
The Molecular Biology Analysis for the Growing and Development of Hydrangea macrophylla ‘Endless Summer’ under Different Light and Temperature Conditions

Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Materials and Procedures
2.2. RNA Extraction, cDNA Library Construction, and Sequencing
2.3. Data Quality Control and Sequence Alignment Analysis
2.4. Gene Expression Quantification and Differential Gene Analysis
2.5. Trend Analysis of Gene Expression Levels
2.6. Protein–Protein Interaction (PPI) Network Construction
2.7. Real-Time Quantitative PCR Verification
3. Results
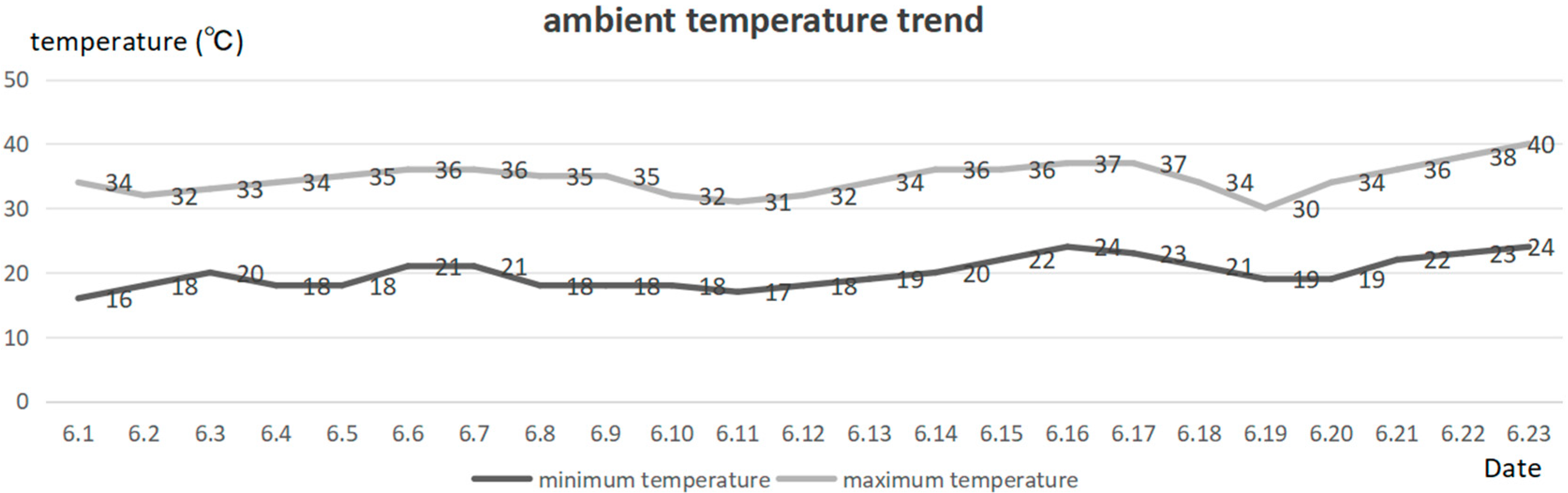
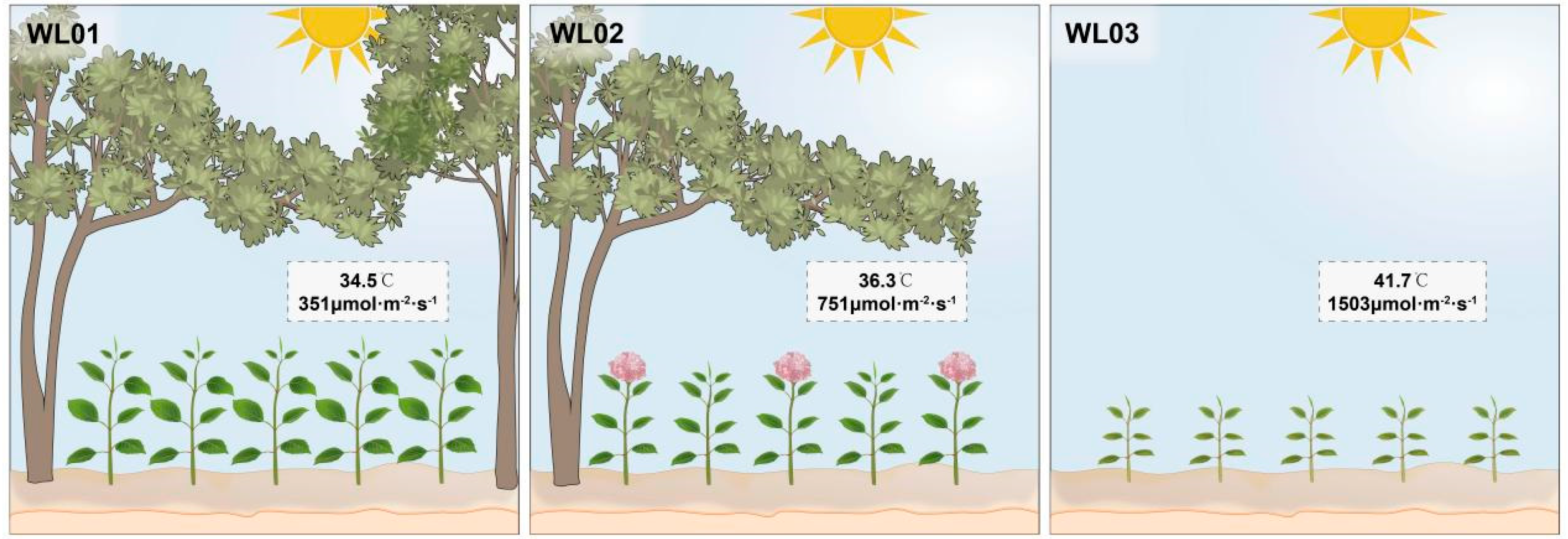
3.1. Growth Status Analysis
3.2. Sequencing Data Assembly Results
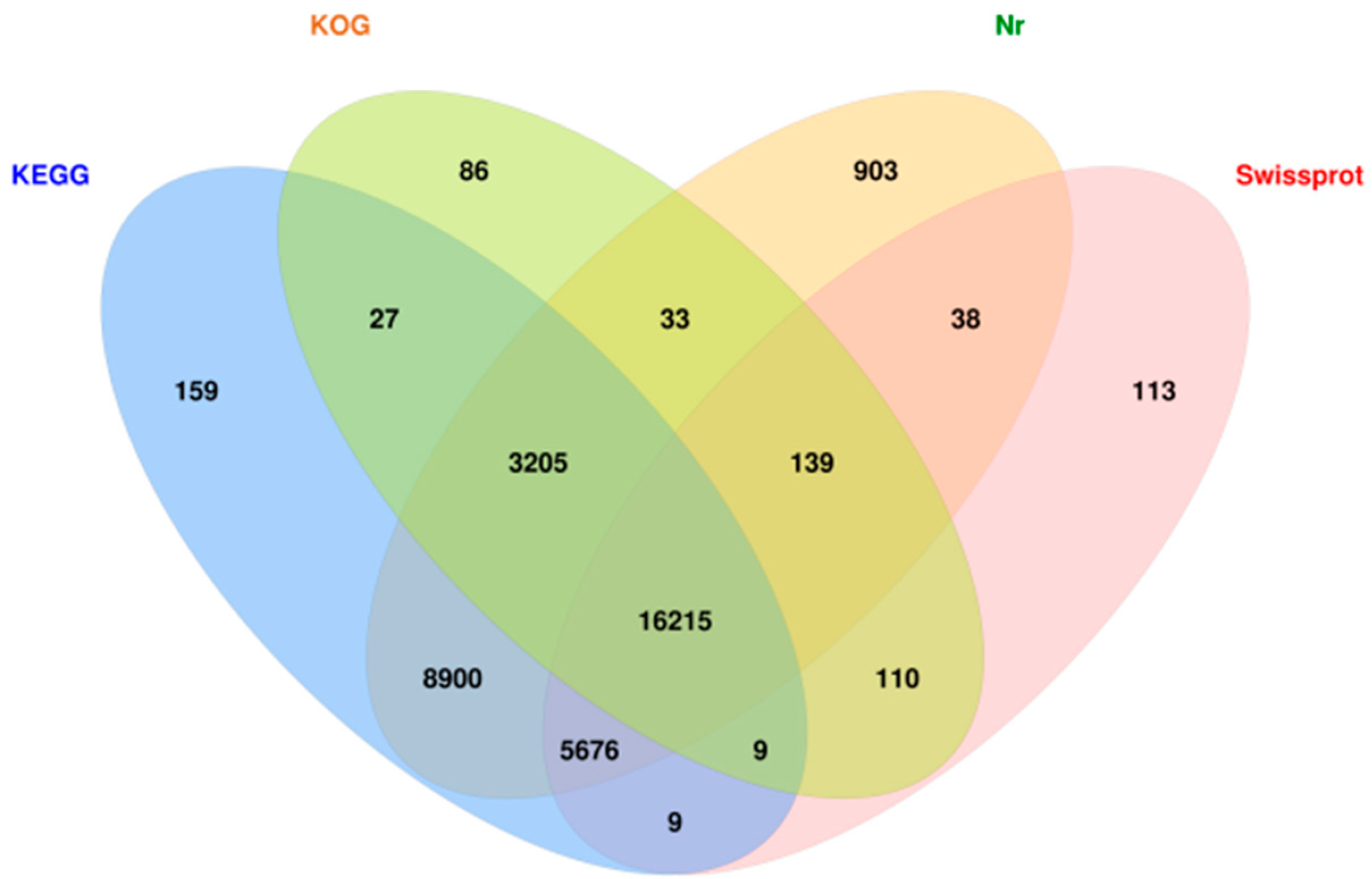
3.3. Functional Annotation and Expression Analysis of Transcriptome Unigenes
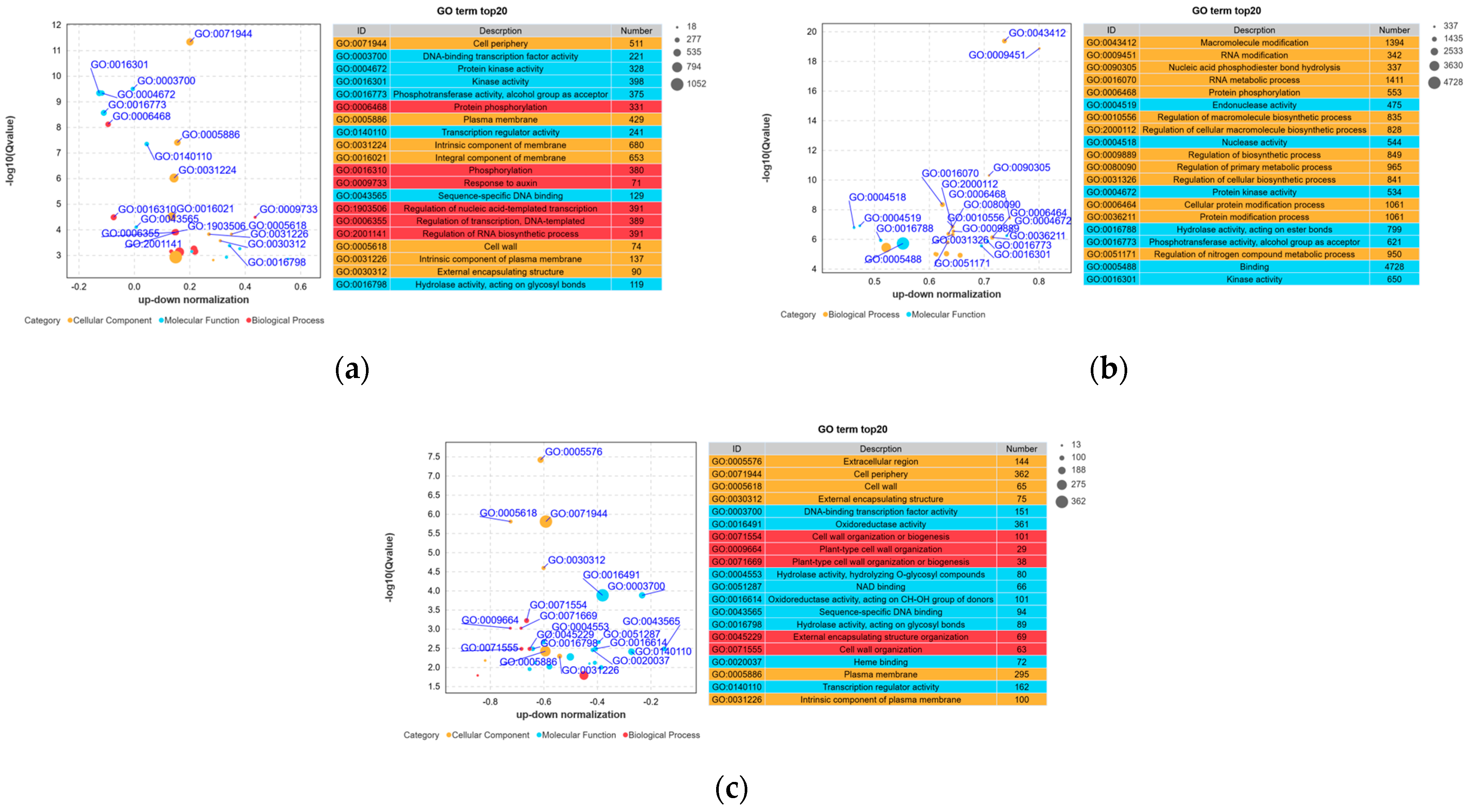
3.4. Gene Ontology Classification of Differentially Expressed Genes
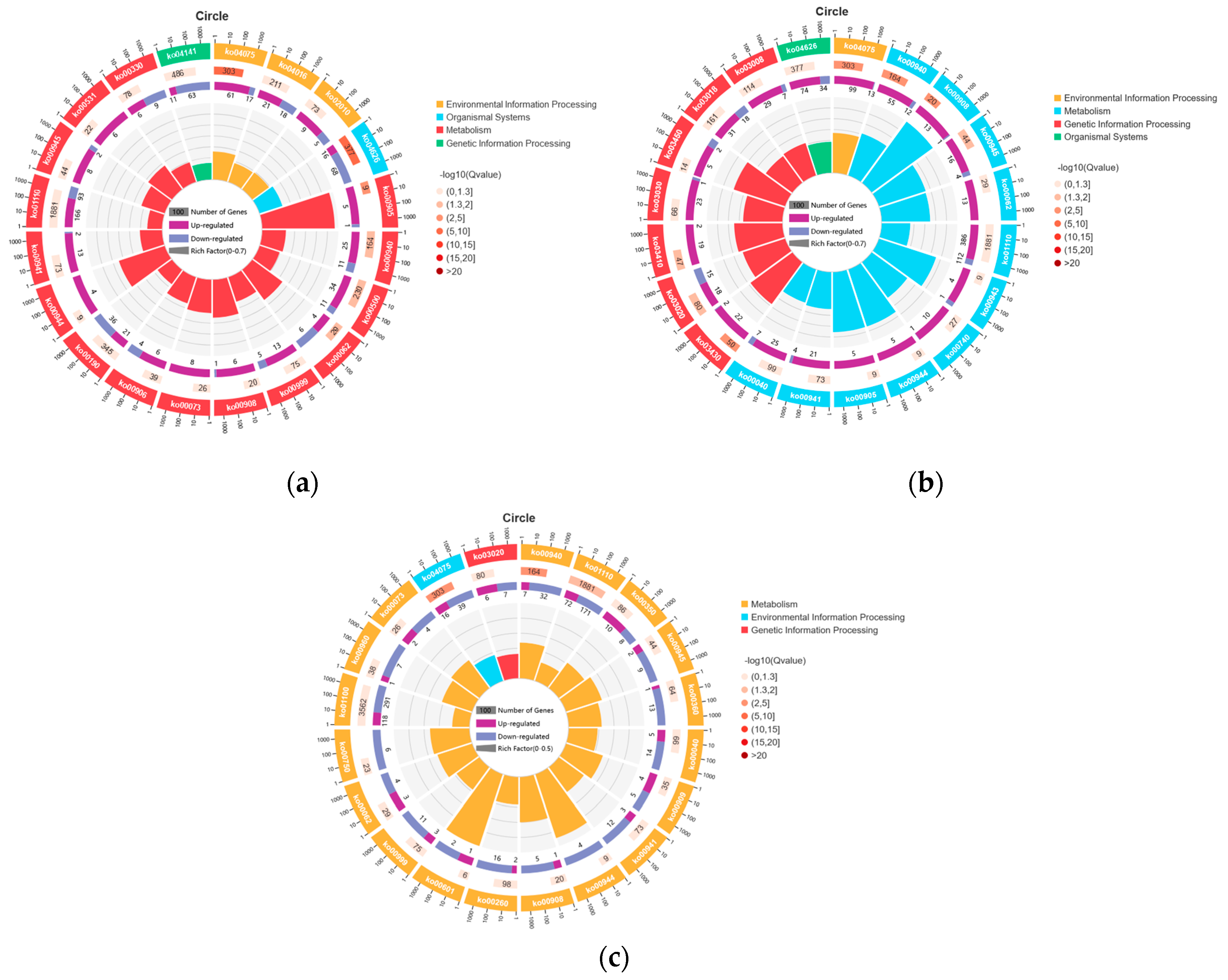
3.5. DEGs Pathway Analysis
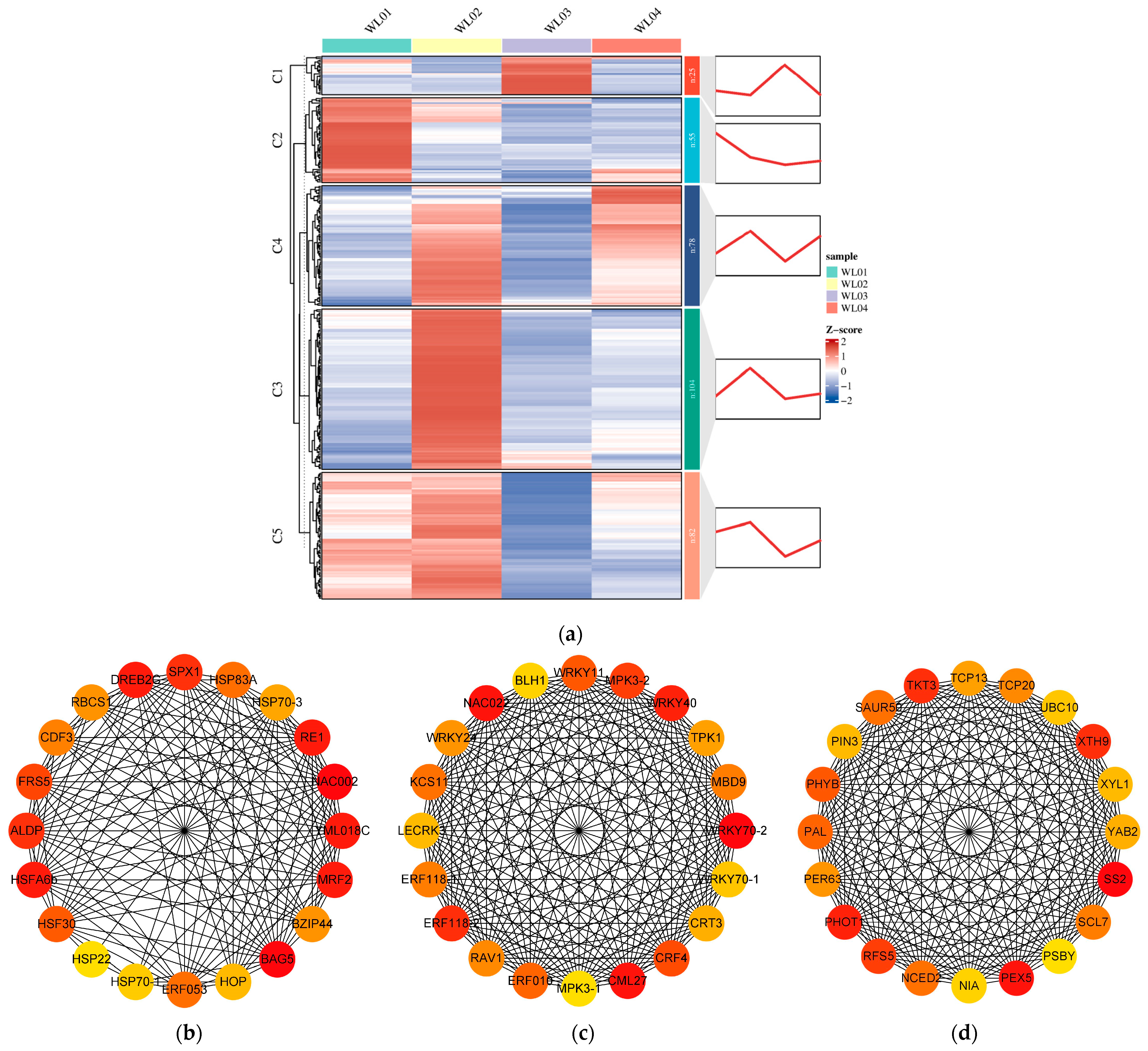
3.6. Trend Analysis of Gene Expression Levels
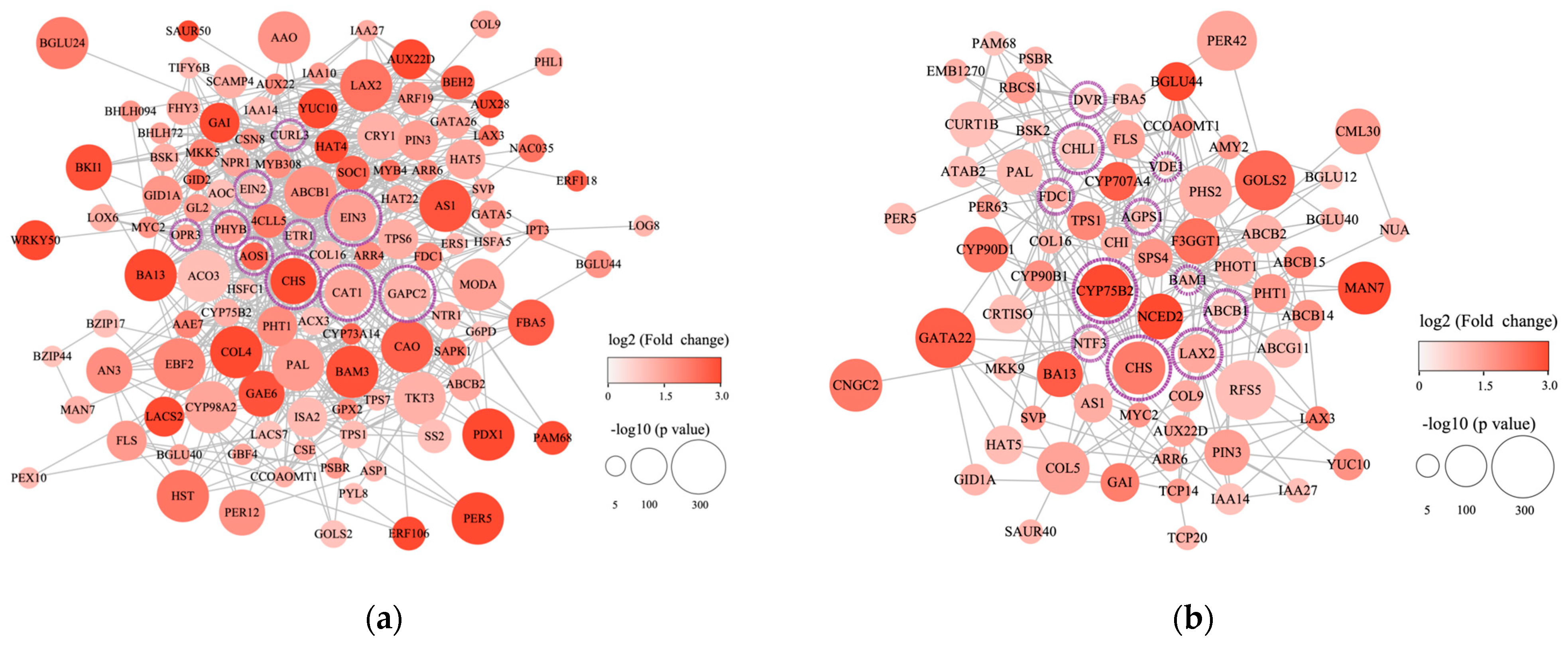
3.7. PPI Analysis
4. Discussion
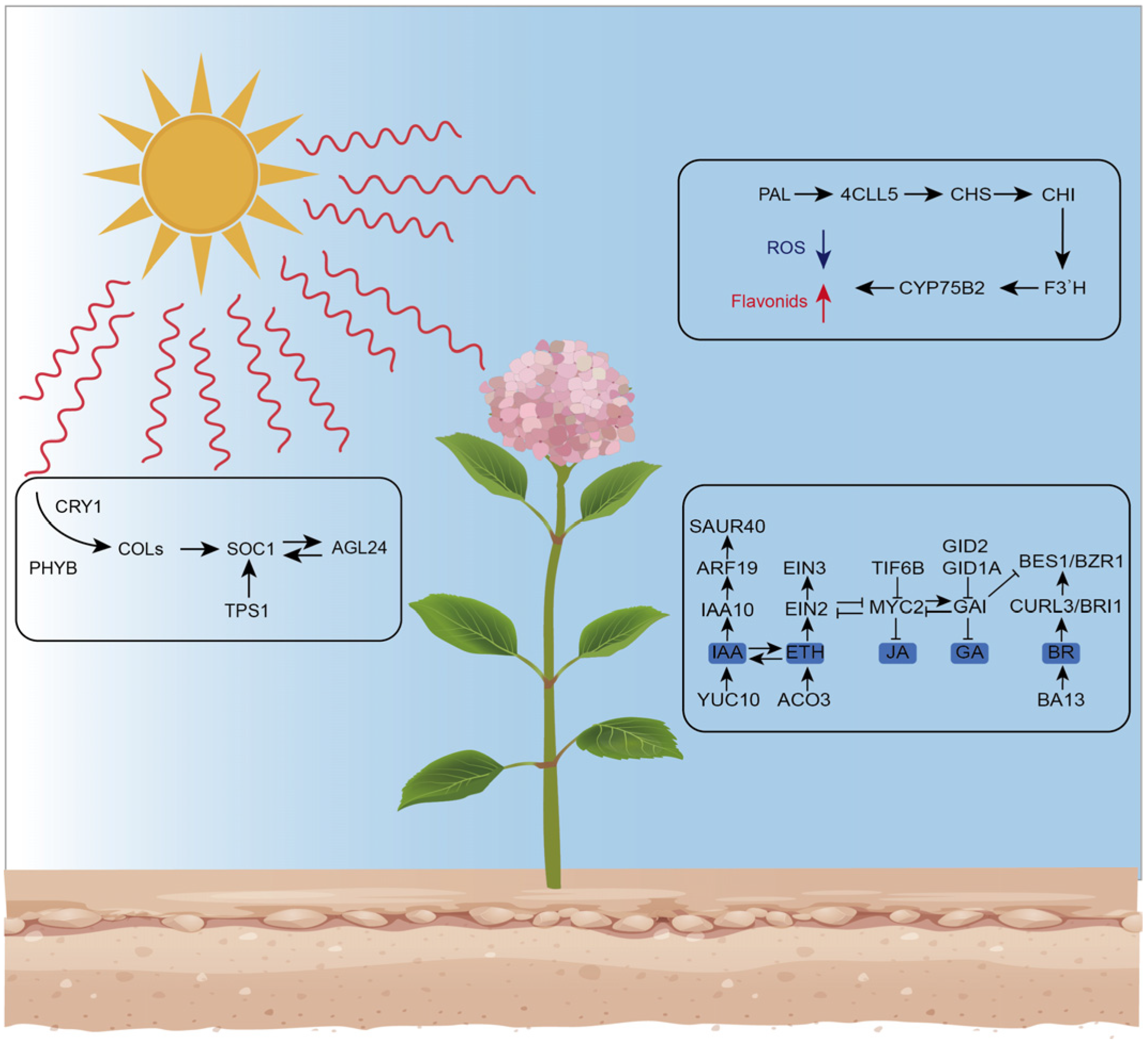
4.1. Phenylpropanoid Metabolism in Response to Semi-Shaded Conditions
4.2. Photosynthesis and Chlorophyll-Related Genes under Semi-Shaded Conditions
4.3. Carbohydrate Metabolism in Response to Semi-Shaded Conditions
4.4. Key Transcription Factors for Normal Growth in Semi-Shade Conditions
4.5. Plant Hormone and Signal Transduction Play Significant Roles in Hydrangea’s Adaptation to Semi-Shaded Conditions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Peng, J.; Dong, X.; Xue, C.; Liu, Z.; Cao, F. Exploring the molecular mechanism of blue flower color formation in Hydrangea macrophylla cv. “Forever Summer”. Front. Plant Sci. 2021, 12, 585665. [Google Scholar] [CrossRef]
- Rahmati, R.; Hamid, R.; Ghorbanzadeh, Z.; Jacob, F.; Azadi, P.; Zeinalabedini, M.; Farsad, L.K.; Kazemi, M.; Ebrahimi, M.A.; Shahinnia, F.; et al. Comparative transcriptome analysis unveils the molecular mechanism underlying sepal colour changes under acidic pH substratum in Hydrangea macrophylla. Int. J. Mol. Sci. 2022, 23, 15428. [Google Scholar] [CrossRef]
- Xu, H.; Liu, C.; Zhong, H.D. The effect of different light intensities on the blooming of Hygrangea macrophylla. Nothern Hortic. 2014, 1, 81–82. [Google Scholar]
- Li, X.; Liang, T.; Liu, H. How plants coordinate their development in response to light and temperature signals. Plant Cell 2021, 34, 955–966. [Google Scholar] [CrossRef]
- Chen, Z.L.; Galli, M.; Gallavotti, A. Mechanisms of temperature-regulated growth and thermotolerance in crop species. Curr. Opin. Plant Biol. 2022, 65, 102134. [Google Scholar] [CrossRef]
