
Analysis of Transcriptome and Expression of C4H and FLS Genes on Four Flower Colors of Impatiens uliginosa

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Transcriptome Sequencing and Analysis
2.2.1. RNA Extraction and Detection
2.2.2. Second-Generation Sequencing and Assembly
2.2.3. Sequence Analysis and Annotation
2.2.4. Screening and Enrichment Analysis of Differentially Expressed Genes (DEGs)
2.2.5. Related Gene Expression and Analysis
3. Results
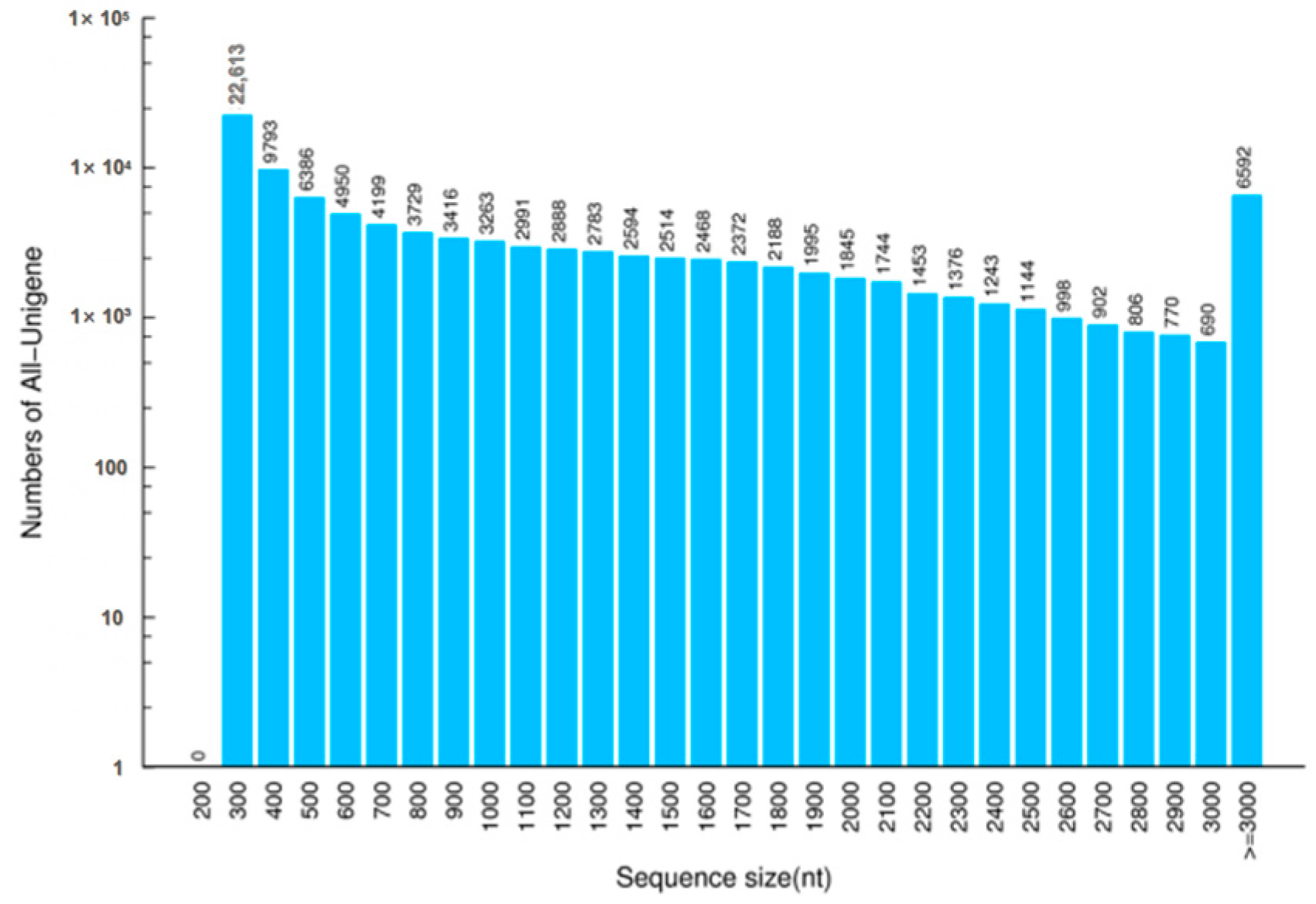
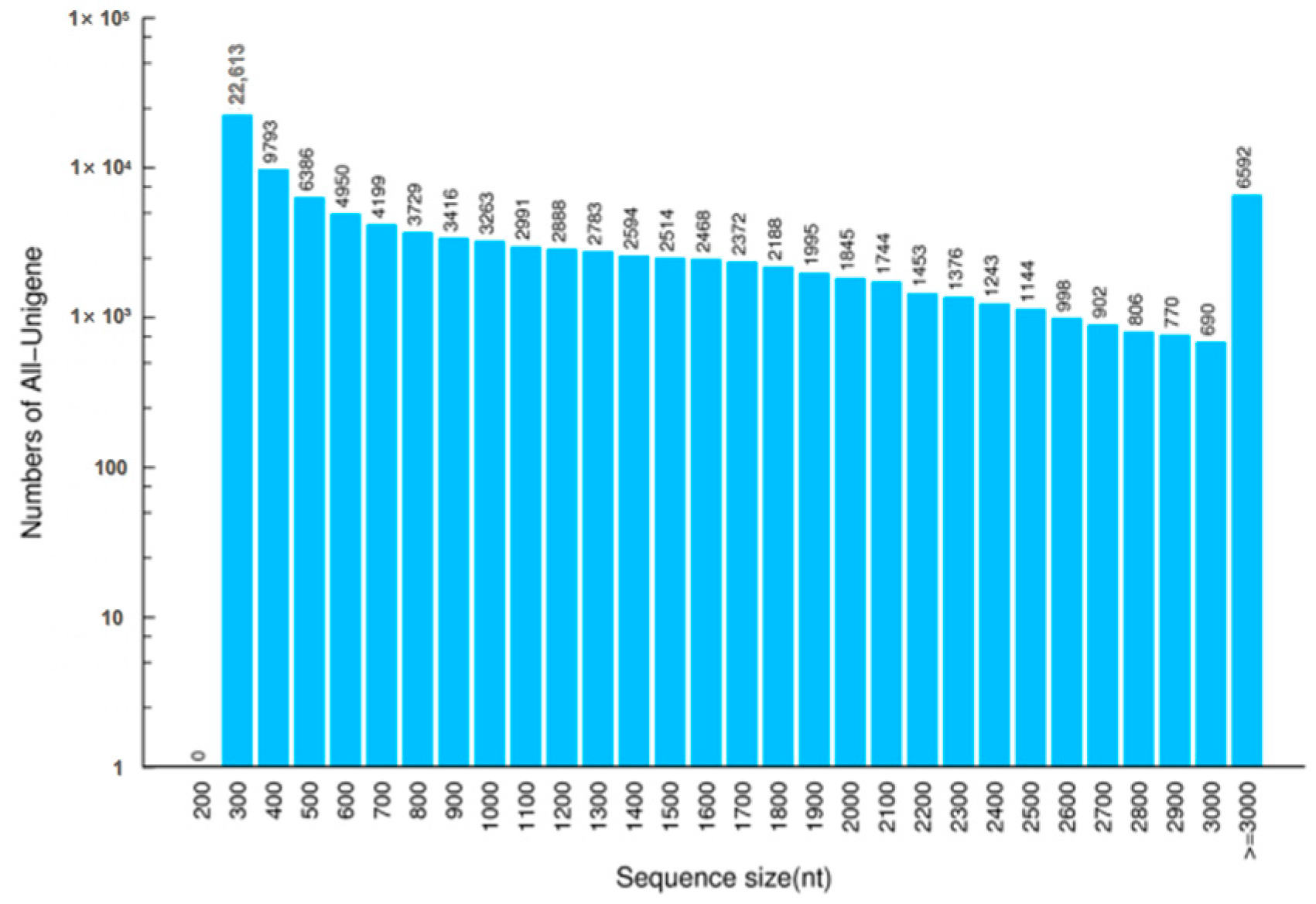
3.1. RNA Sequencing and Transcriptome De Novo Assembly
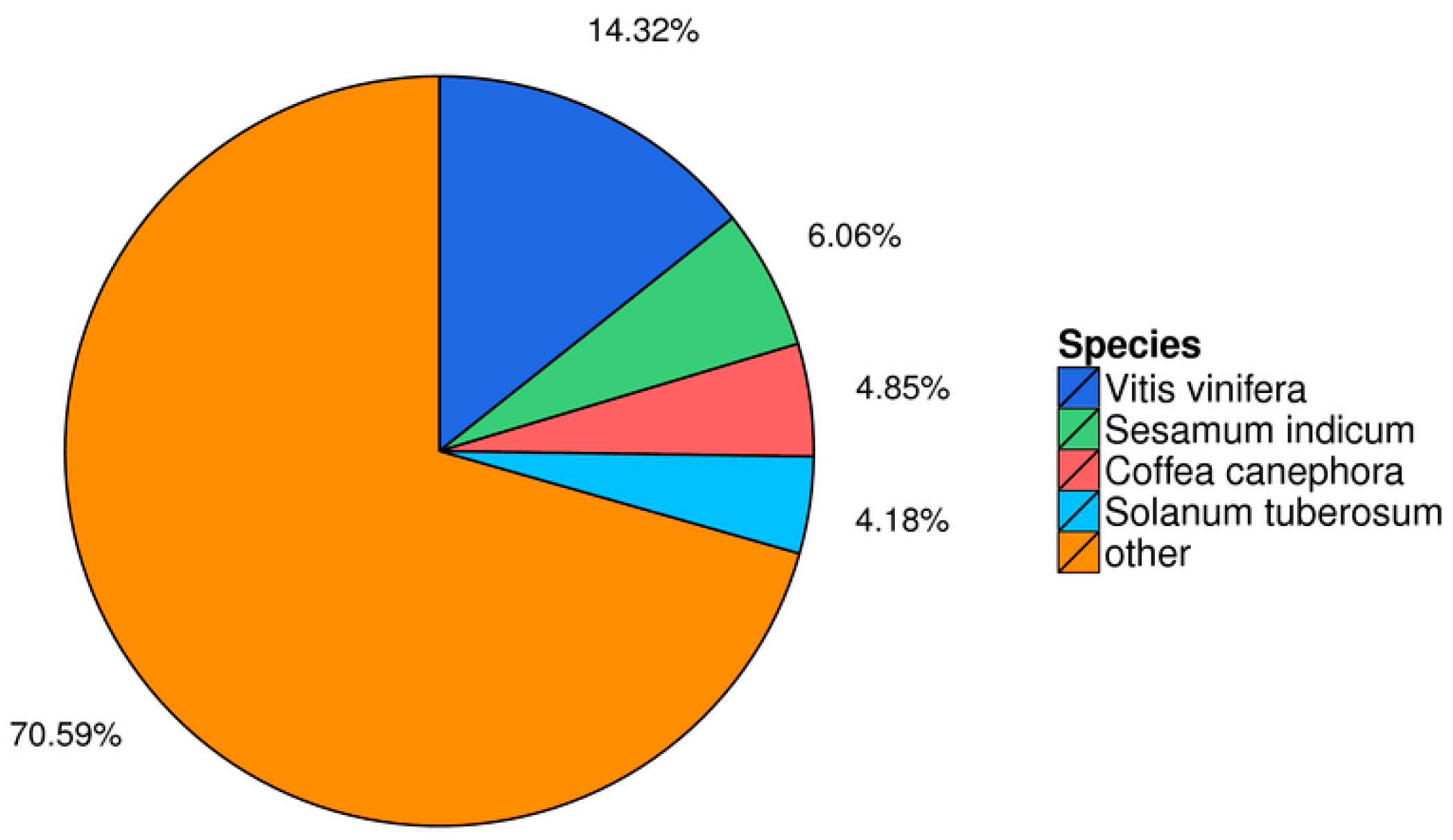
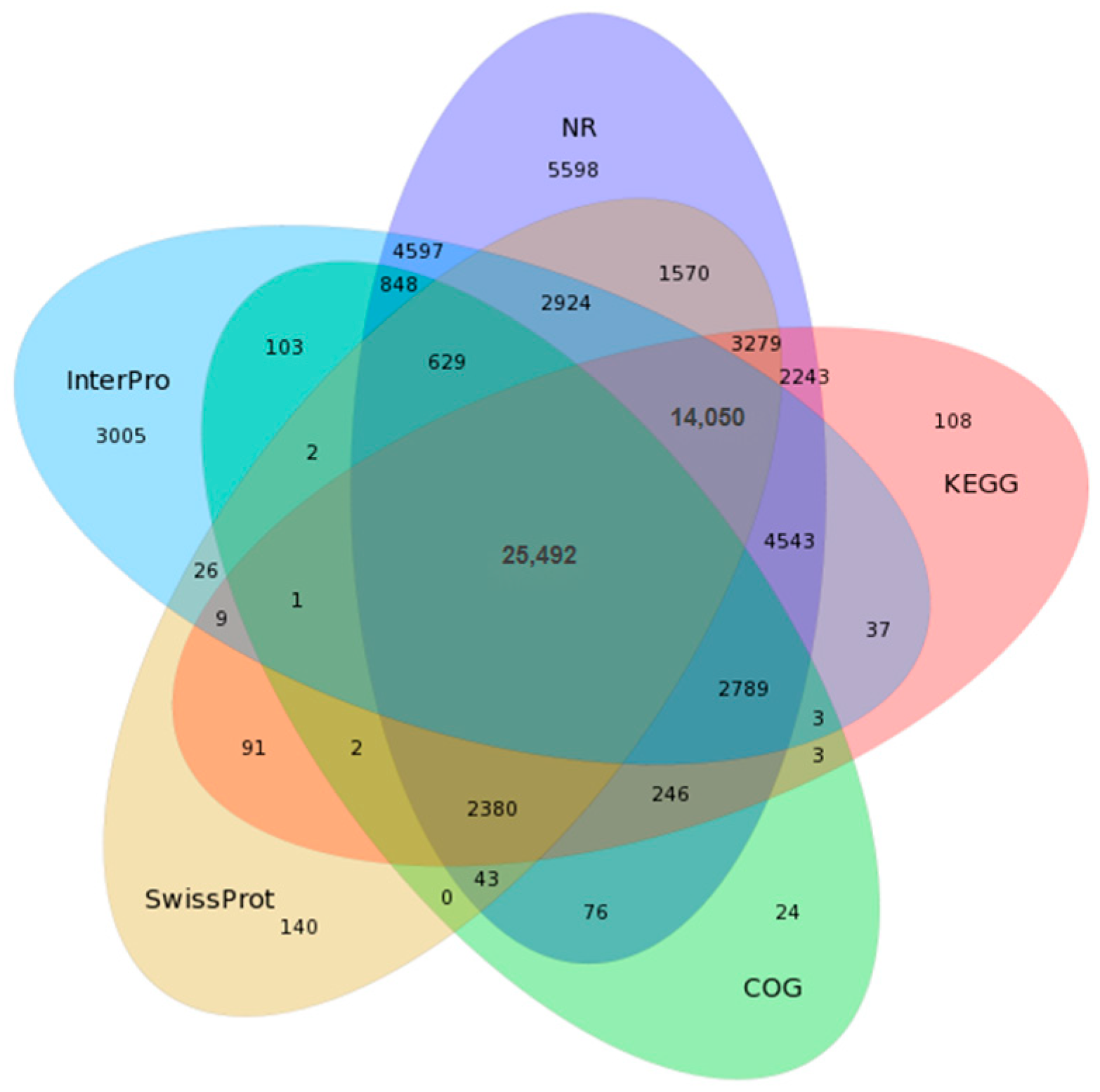
3.2. Functional Annotation of Transcriptome Genes of I. uliginosa
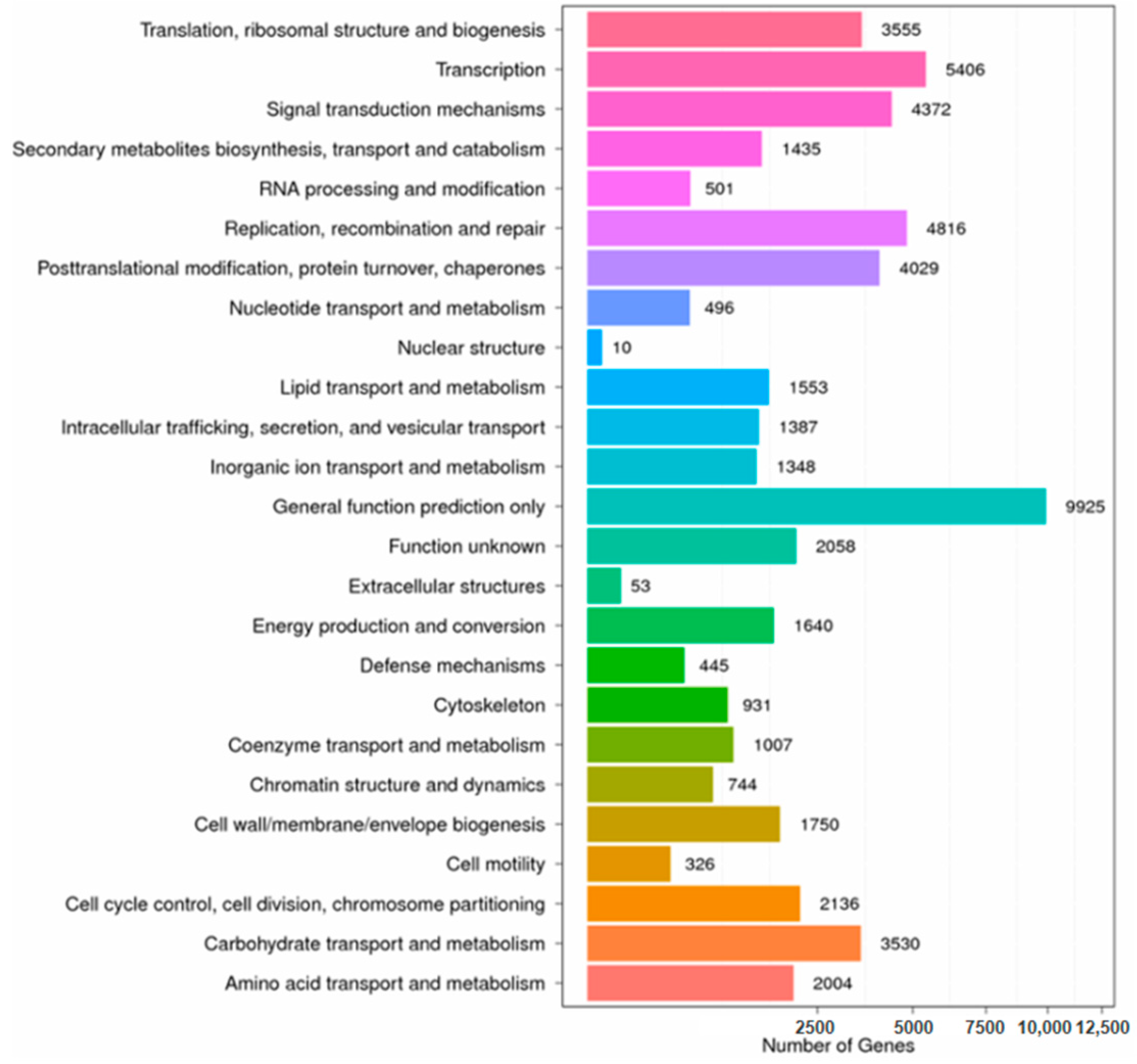
3.3. GO (Gene Ontology) and COG (Clusters of Orthologous Group of Proteins) Annotation Analysis of I. uliginosa Transcriptome Data
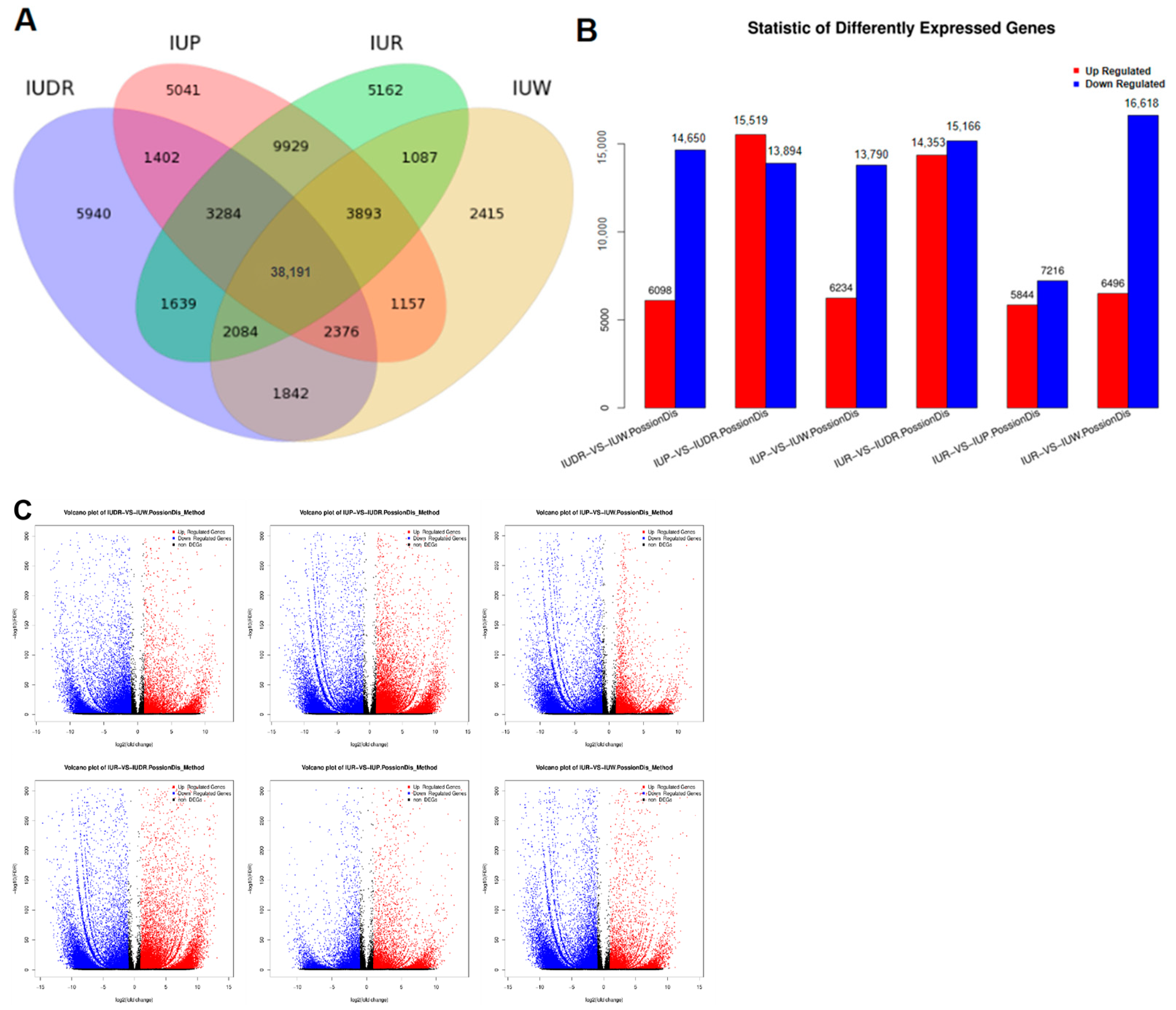
3.4. Identification and Functional Annotation of Differential Genes (DEGs) in I. uliginosa
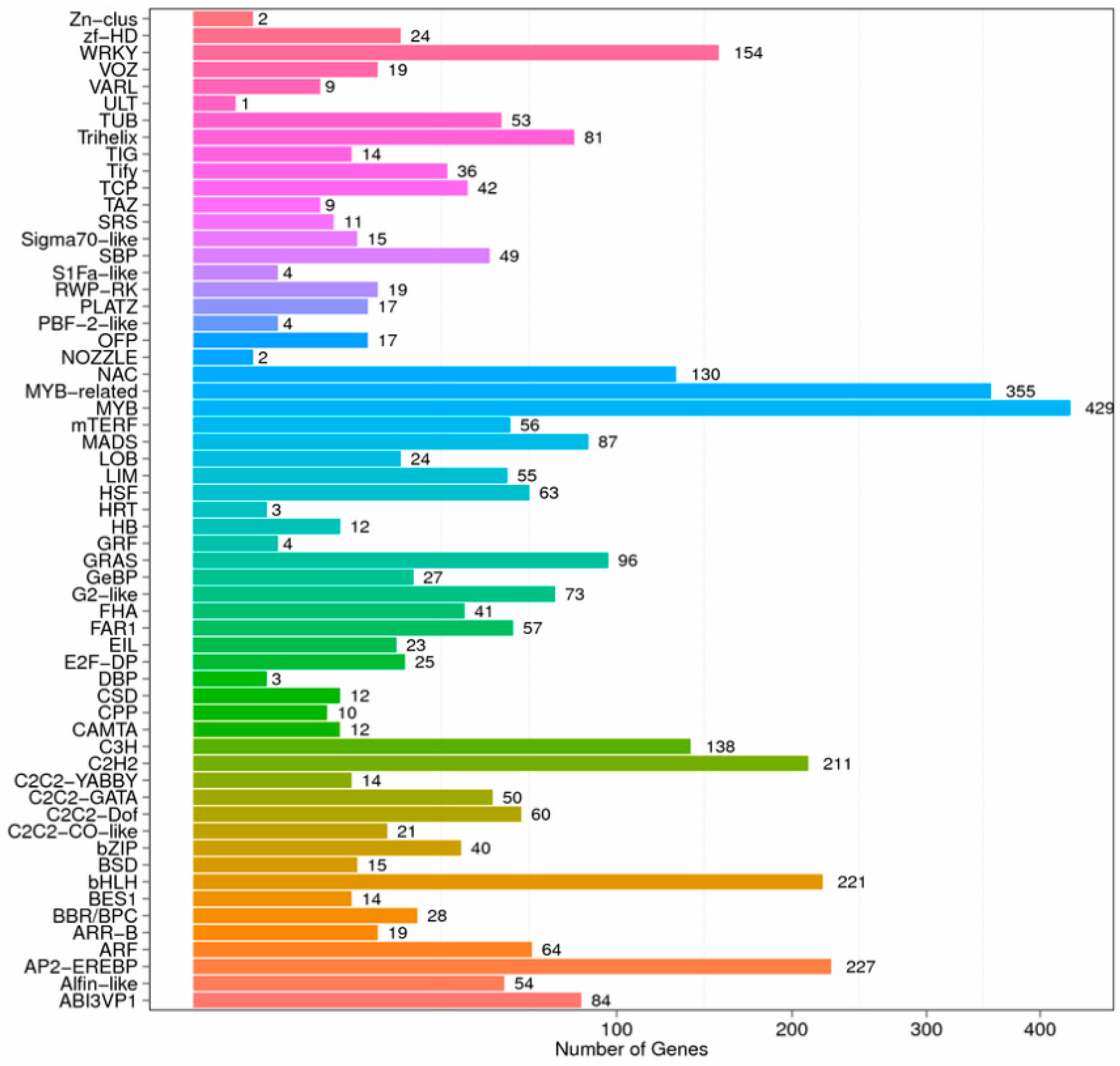
3.5. Identification and Expression Analysis of Transcription Factors in I. uliginosa
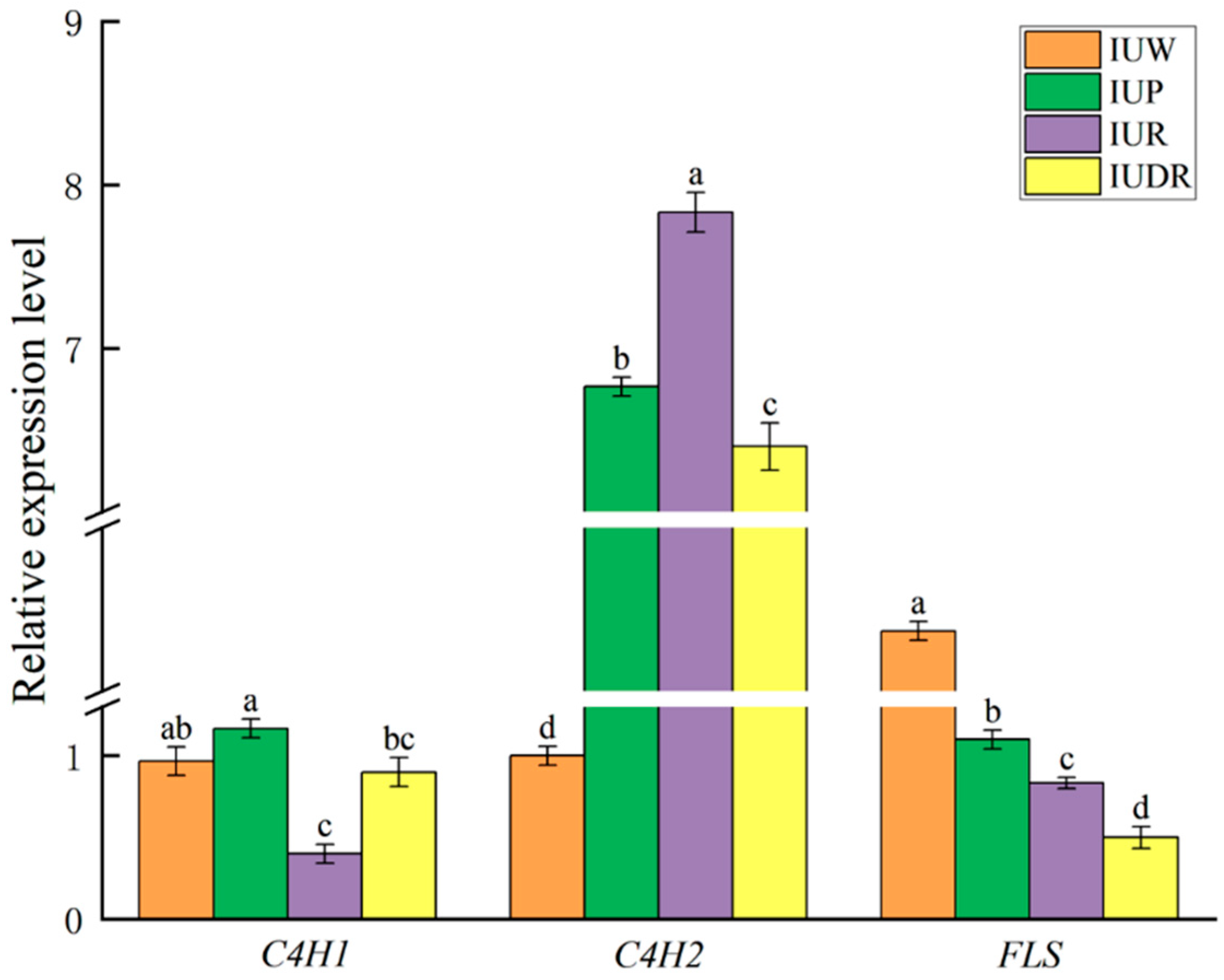
3.6. qRT-PCR Validation of the Candidate Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kellenberger, R.T.; Glover, G.B. The evolution of flower colour. Curr. Biol. 2023, 33, R484–R488. [Google Scholar] [CrossRef] [PubMed]
- Li, H. The of Anthocyanidin, Metal Elementsand pH on the Colour Formation of Blue Flowers from Monocots. Master’s Thesis, North West Agriculture and Forestry University, Xianyang, China, 2013; pp. 52–56. [Google Scholar]
