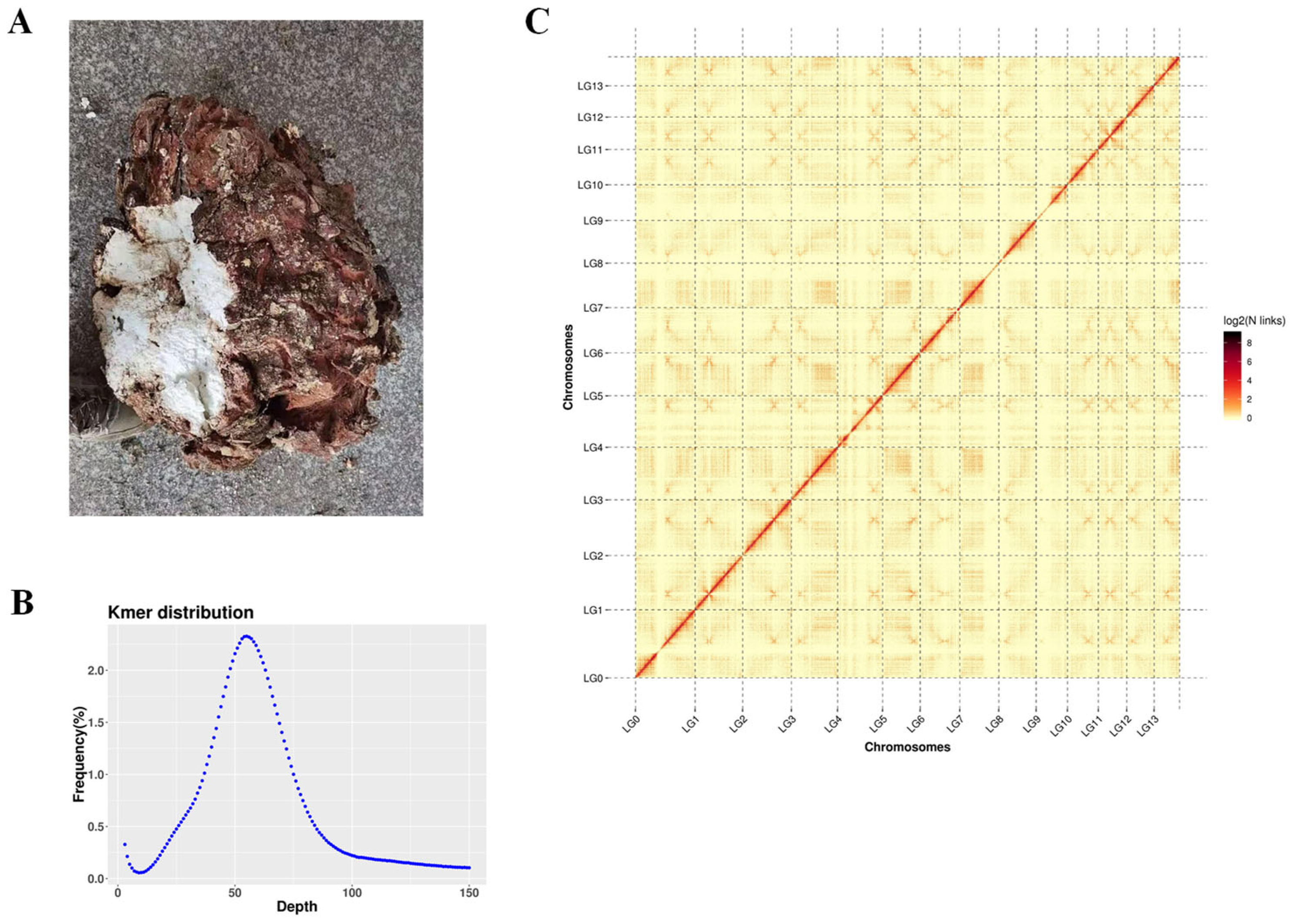
3.1. Sequencing and Assembly of the Genome
3.2. Assessment of Genomic Integrity
3.3. Genome Annotation
3.4. The Growth of Five Edible Mushroom-Forming Fungi on Pine Wood Sawdust
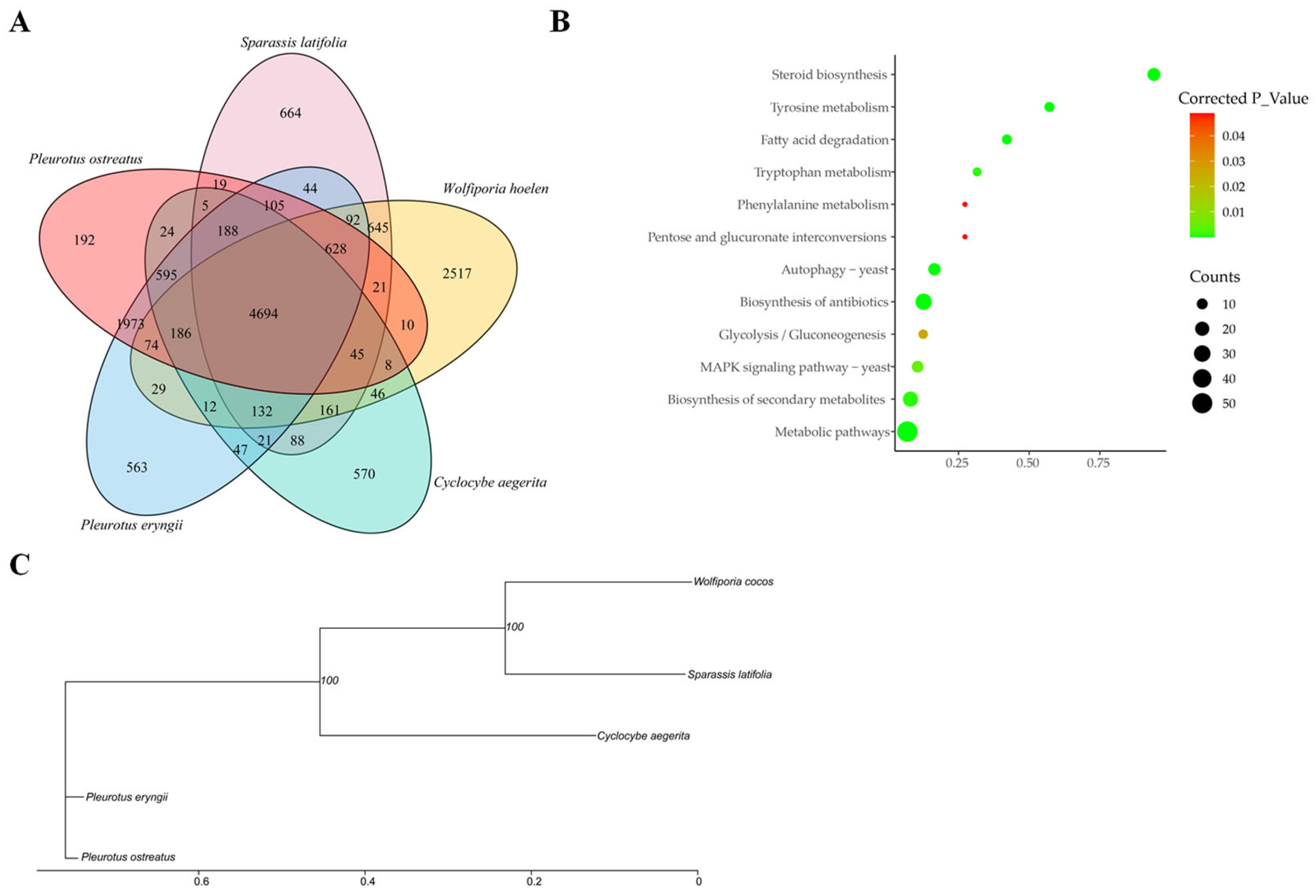
3.5. Employing Comparative Genomics Analysis to Identify Genes Associated with Pine Utilization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Li, M.; Li, H.; Sheng, R.C.; Sun, H.; Sun, S.H.; Chen, F.M. The First Record of Monochamus saltuarius (Coleoptera; Cerambycidae) as Vector of Bursaphelenchus xylophilus and Its New Potential Hosts in China. Insects 2020, 11, 636. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.J.; Wu, X.Q.; Li, H.Y.; Wang, Y.C.; Huang, X.; Wang, Y.; Li, Y. BxCDP1 from the pine wood nematode Bursaphelenchus xylophilus is recognized as a novel molecular pattern. Mol. Plant Pathol. 2020, 21, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.F.; Chen, F.M.; Xie, L.Y.; Pan, H.Y.; Ye, J.R. Genetic diversity of pine-parasitic nematodes Bursaphelenchus xylophilus and Bursaphelenchus mucronatus in China. Forest Pathol. 2017, 47, e12334. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, X.; Li, J.; Ren, J.; Ren, L.; Luo, Y. Pine Wilt Disease in Northeast and Northwest China: A Comprehensive Risk Review. Forests 2023, 14, 174. [Google Scholar] [CrossRef]
- Zhao, J.; Huang, J.; Yan, J.; Fang, G. Economic Loss of Pine Wood Nematode Disease in Mainland China from 1998 to 2017. Forests 2020, 11, 1042. [Google Scholar] [CrossRef]
- Croan, S.C. Conversion of conifer wastes into edible and medicinal mushrooms. For. Prod. J. 2004, 54, 68–76. [Google Scholar]
- Ma, L.; Lin, Y.Q.; Yang, C.; Ying, Z.H.; Jiang, X.L. Production of liquid spawn of an edible mushroom, Sparassis latifolia by submerged fermentation and mycelial growth on pine wood sawdust. Sci. Hortic. 2016, 209, 22–30. [Google Scholar] [CrossRef]
- Xiao, D.L.; Ma, L.; Yang, C.; Lin, Y.Q. Transcriptome analysis of Sparassis latifolia cultivated with different carbon sources. Microbiol. China 2019, 046, 1654–1661. [Google Scholar]
- Yang, C.; Ma, L.; Xiao, D.; Liu, X.; Jiang, X.; Lin, Y. Comparative transcriptomics reveals unique pine wood decay strategies in the Sparassis latifolia. Sci. Rep. 2022, 12, 19875. [Google Scholar] [CrossRef]
- Stalpers, J.A.; Redhead, S.A.; May, T.W.; Rossman, A.Y.; Crouch, J.A.; Cubeta, M.A.; Dai, Y.C.; Kirschner, R.; Langer, G.J.; Larsson, K.H.; et al. Competing sexual-asexual generic names in Agaricomycotina (Basidiomycota) with recommendations for use. IMA Fungus 2021, 12, 22. [Google Scholar] [CrossRef]