- Gururani, M.A.; Venkatesh, J.; Tran, L.S.P. Regulation of photosynthesis during abiotic stress-induced photoinhibition. Mol. Plant 2015, 8, 1304–1320. [Google Scholar] [CrossRef]
- Saibo, N.J.M.; Lourenco, T.; Oliveira, M.M. Transcription factors and regulation of photosynthetic and related metabolism under environmental stresses. Ann. Bot. 2009, 103, 609–623. [Google Scholar] [CrossRef]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. String Tie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Tang, D.D.; Chen, M.J.; Huang, X.H.; Zhang, G.C.; Zeng, L.; Zhang, G.S.; Wu, S.J.; Wang, Y.W. SR plot: A free online platform for data visualization and graphing. PLoS ONE 2023, 18, e0294236. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Liu, Y.; Lyu, T.; Lyu, Y. Study on the flower induction mechanism of Hydrangea macrophylla. Int. J. Mol. Sci. 2023, 24, 7691. [Google Scholar] [CrossRef]
- Krouk, G.; Lacombe, B.; Bielach, A.; Perrine-Walker, F.; Malinska, K.; Mounier, E.; Hoyerova, K.; Tillard, P.; Leon, S.; Ljung, K.; et al. Nitrate-regulated auxin transport by NRT1.1 defines a mechanism for nutrient sensing in plants. Dev. Cell. 2010, 18, 927–937. [Google Scholar] [CrossRef]
- Bao, Y.; Chen, C.; Fu, L.; Chen, Y.Q. Comparative transcriptome analysis of Rosa chinensis ‘Old Blush’ provides insights into the crucial factors and signaling pathways in salt stress response. Agron. J. 2021, 113, 3031–3050. [Google Scholar] [CrossRef]
- Su, N.N.; Zhu, A.Q.; Tao, X.; Ding, Z.J.; Chang, S.H.; Ye, F.; Zhang, Y.; Zhao, C.; Chen, Q.; Wang, J.Q.; et al. Structures and mechanisms of the Arabidopsis auxin transporter PIN3. Nature 2022, 610, 616–621. [Google Scholar] [CrossRef]
- Ma, H.S.; Liang, D.; Shuai, P.; Xia, X.L.; Yin, W.L. The salt- and drought-inducible poplar GRAS protein SCL7 confers salt and drought tolerance in Arabidopsis thaliana. J. Exp. Bot. 2010, 61, 4011–4019. [Google Scholar] [CrossRef]
- Yuan, L.; Zheng, Y.; Nie, L.; Zhang, L.; Wu, Y.; Zhu, S.; Hou, J.; Shan, G.L.; Liu, T.K.; Chen, G.; et al. Transcriptional profiling reveals changes in gene regulation and signaling transduction pathways during temperature stress in wucai (Brassica campestris L.). BMC Genom. 2021, 22, 687. [Google Scholar] [CrossRef]
- Jeon, J.; Rahman, M.M.; Yang, H.W.; Kim, J.; Gam, H.J.; Song, J.Y.; Jeong, S.W.; Kim, J.I.; Choi, M.G.; Shin, D.H.; et al. Modulation of warm temperature-sensitive growth using a phytochrome B dark reversion variant, phyB[G515E], in Arabidopsis and rice. J. Adv. Res. 2023; in press. [Google Scholar] [CrossRef]
- Khan, B.R.; Zolman, B.K. 5 Mutants that differentially disrupt pts1 and pts2 peroxisomal matrix protein import in Arabidopsis. Plant Physiol. 2010, 154, 1602–1615. [Google Scholar] [CrossRef]
- Jie, H.D.; He, P.L.; Zhao, L.; Ma, Y.S.; Jie, Y.C. Molecular mechanisms regulating phenylpropanoid metabolism in exogenously-sprayed ethylene forage ramie based on transcriptomic and metabolomic analyses. Plants 2023, 12, 3899. [Google Scholar] [CrossRef]