- Han, Y.; Huang, K.; Liu, Y.; Jiao, T.; Ma, G.; Qian, Y.; Wang, P.; Dai, X.; Gao, L.; Xia, T. Functional Analysis of Two Flavanone-3-Hydroxylase Genes from Camellia sinensis: A Critical Role in Flavonoid Accumulation. Genes 2017, 8, 300. [Google Scholar] [CrossRef] [PubMed]
- Lou, Q.; Liu, Y.; Qi, Y.; Jiao, S.; Tian, F.; Jiang, L.; Wang, Y. Transcriptome sequencing and metabolite analysis reveals the role of delphinidin metabolism in flower colour in grape hyacinth. J. Exp. Bot. 2014, 65, 3157–3164. [Google Scholar] [CrossRef] [PubMed]
- Hagen, C.W. The differentiation of pigmentation in flower parts I. The flavonoid pigments of Impatiens balsamina, genotype llHHPrPr, and their distribution within the plant. Am. J. Bot. 1966, 53, 46–54. [Google Scholar] [CrossRef]
- Miles, C.D.; Hagen, C.W. The differentiation of pigmentation in flower parts, IV. Flavonoid elaborating enzymes from petals of Impatiens balsamina. Plant Physiol. 1968, 43, 1347–1354. [Google Scholar] [PubMed]
- Clevenger, S. Anthocyanidins of some Impatiens species. Evolution 1971, 25, 669–677. [Google Scholar] [PubMed]
- Hasan, A.; Tahir, M.N. Flavonoids from the leaves of Impatiens bicolor. Turk. J. Chem. 2005, 29, 65–70. [Google Scholar]
- Tatsuzawa, F.; Saito, N.; Mikanagi, Y.; Shinoda, K.; Toki, K.; Shigihara, A.; Honda, T. An unusual acylated malvidin 3-glucoside from flowers of Impatiens textori Miq. (Balsaminaceae). Phytochemistry 2009, 70, 672–674. [Google Scholar] [CrossRef]
- Lei, J.; Qian, S.H.; Jiang, J.Q. Two new flavone glycosides from the seeds of Impatiens balsamina L. J. Asian Nat. Prod. Res. 2010, 12, 1033–1037. [Google Scholar] [CrossRef]