- Liu, J.; Yu, J.; Peng, X. Poria cocos Polysaccharides Alleviates Chronic Nonbacterial Prostatitis by Preventing Oxidative Stress, Regulating Hormone Production, Modifying Gut Microbiota, and Remodeling the DNA Methylome. J. Agric. Food Chem. 2020, 68, 12661–12670. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Dong, X.; Li, B.; Chen, T.; Yu, B.; Wang, X.; Dou, X.; Peng, B.; Hu, Q. Poria cocos polysaccharide-functionalized graphene oxide nanosheet induces efficient cancer immunotherapy in mice. Front. Bioeng. Biotechnol. 2022, 10, 1050077. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Huang, J.; Sun, M.; Jiang, Y.; Wang, S.; Wang, L.; Yu, N.; Peng, D.; Wang, Y.; Chen, W.; et al. Poria cocos polysaccharide improves intestinal barrier function and maintains intestinal homeostasis in mice. Int. J. Biol. Macromol. 2023, 249, 125953. [Google Scholar] [CrossRef]
- Lee, S.R.; Lee, S.; Moon, E.; Park, H.J.; Park, H.B.; Kim, K.H. Bioactivity-guided isolation of anti-inflammatory triterpenoids from the sclerotia of Poria cocos using LPS-stimulated Raw264.7 cells. Bioorg. Chem. 2017, 70, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Li, S.; Tian, M.; Yang, P.; Ding, Y.; Wang, Y.; Duan, G.; Zhang, D.; Chen, B.; Tan, Q. Facile preparation of a novel nanoemulsion based hyaluronic acid hydrogel loading with Poria cocos triterpenoids extract for wound dressing. Int. J. Biol. Macromol. 2023, 226, 1490–1499. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.L.; Kuo, H.P.; Huang, H.W.; Cheng, M.Y.; Chao, H.F.; Lu, S.M.; Lin, H.C.; Wang, C.J.; Chang, T.C.; Wu, C.R. Poria cocos Lanostane Triterpenoids Extract Promotes Collagen and Hyaluronic Acid Production in D-Galactose-Induced Aging Rats. Life 2023, 13, 2130. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.L.; Huang, H.W.; Su, M.H.; Lin, H.C.; Wu, W.M. The Lanostane Triterpenoids in Poria cocos Play Beneficial Roles in Immunoregulatory Activity. Life 2021, 11, 111. [Google Scholar] [CrossRef]
- Zhu, W.; Liu, Y.; Tang, J.; Liu, H.; Jing, N.; Li, F.; Xu, R.; Shu, S. Functional Analysis of Sterol O-Acyltransferase Involved in the Biosynthetic Pathway of Pachymic Acid in Wolfiporia cocos. Molecules 2021, 27, 143. [Google Scholar] [CrossRef]
- Zeng, G.; Li, Z.; Zhao, Z. Metabolome analysis of key genes for synthesis and accumulation of triterpenoids in Wolfiporia cocos. Sci. Rep. 2022, 12, 1574. [Google Scholar] [CrossRef]