- Dong, C.J.; Cao, N.; Zhang, Z.G.; Shang, Q.M. Phenylalanine ammonia-lyase gene families in cucurbit species: Structure, evolution, and expression. J. Integr. Agr. 2016, 15, 1239–1255. [Google Scholar] [CrossRef]
- Wang, Q.Q.; Li, Y.Y.; Chen, J.T.; Zhu, M.J.; Liu, X.D.; Zhou, Z.; Zhang, D.Y.; Liu, Z.J.; Lan, S.R. Genome-wide identification of YABBY genes in three Cymbidium species and expression patterns in C. ensifolium (Orchidaceae). Front. Plant Sci. 2022, 13, 995734. [Google Scholar] [CrossRef]
- Wimalagunasekara, S.S.; Weeraman, J.W.J.K.; Tirimanne, S.; Fernando, P.C. Protein-protein interaction (PPI) network analysis reveals important hub proteins and sub-network modules for root development in rice (Oryza sativa). J. Genet. Eng. Biotechn. 2023, 21, 69. [Google Scholar] [CrossRef]
- Struk, S.; Jacobs, A.; Martín-Fontecha, E.S.; Gevaert, K.; Cubas, P.; Goormachtig, S. Exploring the protein-protein interaction landscape in plants. Plant Cell Environ. 2019, 42, 387–409. [Google Scholar] [CrossRef]
- Dong, N.Q.; Lin, H.X. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. J. Integr. Plant Biol. 2021, 63, 180–209. [Google Scholar] [CrossRef]
- Rahim, M.A.; Zhang, X.B.; Busatto, N. Editorial: Phenylpropanoid biosynthesis in plants. Front. Plant Sci. 2023, 14, 1230664. [Google Scholar] [CrossRef]
- Jiang, N.; Doseff, A.I.; Grotewold, E. Flavones: From biosynthesis to health benefits. Plants 2016, 5, 27. [Google Scholar] [CrossRef]
- Wang, P.R.; Wan, C.M.; Xu, Z.J.; Wang, P.Y.; Wang, W.M.; Sun, C.H.; Ma, X.Z.; Xiao, Y.H.; Zhu, J.Q.; Gao, X.L.; et al. One divinyl reductase reduces the 8-vinyl groups in various intermediates of chlorophyll biosynthesis in a given higher plant species, but the isozyme differs between species. Plant Physiol. 2013, 161, 521–534. [Google Scholar] [CrossRef]
- Bucinská, L.; Kiss, É.; Koník, P.; Knoppová, J.; Komenda, J.; Sobotka, R. The ribosome-bound protein Pam68 promotes insertion of chlorophyll into the CP47 subunit of photosystem II. Plant Physiol. 2018, 176, 2931–2942. [Google Scholar] [CrossRef]
- Armbruster, U.; Rühle, T.; Kreller, R.; Strotbek, C.; Zühlke, J.; Tadini, L.; Blunder, T.; Hertle, A.P.; Qi, Y.F.; Rengstl, B.; et al. The PHOTOSYNTHESIS AFFECTED MUTANT68-LIKE protein Evolved from a PSII assembly factor to mediate assembly of the chloroplast NAD(P)H dehydrogenase complex in Arabidopsis. Plant Cell 2013, 25, 3926–3943. [Google Scholar] [CrossRef]
- Van Rooijen, R.; Harbinson, J.; Aarts, M.G.M. Photosynthetic response to increased irradiance correlates to variation in transcriptional response of lipid-remodeling and heat-shock genes. Plant Direct 2018, 2, e00069. [Google Scholar] [CrossRef]