- Mariana, N.V.; Peter, W.; Gerold, J. Flavonoids from the flowers of Impatiens glandulifera Royle isolated by high performance countercurrent chromatography. Phytochem. Anal. PCA 2016, 27, 116–125. [Google Scholar]
- Zhao, L.Q.; Liu, Y.; Huang, Q.; Gao, S.; Huang, M.J.; Huang, H.Q. Effects of cell morphology, physiology, biochemistry and CHS genes on four flower colors of Impatiens uliginosa. Front. Plant Sci. 2024, 15, 151343830. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zhao, Z.; Gao, A.; Chen, Y.; Huang, J.; Dang, Z.; Luo, R. Cloning of mango C4H gene and its expression analysis. Jiangsu Agric. Sci. 2017, 45, 8–12. [Google Scholar]
- Ma, J.; Su, L.; Yuan, M.; Ji, H.; Li, A.; Wang, X. Cloning and expression studies of peanut C4H and ANR genes. J. Nucl. Agric. Sci. 2012, 26, 43–48. [Google Scholar]
- Yao, S.; Wang, W.; Li, M.; Xu, Y.; Wang, Y.; Liu, Y.; Gao, L.; Xia, T. The Gene Cloning and expression analysis of tea tree cinnamic acid 4-hydroxylase gene. J. Tea Sci. 2015, 35, 35–44. [Google Scholar]
- Zhao, L.; Ma, L.; Yang, Z.; Feng, W.; Zheng, X. Cloning and expression analysis of the C4H gene of Solanum vulgare. Acta Pharm. Sin. 2017, 52, 821–831. [Google Scholar]
- Li, L.; Zhao, Y.; Ma, L. New advances in the study of key enzymes of the phenylalanine metabolic pathway: PAL, C4H, 4CL. Chin. J. Bioinform. 2007, 20, 187–189. [Google Scholar]
- Liang, L.; Han, X.; Zhang, Z.; Guo, Q.; Xu, Y.; Liu, J.; Lian, Y. Cloning and expression analysis of the cinnamic acid-4-hydroxylase (C4H) gene from Cinnamomum album. Chin. J. Tradit. Chin. Med. 2014, 39, 1767–1771. [Google Scholar]