- Cai, D.F.; Chen, D.H.; Zheng, P.W.; Yang, J.; Huang, X.; Lin, Y.Q. High yield cultivation and management techniques of Minling A5 on pine stump. Edible Fungi China 2016, 35, 74–77. [Google Scholar] [CrossRef]
- Gaskell, J.; Blanchette, R.A.; Stewart, P.E.; BonDurant, S.S.; Adams, M.; Sabat, G.; Kersten, P.; Cullen, D. Transcriptome and Secretome Analyses of the Wood Decay Fungus Wolfiporia cocos Support Alternative Mechanisms of Lignocellulose Conversion. Appl. Environ. Microbiol. 2016, 82, 3979–3987. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Min, B.; Han, J.G.; Park, H.; Baek, S.; Jeong, S.; Choi, I.G. Draft Genome Sequence of the Reference Strain of the Korean Medicinal Mushroom Wolfiporia cocos KMCC03342. Mycobiology 2022, 50, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Yang, Y.; Bi, G.; Nelson, D.; Hu, S.; Makunga, N.P.; Yu, B.; Liu, X.; Li, X.; Hu, X. Genomic and Transcriptomic Insight of Giant Sclerotium Formation of Wood-Decay Fungi. Front. Microbiol. 2021, 12, 746121. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Meng, G.; Dong, C. Homokaryotic High-Quality Genome Assembly of Medicinal Fungi Wolfiporia hoelen Reveals Auto-Regulation and High-Temperature Adaption of Probable Two-Speed Genome. Int. J. Mol. Sci. 2022, 23, 10484. [Google Scholar] [CrossRef]
- Luo, H.; Qian, J.; Xu, Z.; Liu, W.; Xu, L.; Li, Y.; Xu, J.; Zhang, J.; Xu, X.; Liu, C.; et al. The Wolfiporia cocos Genome and Transcriptome Shed Light on the Formation of Its Edible and Medicinal Sclerotium. Genom. Proteom. Bioinform. 2020, 18, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Floudas, D.; Binder, M.; Riley, R.; Barry, K.; Blanchette, R.A.; Henrissat, B.; Martínez, A.T.; Otillar, R.; Spatafora, J.W.; Yadav, J.S.; et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 2012, 336, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Biel, S.W.; Parrish, F.W. Isolation of DNA from fungal mycelia and sclerotia without use of density gradient ultracentrifugation. Anal. Biochem. 1986, 154, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Zhao, L.; Yuan, Z.; Canario, A.; Liu, Q.; Chen, S.; Guo, J.; Luo, W.; Yan, H.; Zhang, D.; et al. Chromosome-level genome assembly of largemouth bass (Micropterus salmoides) using PacBio and Hi-C technologies. Sci. Data 2022, 9, 482. [Google Scholar] [CrossRef]
- Korlach, J.; Bjornson, K.P.; Chaudhuri, B.P.; Cicero, R.L.; Flusberg, B.A.; Gray, J.J.; Holden, D.; Saxena, R.; Wegener, J.; Turner, S.W. Real-time DNA sequencing from single polymerase molecules. Methods Enzymol. 2010, 472, 431–455. [Google Scholar] [CrossRef]