- An, J.; Wei, X.; Huo, H. Transcriptome analysis reveals the accelerated expression of genes related to photosynthesis and chlorophyll biosynthesis contribution to shade-tolerant in Phoebe bournei. BMC Plant Biol. 2022, 22, 268. [Google Scholar] [CrossRef]
- Bolouri Moghaddam, M.R.; Van den Ende, W. Sugars, the clock and transition to flowering. Front. Plant Sci. 2013, 4, 22. [Google Scholar] [CrossRef]
- Wei, J.; Chen, Q.; Lin, J.; Chen, F.; Chen, R.; Liu, H.; Chu, P.; Lu, Z.; Li, S.; Yu, G. Genome-wide identification and expression analysis of tomato glycoside hydrolase family 1 β-glucosidase genes in response to abiotic stresses. Biotechnol. Biotechnol. Equip. 2022, 36, 268–280. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, T.; Xu, X.; Sun, Y.; Zhang, Y.; Hou, M.; Huang, S.; Yuan, H.; Tong, H. Identification of GH1 gene family fgt members in Stevia rebaudiana and their expression when grown in darkness. Mol. Biol. Rep. 2020, 47, 8739–8746. [Google Scholar] [CrossRef]
- Ma, Y.; Han, Y.; Feng, X.; Gao, H.; Cao, B.; Song, L. Genome-wide identification of BAM (β-amylase) gene family in jujube (Ziziphus jujuba Mill.) and expression in response to abiotic stress. BMC Genom. 2022, 23, 438. [Google Scholar] [CrossRef]
- Zanella, M.; Borghi, G.L.; Pirone, C.; Thalmann, M.; Pazmino, D.; Costa, A.; Santelia, D.; Trost, P.; Sparla, F. β-amylase 1 (BAM1) degrades transitory starch to sustain proline biosynthesis during drought stress. J. Exp. Bot. 2016, 67, 1819–1826. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, J.; Khan, M.; Wang, Y.; Xiao, W.; Fang, T.; Qu, J.; Xiao, P.; Li, C.L.; Liu, J.H. Transcription factors ABF4 and ABR1 synergistically regulate amylase-mediated starch catabolism in drought tolerance. Plant Physiol. 2023, 191, 591–609. [Google Scholar] [CrossRef]
- Erpen, L.; Devi, H.S.; Grosser, J.W.; Dutt, M. Potential use of the DREB/ERF, MYB, NAC and WRKY transcription factors to improve abiotic and biotic stress in transgenic plants. Plant Cell Tiss. Org. 2018, 132, 1–25. [Google Scholar] [CrossRef]
- Adem, M.; Gadissa, F.; Zhao, K.; Beyene, D. Transcriptome-wide identification and expression analysis of the NAC gene family in lowland bamboo [Oxytenanthera abyssinica (A.Rich) Munro] under abiotic stresses. Plant Sci. Today 2023, 10, 250–259. [Google Scholar] [CrossRef]
- Wu, Z.; Li, T.; Xiang, J.; Teng, R.D.; Zhang, D.H.; Teng, N.J. A lily membrane-associated NAC transcription factor LlNAC014 is involved in thermotolerance via activation of the DREB2-HSFA3 module. J. Exp. Bot. 2023, 74, 945–963. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.N.; Liu, H.Y.; Mei, Q.L.; Yang, J.; Ma, F.W.; Mao, K. Characteristics of bHLH transcription factors and their roles in the abiotic stress responses of horticultural crops. Sci. Hortic. 2023, 310, 111710. [Google Scholar] [CrossRef]