- Baek, M.H.; Chung, B.Y.; Kim, J.H.; Kim, J.S.; Lee, S.S.; An, B.C.; Lee, I.J.; Kim, T.H. cDNA cloning and expression pattern of cinnamate-4-hydroxylase in the Korean black raspberry. BMB Rep. 2008, 41, 529–536. [Google Scholar] [CrossRef]
- Tao, S.; Wang, D.; Jin, C.; Sun, W.; Liu, X.; Zhang, S.; Gao, F.; Khanizadeh, S. Cinnamate-4-hydroxylase gene is involved in the step of lignin biosynthesis in Chinese white pear. J. Am. Soc. Hortic. Sci. 2015, 140, 573–579. [Google Scholar] [CrossRef]
- Li, Y.; Ran, L.; Mo, T.; Liu, N.; Zeng, J.; Liang, A.; Wang, C.; Suo, Q.; Chen, Z.; Wang, Y.; et al. Yellow Petal locus GaYP promotes flavonol biosynthesis and yellow coloration in petals of Asiatic cotton (Gossypium arboreum). TAG Theor. Appl. Genet. 2023, 136, 98. [Google Scholar] [CrossRef]
- Schwinn, K.E.; Boase, M.R.; Bradley, J.M.; Lewis, D.H.; Deroles, S.C.; Martin, C.R.; Davies, K.M. MYB and bHLH transcription factor transgenes increase anthocyanin pigmentation in petunia and lisianthus plants, and the petunia phenotypes are strongly enhanced under field conditions. Front. Plant Sci. 2014, 5, 603. [Google Scholar] [CrossRef]
- Tanaka, Y. Cloning and expression of flavonol synthase from Petunia hybrida. Plant J. 2010, 4, 1003–1010. [Google Scholar]
- Pelletier, M.K.; Shirley, B.W. Characterization of flavonol synthase and leucoanthocyanidin dioxygenase genes in Arabidopsis. Further evidence for differential regulation of “early” and “late” genes. Plant Physiol. 1997, 113, 1437. [Google Scholar] [CrossRef]
- Nielsen, K.; Deroles, S.C.; Markham, K.R.; Bradley, M.J.; Podivinsky, E.; Manson, D. Antisense flavonol synthase alters copigmentation and flower color in lisianthus. Mol. Breed. 2002, 9, 217–229. [Google Scholar] [CrossRef]