- Yang, C.; Ma, L.; Xiao, D.L.; Liu, X.Y.; Jiang, X.L.; Ying, Z.H.; Lin, Y.Q. Chromosome-scale assembly of the Sparassis latifolia genome obtained using long-read and Hi-C sequencing. G3 Genes|Genomes|Genet. 2021, 11, jkab173. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Zdobnov, E.M. BUSCO: Assessing Genomic Data Quality and Beyond. Curr. Protoc. 2021, 1, e323. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Roach, M.J.; Schmidt, S.A.; Borneman, A.R. Purge Haplotigs: Allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 2018, 19, 460. [Google Scholar] [CrossRef]
- Servant, N.; Varoquaux, N.; Lajoie, B.R.; Viara, E.; Chen, C.J.; Vert, J.P.; Heard, E.; Dekker, J.; Barillot, E. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015, 16, 259. [Google Scholar] [CrossRef]
- Burton, J.N.; Adey, A.; Patwardhan, R.P.; Qiu, R.; Kitzman, J.O.; Shendure, J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013, 31, 1119–1125. [Google Scholar] [CrossRef]
- Han, Y.; Wessler, S.R. MITE-Hunter: A program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 2010, 38, e199. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21 (Suppl. S1), i351–i358. [Google Scholar] [CrossRef]
- Edgar, R.C.; Myers, E.W. PILER: Identification and classification of genomic repeats. Bioinformatics 2005, 21 (Suppl. S1), i152–i158. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 19 (Suppl. S2), ii215–ii225. [Google Scholar] [CrossRef] [PubMed]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Alioto, T.; Blanco, E.; Parra, G.; Guigó, R. Using geneid to Identify Genes. Curr. Protoc. Bioinform. 2018, 64, e56. [Google Scholar] [CrossRef] [PubMed]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef] [PubMed]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Keilwagen, J.; Wenk, M.; Erickson, J.L.; Schattat, M.H.; Grau, J.; Hartung, F. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016, 44, e89. [Google Scholar] [CrossRef]
- Keilwagen, J.; Hartung, F.; Grau, J. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. Methods Mol. Biol. 2019, 1962, 161–177. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.A.; Haas, B.J.; Hamilton, J.P.; Mount, S.M.; Buell, C.R. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genomics 2006, 7, 327. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Sperschneider, J.; Gardiner, D.M.; Dodds, P.N.; Tini, F.; Covarelli, L.; Singh, K.B.; Manners, J.M.; Taylor, J.M. EffectorP: Predicting fungal effector proteins from secretomes using machine learning. New Phytol. 2016, 210, 743–761. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Liu, Y.-X.; Huang, L. ImageGP: An easy-to-use data visualization web server for scientific researchers. iMeta 2022, 1, e5. [Google Scholar] [CrossRef] [PubMed]