- Ohashi-Ito, K.; Bergmann, D.C. Arabidopsis FAMA controls the final proliferation/differentiation switch during stomatal development. Plant Cell 2006, 18, 2493–2505. [Google Scholar] [CrossRef] [PubMed]
- Nemie-Feyissa, D.; Heidari, B.; Blaise, M.; Lillo, C. Analysis of interactions between heterologously produced bHLH and MYB proteins that regulate anthocyanin biosynthesis: Quantitative interaction kinetics by microscale thermophoresis. Phytochemistry 2015, 111, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Ning, G.X.; Li, W.F.; Chu, M.Y.; Ma, Z.H.; Wang, P.; Mao, J.; Chen, B.H. MdbHLH51 plays a positive role in anthocyanin accumulation in ‘Red Delicious’ apples. Trees 2022, 36, 1687–1695. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Zhang, Y.M.; Hu, H.F.; Chen, L.; Zhang, H.F.; Chen, R.G. CabHLH79 acts upstream of to CaNAC035 regulate cold stress in Pepper. Int. J. Mol. Sci. 2022, 23, 2537. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, I. Regulation and function of SOC1, a flowering pathway integrator. J. Exp. Bot. 2010, 61, 2247–2254. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Beruto, M.; Xue, J.; Zhu, F.; Liu, C.; Yan, Y.; Zhang, X. Molecular cloning and potential function prediction of homologous SOC1 genes in tree peony. Plant Cell Rep. 2015, 34, 1459–1471. [Google Scholar] [CrossRef]
- Suárez-López, P.; Wheatley, K.; Robson, F.; Onouchi, H.; Valverde, F.; Coupland, G. CONSTANS mediates between the circadian clock and the control of flowering in Arabidopsis. Nature 2001, 410, 1116–1120. [Google Scholar] [CrossRef]
- Datta, S.; Hettiarachchi, G.H.C.M.; Holm, D.M. Arabidopsis CONSTANS-LIKE is a positive regulator of red light signaling and root growth. Plant Cell 2006, 18, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Sun, J.J.; Jiang, A.Q.; Bai, M.J.; Fan, C.G.; Liu, J.Y.; Ning, G.G.; Wang, C.Q. Alternate expression of Constans-Like 4 in short days and CONSTANS in long days facilitates day-neutral response in Rosa chinensis. J. Exp. Bot. 2020, 71, 4057–4068. [Google Scholar] [CrossRef] [PubMed]
- Min, J.H.; Chung, J.S.; Lee, K.H.; Kim, C.S. The CONSTANS-like 4 transcription factor, AtCOL4, positively regulates abiotic stress tolerance through an abscisic acid-dependent manner in Arabidopsis. J. Integr. Plant Biol. 2015, 57, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Li, F.F.; Niu, J.H.; Yu, X.; Kong, Q.H.; Wang, R.F.; Qin, L.; Chen, E.Y.; Yang, Y.B.; Liu, Z.Y.; Lang, L.N.; et al. Isolation and identification of SiCOL5, which is involved in photoperiod response, based on the quantitative trait locus mapping of. Front. Plant Sci. 2022, 13, 969604. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.X.; He, D.X.; Wang, N.; Yao, X.D.; Xie, F.T. Transcriptome and metabolome jointly revealed the regulation and pathway of flower and pod abscission caused by shading in soybean (Glycine max L.). Agronomy 2024, 14, 106. [Google Scholar] [CrossRef]