- Almeida, J.R.; D’Amico, E.; Preuss, A.; Carbone, F.; de Vos, C.H.; Deiml, B.; Mourgues, F.; Perrotta, G.; Fischer, T.C.; Bovy, A.G.; et al. Characterization of major enzymes and genes involved in flavonoid and proanthocyanidin biosynthesis during fruit development in strawberry (Fragaria xananassa). Arch. Biochem. Biophys. 2007, 465, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Githiri, S.M.; Hatayama, K.; Dubouzet, E.G.; Shimada, N.; Aoki, T.; Ayabe, S.; Iwashina, T.; Toda, K.; Matsumura, H. A single-base deletion in soybean flavonol synthase gene is associated with magenta flower color. Plant Mol. Biol. 2007, 63, 125–135. [Google Scholar] [CrossRef]
- Luo, P. Molecular Cloning and Functional Analysis of Fiavonoids Biosynthesis Related Genes from Rosa rugosa. Ph.D. Thesis, Huazhong Agricultural University, Wuhan, China, 2016; pp. 2–4. [Google Scholar]
- Lukačin, R.; Wellmann, F.; Britsch, L.; Martens, S.; Matern, U. Flavonol synthase from Citrus unshiu is a bifunctional dioxygenase. Phytochemistry 2003, 62, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wei, C.M.; Li, X.Y.; Meng, D.C.; Gu, Z.J.; Qu, S.P.; Huang, M.J.; Huang, H.Q. De novo transcriptome sequencing of Impatiens uliginosa and the analysis of candidate genes related to spur development. BMC Plant Biol. 2022, 22, 553. [Google Scholar] [CrossRef]
- Allan, A.C.; Hellens, R.P.; Laing, W.A. MYB transcription factors that colour our fruit. Trends Plant Sci. 2008, 13, 99–102. [Google Scholar] [CrossRef]
- Nie, S.; Zhao, S.W.; Shi, T.L.; Zhao, W.; Zhang, R.G.; Tian, X.C.; Guo, J.F.; Yan, X.M.; Bao, Y.T.; Li, Z.C.; et al. Gapless genome assembly of azalea and multi-omics investigation into divergence between two species with distinct flower color. Hortic. Res. 2023, 10, 241. [Google Scholar] [CrossRef]