- Farjon, A.; Filer, D. An Atlas of the World’s Conifers: An Analysis of Their Distribution, Biogeography, Diversity and Conservation Status; Brill: Leiden, The Netherlands, 2013. [Google Scholar]
- Webster, J.; Mota, M. Pine wilt disease: Global issues, trade and economic impact. In Pine Wilt Disease: A Worldwide Threat to Forest Ecosystems; Springer: Dordrecht, The Netherlands, 2008. [Google Scholar]
- Hao, Z.; Huang, J.; Li, X.; Sun, H.; Fang, G. A multi-point aggregation trend of the outbreak of pine wilt disease in China over the past 20 years. Forest Ecol. Manag. 2022, 505, 119890. [Google Scholar] [CrossRef]
- Goodell, B.; Nicholas, D.D.; Schultz, T.P. (Eds.) Wood Deterioration and Preservation: Advances in Our Changing World; American Chemical Society: Washington, DC, USA, 2003. [Google Scholar]
- Almási, É.; Sahu, N.; Krizsán, K.; Bálint, B.; Kovács, G.M.; Kiss, B.; Cseklye, J.; Drula, E.; Henrissat, B.; Nagy, I.; et al. Comparative genomics reveals unique wood-decay strategies and fruiting body development in the Schizophyllaceae. New Phytol. 2019, 224, 902–915. [Google Scholar] [CrossRef] [PubMed]
- Belt, T.; Altgen, M.; Makela, M.; Hanninen, T.; Rautkari, L. Cellular level chemical changes in Scots pine heartwood during incipient brown rot decay. Sci. Rep. 2019, 9, 5188. [Google Scholar] [CrossRef] [PubMed]
- Nie, A.; Chao, Y.; Zhang, X.; Jia, W.; Zhou, Z.; Zhu, C. Phytochemistry and Pharmacological Activities of Wolfiporia cocos (F.A. Wolf) Ryvarden & Gilb. Front. Pharmacol. 2020, 11, 505249. [Google Scholar] [CrossRef]
- Hu, G.S.; Huang, C.G.; Zhang, Y.; Xiao, W.; Jia, J.M. Accumulation of biomass and four triterpenoids in two-stage cultured Poria cocos mycelia and diuretic activity in rats. Chin. J. Nat. Med. 2017, 15, 265–270. [Google Scholar] [CrossRef]
- Wu, Y. A Preliminary Study on Cultivation of Poria cocos on Nematode affected Pine Wood. J. Fujian For. Sci. Technol. 2013, 40, 51–55. [Google Scholar]
- Raza, Q.; Rashid, M.A.R.; Waqas, M.; Ali, Z.; Rana, I.A.; Khan, S.H.; Khan, I.A.; Atif, R.M. Genomic diversity of aquaporins across genus Oryza provides a rich genetic resource for development of climate resilient rice cultivars. BMC Plant Biol. 2023, 23, 172. [Google Scholar] [CrossRef] [PubMed]