- Chen, W.; Hao, W.J.; Xu, Y.X.; Zheng, C.; Ni, D.J.; Yao, M.Z.; Chen, L. Isolation and characterization of CsWRKY7, a subgroup IId WRKY transcription factor from camellia sinensis, linked to development in Arabidopsis. Int. J. Mol. Sci. 2019, 20, 2815. [Google Scholar] [CrossRef]
- Jia, P.; Sharif, R.; Li, Y.M.; Sun, T.B.; Li, S.K.; Zhang, X.M.; Dong, Q.L.; Luan, H.A.; Guo, S.P.; Ren, X.L.; et al. The BELL1-like homeobox gene MdBLH14 from apple controls flowering and plant height via repression of MdGA20ox3. Int. J. Biol. Macromol. 2023, 242, 124790. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Xuan, L.; Xu, L.-A.; Huang, M.-R.; Xu, M. Identification and characterization of nine PAT1 branch genes in poplar. Plant Growth Regul. 2017, 81, 355–364. [Google Scholar] [CrossRef]
- Bolle, C.; Koncz, C.; Chua, N.H. PAT1, a new member of the GRAS family, is involved in phytochrome A signal transduction. Genes Dev. 2000, 14, 1269–1278. [Google Scholar] [CrossRef]
- Muntha, S.T.; Zhang, L.L.; Zhou, Y.F.; Zhao, X.; Hu, Z.Y.; Yang, J.H.; Zhang, M.F. Phytochrome a signal transduction 1 and CONSTANS-LIKE 13 coordinately orchestrate shoot branching and flowering in leafy Brassica juncea. Plant Biotechnol. J. 2019, 17, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Baek, J.; Choe, U.; Lim, C.O. Overexpression of DREB2C delays flowering in arabidopsis thaliana via the activation of FLC. J. Plant Biol. 2022, 65, 133–143. [Google Scholar] [CrossRef]
- Lin, R.C.; Wang, H.Y. Arabidopsis FHY3/FAR1 gene family and distinct roles of its members in light control of arabidopsis development. Plant Physiol. 2004, 136, 4010–4022. [Google Scholar] [CrossRef] [PubMed]
- Paik, I.; Huq, E. Plant photoreceptors: Multi-functional sensory proteins and their signaling networks. Semin. Cell Dev. Biol. 2019, 92, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Amor, M.B.; Guis, M.; Latché, A.; Bouzayen, M.; Pech, J.C.; Roustan, J.P. Expression of an antisense 1-aminocyclopropane-1-carboxylate oxidase gene stimulates shoot regeneration in Cucumis melo. Plant Cell Rep. 1998, 17, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.E.; Findell, J.L.; Schaller, G.E.; Sisler, E.C.; Bleecker, A.B. Ethylene perception by the ERS1 protein in Arabidopsis. Plant Physiol. 2000, 123, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Catala, R.; Lopez-Cobollo, R.; Mar Castellano, M.; Angosto, T.; Alonso, J.M.; Ecker, J.R.; Salinas, J. The Arabidopsis 14-3-3 protein RARE COLD INDUCIBLE 1A links low-temperature response and ethylene biosynthesis to regulate freezing tolerance and cold acclimation. Plant Cell 2014, 26, 3326–3342. [Google Scholar] [CrossRef]
- Jensen, L.; Hegelund, J.N.; Olsen, A.; Lutken, H.; Muller, R. A natural frameshift mutation in Campanula EIL2 correlates with ethylene insensitivity in flowers. BMC Plant Biol. 2016, 16, 117. [Google Scholar] [CrossRef]