- Nie, S.; Ma, H.Y.; Shi, T.L.; Tian, X.C.; El-Kassaby, Y.A.; Porth, I.; Yang, F.S.; Mao, J.F. Progress in phylogenetics, multi-omics and flower coloration studies in Rhododendron. Ornam. Plant Res. 2024, 4, e003. [Google Scholar] [CrossRef]
- Song, X.; Tian, Y.; Gao, K.; Li, J.; Li, Y.; Wang, J.; Deng, C.; Zhang, F.; Kong, D.; Fan, G.; et al. Genetic and QTL analysis of flower color and pigments in small-flowered chrysanthemum based on high-density genetic map. Ornam. Plant Res. 2023, 3, 17. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, W.; Liu, J.; Liu, H.; Lv, Z.; Zhang, C.; Chen, D.; Jiao, Z. Postharvest UV-C irradiation increased the flavonoids and anthocyanins accumulation, phenylpropanoid pathway gene expression, and antioxidant activity in sweet cherries (Prunus avium L.). Postharvest Biol. Technol. 2021, 175, 111490. [Google Scholar] [CrossRef]
- Peng, H.; Yang, T.; Whitaker, B.D.; Trouth, F.; Shangguan, L.; Dong, W.; Jurick, W.M. Characterization of spermidine hydroxycinnamoyl transferases from eggplant (Solanum melongena L.) and its wild relative Solanum richardii Dunal. Hortic. Res. 2016, 3, 16062. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zheng, R.; Luo, J.; Wang, C. Cloning and Characterization of Cinnamate 4-hydroxylase (C4H) Genes from Osmanthus fragrans. J. Hortic. 2016, 43, 525–537. [Google Scholar]