- Adomako, M.; Ernst, D.; Simkovsky, R.; Chao, Y.Y.; Wang, J.; Fang, M.; Bouchier, C.; Lopez-Igual, R.; Mazel, D.; Gugger, M.; et al. Comparative Genomics of Synechococcus elongatus Explains the Phenotypic Diversity of the Strains. mBio 2022, 13, e0086222. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Tang, J.; Chen, J.J.; Peng, A.Y.; Wang, Q.M.; Rao, L.Q.; Yang, H.; Zhang, X.W.; Yang, H.Z.; Zhang, C.; et al. Transcriptome analysis of three cultivars of Poria cocos reveals genes related to the biosynthesis of polysaccharides. J. Asian Nat. Prod. Res. 2019, 21, 462–475. [Google Scholar] [CrossRef] [PubMed]
- Elkady, W.M.; Gonaid, M.H.; Yousif, M.F.; El-Sayed, M.; Omar, H.A.N. Impact of Altitudinal Variation on the Phytochemical Profile, Anthelmintic and Antimicrobial Activity of Two Pinus Species. Molecules 2021, 26, 3170. [Google Scholar] [CrossRef] [PubMed]
- Gad, H.; Al-Sayed, E.; Ayoub, I. Phytochemical discrimination of Pinus species based on GC-MS and ATR-IR analyses and their impact on Helicobacter pylori. Phytochem. Anal. 2021, 32, 820–835. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Xu, R.; Gu, S.; Chen, K.; Li, J.; He, X.; Shang, S.; Song, Z.; Song, J. Discovery of Natural Rosin Derivatives Containing Oxime Ester Moieties as Potential Antifungal Agents to Control Tomato Gray Mold Caused by Botrytis cinerea. J. Agric. Food Chem. 2022, 70, 5551–5560. [Google Scholar] [CrossRef] [PubMed]
- Tao, P.; Wu, C.; Hao, J.; Gao, Y.; He, X.; Li, J.; Shang, S.; Song, Z.; Song, J. Antifungal Application of Rosin Derivatives from Renewable Pine Resin in Crop Protection. J. Agric. Food Chem. 2020, 68, 4144–4154. [Google Scholar] [CrossRef]
- Kugler, S.; Ossowicz, P.; Malarczyk-Matusiak, K.; Wierzbicka, E. Advances in Rosin-Based Chemicals: The Latest Recipes, Applications and Future Trends. Molecules 2019, 24, 1651. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Library Type | Sequencing Mode | Clean Data (Gb) | Application |
|---|---|---|---|
| Illumina | Pair end 150 bp | 4.15 | Genome survey and correction |
| PacBio | Sequel II HiFi | 84.4 | Genome assembly |
| Hi-C | Pair end 150 bp | 8.47 | Assisted assembly at the chromosomal level |
| Transcriptome | Pair end 150 bp | 7.69 | gene annotation |
| Genome assembly and scaffolding at chromosomal level | |||
| Contig number | 112 | ||
| Contig length (bp) | 62,945,509 | ||
| Contig N50 (bp) | 4,211,296 | ||
| Contig N90 (bp) | 2,386,851 | ||
| Scaffold number | 96 | ||
| Scaffold length (bp) | 62,947,109 | ||
| Scaffold N50 (bp) | 4,456,852 | ||
| Scaffold N90 (bp) | 3,064,705 | ||
| GC content (%) | 51.89 | ||
| Anchored chromosomes size (bp) | 61,059,735 | ||
| Species | Total Gene Number | Cluster Gene Number | Total Gene Family Number | Unique Gene Family Number |