- Liu, C.; Li, J.; Zhu, P.; Yu, J.; Hou, J.; Wang, C.; Long, D.; Yu, M.; Zhao, A. Mulberry EIL3 confers salt and drought tolerances and modulates ethylene biosynthetic gene expression. PeerJ 2019, 7, e6391. [Google Scholar] [CrossRef]
- Dolgikh, V.A.; Pukhovaya, E.M.; Zemlyanskaya, E.V. Shaping ethylene response: The role of EIN3/EIL1 transcription factors. Front. Plant Sci. 2019, 10, 1030. [Google Scholar] [CrossRef]
- Nieuwenhuizen, N.J.; Chen, X.; Wang, M.Y.; Matich, A.J.; Perez, R.L.; Allan, A.C.; Green, S.A.; Atkinson, R.G. Natural ariation in monoterpene synthesis in kiwifruit: Transcriptional regulation of eerpene synthases by NAC and ETHYLENE-INSENSITIVE3-like transcription factors. Plant Physiol. 2015, 167, 1243–1258. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.R.; Allan, A.C.; Chen, K.S.; Ferguson, I.B. Kiwifruit EIL and ERF genes involved in regulating fruit ripening. Plant Physiol. 2010, 153, 1280–1292. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhang, B.; Li, X.; Xu, C.; Yin, X.; Shan, L.; Ferguson, I.; Chen, K. Ethylene signal transduction elements involved in chilling injury in non-climacteric loquat fruit. J. Exp. Bot. 2010, 61, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Liu, R.L.; Li, Y.; Shen, X.; Zhong, S.W.; Shi, H. EIN3 and PIF3 form an interdependent module that represses chloroplast development in buried seedlings. Plant Cell 2017, 29, 3051–3067. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Sudhakar, D.; Peng, J.; Richards, D.E.; Christou, P.; Harberd, N.P. Expression of Arabidopsis GAI in transgenic rice represses multiple gibberellin responses. Plant Cell 2001, 13, 1791–1802. [Google Scholar] [CrossRef] [PubMed]
- Arain, S.; Meer, M.; Sajjad, M.; Yasmin, H. Light contributes to salt resistance through GAI protein regulation in Arabidopsis thaliana. Plant Physiol. Bioch. 2021, 159, 1–11. [Google Scholar] [CrossRef]
- Wang, H.P.; Pan, J.J.; Li, Y.; Lou, D.J.; Hu, Y.R.; Yu, D.Q. The DELLA-CONSTANS transcription factor cascade integrates gibberellic acid and photoperiod signaling to regulate flowering. Plant Physiol. 2016, 172, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Fukazawa, J.; Ohashi, Y.; Takahashi, R.; Nakai, K.; Takahashi, Y. DELLA degradation by gibberellin promotes flowering via GAF1-TPR-dependent repression of floral repressors in Arabidopsis. Plant Cell 2021, 33, 2258–2272. [Google Scholar] [CrossRef]
- Ogawa, S.; Miyamoto, K.; Nemoto, K.; Sawasaki, T.; Yamane, H.; Nojiri, H.; Okada, K. OsMYC2, an essential factor for JA-inductive sakuranetin production in rice, interacts with MYC2-like proteins that enhance its transactivation ability. Sci. Rep. 2017, 7, 40175. [Google Scholar] [CrossRef]