- Liu, H.; Su, B.; Zhang, H.; Gong, J.; Zhang, B.; Liu, Y.; Du, L. Identification and Functional Analysis of a Flavonol Synthase Gene from Grape Hyacinth. Molecules 2019, 24, 1579. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Ning, G.; Wang, Z.; Shen, Y.; Jin, H.; Li, P.; Huang, S.; Zhao, J.; Bao, M. Disequilibrium of Flavonol Synthase and Dihydroflavonol-4-Reductase Expression Associated Tightly to White vs. Red Color Flower Formation in Plants. Front. Plant Sci. 2016, 6, 1257. [Google Scholar] [CrossRef]
- Davies, K.M.; Schwinn, K.E.; Deroles, S.C.; Manson, D.G.; Lewis, D.H.; Bloor, S.J.; Bradley, J.M. Enhancing anthocyanin production by altering competition for substrate between flavonol synthase and dihydroflavonol 4-reductase. Euphytica 2003, 131, 259–268. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Raw Reads (Mb) | Total Clean Reads (Mb) | Total Clean Bases (Gb) | Clean Reads Q20 (%) | Clean Reads Q30 (%) | GC (%) | Clean Reads Ratio (%) |
|---|---|---|---|---|---|---|---|
| IUDR | 52.69 | 44.62 | 6.69 | 98.78 | 96.09 | 41.46 | 84.68 |
| IUP | 54.33 | 45.11 | 6.77 | 98.75 | 96 | 42.77 | 83.02 |
| IUR | 52.69 | 44.35 | 6.65 | 98.78 | 96.11 | 42.2 | 84.17 |
| IUW | 51.04 | 44.77 | 6.71 | 98.72 | 95.94 | 41.71 | 87.71 |
| Sample | Total Number | Total Length | Mean Length | N50 | N70 | N90 | GC (%) |
|---|---|---|---|---|---|---|---|
| IUDR | 79,277 | 69,699,393 | 879 | 1545 | 946 | 336 | 41.46 |
| IUP | 91,109 | 76,919,883 | 844 | 1521 | 868 | 318 | 42.77 |
| IUR | 93,522 | 80,820,572 | 864 | 1530 | 903 | 329 | 42.2 |
| IUW | 64,723 | 59,663,736 | 921 | 1600 | 982 | 361 | 41.71 |
| Sample | Total Number | Total Length | Mean Length | N50 | N70 | N90 | GC (%) |
|---|---|---|---|---|---|---|---|
| IUDR | 55,003 | 58,568,930 | 1064 | 1689 | 1135 | 462 | 41.44 |
| IUP | 67,735 | 66,343,559 | 979 | 1646 | 1016 | 390 | 42.76 |