|---|---|---|---|---|
| C. aegerita | 13,341 | 10,574 | 6822 | 570 |
| P. eryngii | 15,954 | 12,632 | 9383 | 563 |
| P. ostreatus | 11,718 | 10,504 | 8769 | 192 |
| S. latifolia | 15,016 | 12,972 | 7552 | 664 |
| W. hoelen | 12,670 | 11,763 | 9300 | 2517 |
| Strains | Sequencing Strategy | Genome Size (Mb) | Number of Scaffolds | N50 of Scaffolds (kb) | Anchored to Chromosome (Mb) | Number of Protein-Coding Genes | Average Gene Length (bp) | Percentage of Repeat Sequences (%) | Transposable Elements (%) | GC Content (%) | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CGMCC 5.78 | HiSeq 2000 Illumina and a fosmid-to-fosmid strategy | 50.6 | 351 | 835 | 10,908 | 1829 | - | 33.5 | 51.7 | [23] | |
| WCLT | HiSeq2500 Illumina and SMRT technology on the PacBio | 62 | 145 | 1599.1 | 61.127 | 11,906 | 1332.76 | 46.6 | - | 51.86 | [21] |
| SS20 | Novaseq6000 Illumina and SMRT technology on the PacBio | 64.44 | 78 | 3760 | 58.26 | 10,567 | 2004 | 48.56 | 46.26 | 50.15 | [22] |
| KMCC03342 | Oxford Nanopore and SMRT technology on the PacBio | 55.5 | 14,296 | 52.2 | [20] | ||||||
| GDMCC 5.219 | PacBio Sequel II | 60.2 | 183 | 1300 | 52 | https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_034769205.1/, accessed on 5 June 2024 | |||||
| Minling A5 | Novaseq6000 Illumina and SMRT technology on the PacBio | 62.95 | 96 | 4456.8 | 61.06 | 12,670 | 2062.38 | 44.37 | 51.89 | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.; Xiao, D.; Jiang, X.; Li, Y.; Liu, X.; Lin, H.; Liu, C.; Ma, L. Chromosome-Scale Genome Assembly Provides Insights into Fresh Pine Wood Decay Strategies of the Wolfiporia hoelen. Horticulturae 2024, 10, 703. https://doi.org/10.3390/horticulturae10070703
Yang C, Xiao D, Jiang X, Li Y, Liu X, Lin H, Liu C, Ma L. Chromosome-Scale Genome Assembly Provides Insights into Fresh Pine Wood Decay Strategies of the Wolfiporia hoelen. Horticulturae. 2024; 10(7):703. https://doi.org/10.3390/horticulturae10070703
Chicago/Turabian StyleYang, Chi, Donglai Xiao, Xiaoling Jiang, Yaru Li, Xiaoyu Liu, Hui Lin, Chuansen Liu, and Lu Ma. 2024. "Chromosome-Scale Genome Assembly Provides Insights into Fresh Pine Wood Decay Strategies of the Wolfiporia hoelen" Horticulturae 10, no. 7: 703. https://doi.org/10.3390/horticulturae10070703
APA StyleYang, C., Xiao, D., Jiang, X., Li, Y., Liu, X., Lin, H., Liu, C., & Ma, L. (2024). Chromosome-Scale Genome Assembly Provides Insights into Fresh Pine Wood Decay Strategies of the Wolfiporia hoelen. Horticulturae, 10(7), 703. https://doi.org/10.3390/horticulturae10070703