- Verma, D.; Jalmi, S.K.; Bhagat, P.K.; Verma, N.; Sinha, A.K. A bHLH transcription factor, MYC2, imparts salt intolerance by regulating proline biosynthesis in Arabidopsis. FEBS J. 2020, 287, 2560–2576. [Google Scholar] [CrossRef]
- Song, R.F.; Li, T.T.; Liu, W.C. Jasmonic acid impairs arabidopsis seedling salt stress tolerance through MYC2-mediated repression of expression. Front. Plant Sci. 2021, 12, 730228. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Zha, S.X.; Luo, Y.Y.; Li, L.; Wang, S.Y.; Wu, S.; Cheng, S.Y.; Li, L.L. JAZ1-3 and MYC2-1 synergistically regulate the transformation from completely mixed flower buds to female flower buds in Castanea mollisima. Int. J. Mol. Sci. 2022, 23, 6452. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.Y.; Lan, Y.H.; Shi, T.L.; Zhu, Z.Q. Diverse contributions of MYC2 and EIN3 in the regulation of Arabidopsis jasmonate-responsive gene expression. Plant Direct 2017, 1, e00015. [Google Scholar] [CrossRef] [PubMed]
- Chini, A.; Fonseca, S.; Fernández, G.; Adie, B.; Chico, J.M.; Lorenzo, O.; García-Casado, G.; López-Vidriero, I.; Lozano, F.M.; Ponce, M.R.; et al. The JAZ family of repressors is the missing link in jasmonate signalling. Nature 2007, 448, 666–671. [Google Scholar] [CrossRef]
- Swarup, R.; Péret, B. AUX/LAX family of auxin influx carriers—An overview. Front. Plant Sci. 2012, 3, 225. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Raw Bases | Clean Reads | Clean Bases | Q20 | Q30 | GC |
|---|---|---|---|---|---|---|---|
| WL01 | 77.23 M | 11.58 G | 76.77 M | 11.48 G | 97.56% | 93.33% | 45.14% |
| WL02 | 63.58 M | 9.54 G | 63.14 M | 9.44 G | 97.20% | 92.46% | 45.07% |
| WL03 | 71.22 M | 10.68 G | 70.77 M | 10.58 G | 97.47% | 93.15% | 45.27% |
| WL04 | 66.07 M | 9.91 G | 65.61 M | 98.17 G | 97.05% | 92.14% | 45.14% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Lyu, T.; Lyu, Y. The Molecular Biology Analysis for the Growing and Development of Hydrangea macrophylla ‘Endless Summer’ under Different Light and Temperature Conditions. Horticulturae 2024, 10, 586. https://doi.org/10.3390/horticulturae10060586
Li Z, Lyu T, Lyu Y. The Molecular Biology Analysis for the Growing and Development of Hydrangea macrophylla ‘Endless Summer’ under Different Light and Temperature Conditions. Horticulturae. 2024; 10(6):586. https://doi.org/10.3390/horticulturae10060586
Chicago/Turabian StyleLi, Zheng, Tong Lyu, and Yingmin Lyu. 2024. "The Molecular Biology Analysis for the Growing and Development of Hydrangea macrophylla ‘Endless Summer’ under Different Light and Temperature Conditions" Horticulturae 10, no. 6: 586. https://doi.org/10.3390/horticulturae10060586
APA StyleLi, Z., Lyu, T., & Lyu, Y. (2024). The Molecular Biology Analysis for the Growing and Development of Hydrangea macrophylla ‘Endless Summer’ under Different Light and Temperature Conditions. Horticulturae, 10(6), 586. https://doi.org/10.3390/horticulturae10060586