| IUR | 70,685 | 70,639,298 | 999 | 1640 | 1046 | 405 | 42.2 |
| IUW | 48,345 | 51,364,234 | 1062 | 1702 | 1121 | 453 | 41.68 |
| All-Unigene | 100,705 | 115,842,379 | 1150 | 1890 | 1267 | 495 | 41.98 |
| Values | Number | Percentage |
|---|---|---|
| NR-Annotated | 71,307 | 70.81% |
| Nt-Annotated | 59,001 | 58.59% |
| Swissprot-Annotated | 50,638 | 50.28% |
| KEGG-Annotated | 55,276 | 54.89% |
| COG-Annotated | 32,641 | 32.41% |
| Interpro-Annotated | 59,058 | 58.64% |
| GO-Annotated | 29,473 | 29.27% |
| Overall | 77,877 | 77.33% |
| Total | 100,705 | 100% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Tan, Y.; Li, X.; Liu, Z.; Li, F.; Huang, H.; Huang, M. Analysis of Transcriptome and Expression of C4H and FLS Genes on Four Flower Colors of Impatiens uliginosa. Horticulturae 2024, 10, 415. https://doi.org/10.3390/horticulturae10040415
Zhang X, Tan Y, Li X, Liu Z, Li F, Huang H, Huang M. Analysis of Transcriptome and Expression of C4H and FLS Genes on Four Flower Colors of Impatiens uliginosa. Horticulturae. 2024; 10(4):415. https://doi.org/10.3390/horticulturae10040415
Chicago/Turabian StyleZhang, Xiaoli, Yi Tan, Xinyi Li, Zengdong Liu, Fan Li, Haiquan Huang, and Meijuan Huang. 2024. "Analysis of Transcriptome and Expression of C4H and FLS Genes on Four Flower Colors of Impatiens uliginosa" Horticulturae 10, no. 4: 415. https://doi.org/10.3390/horticulturae10040415
APA StyleZhang, X., Tan, Y., Li, X., Liu, Z., Li, F., Huang, H., & Huang, M. (2024). Analysis of Transcriptome and Expression of C4H and FLS Genes on Four Flower Colors of Impatiens uliginosa. Horticulturae, 10(4), 415. https://doi.org/10.3390/horticulturae10040415