
Genome-Wide Analysis of SPL Gene Family and Functional Identification of JrSPL02 Gene in the Early Flowering of Walnut

Abstract
1. Introduction
2. Results
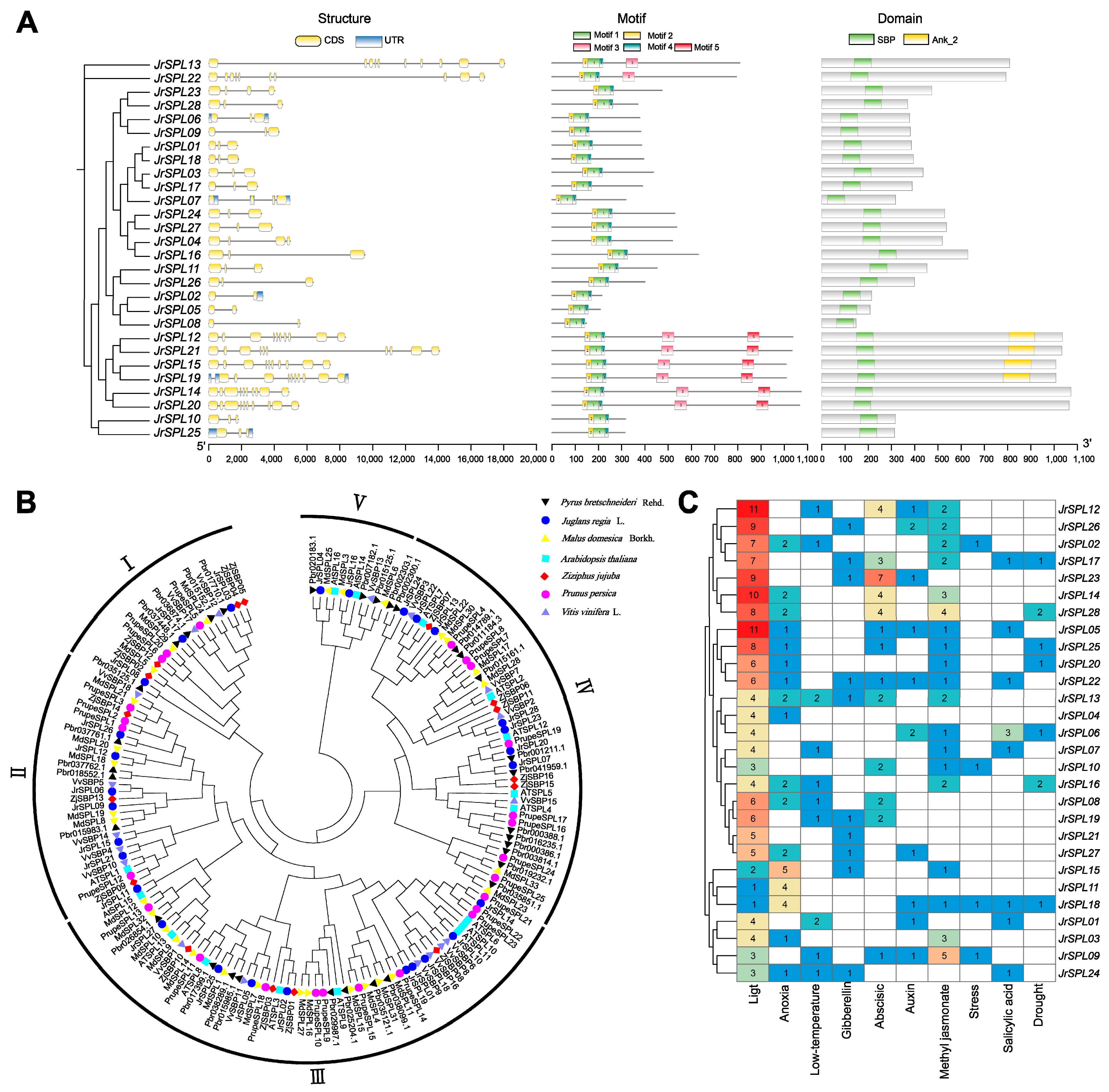
2.1. Identification of Walnut SPL Genes
2.2. Phylogenetic Tree Analysis of Walnut JrSPL Gene Family Members
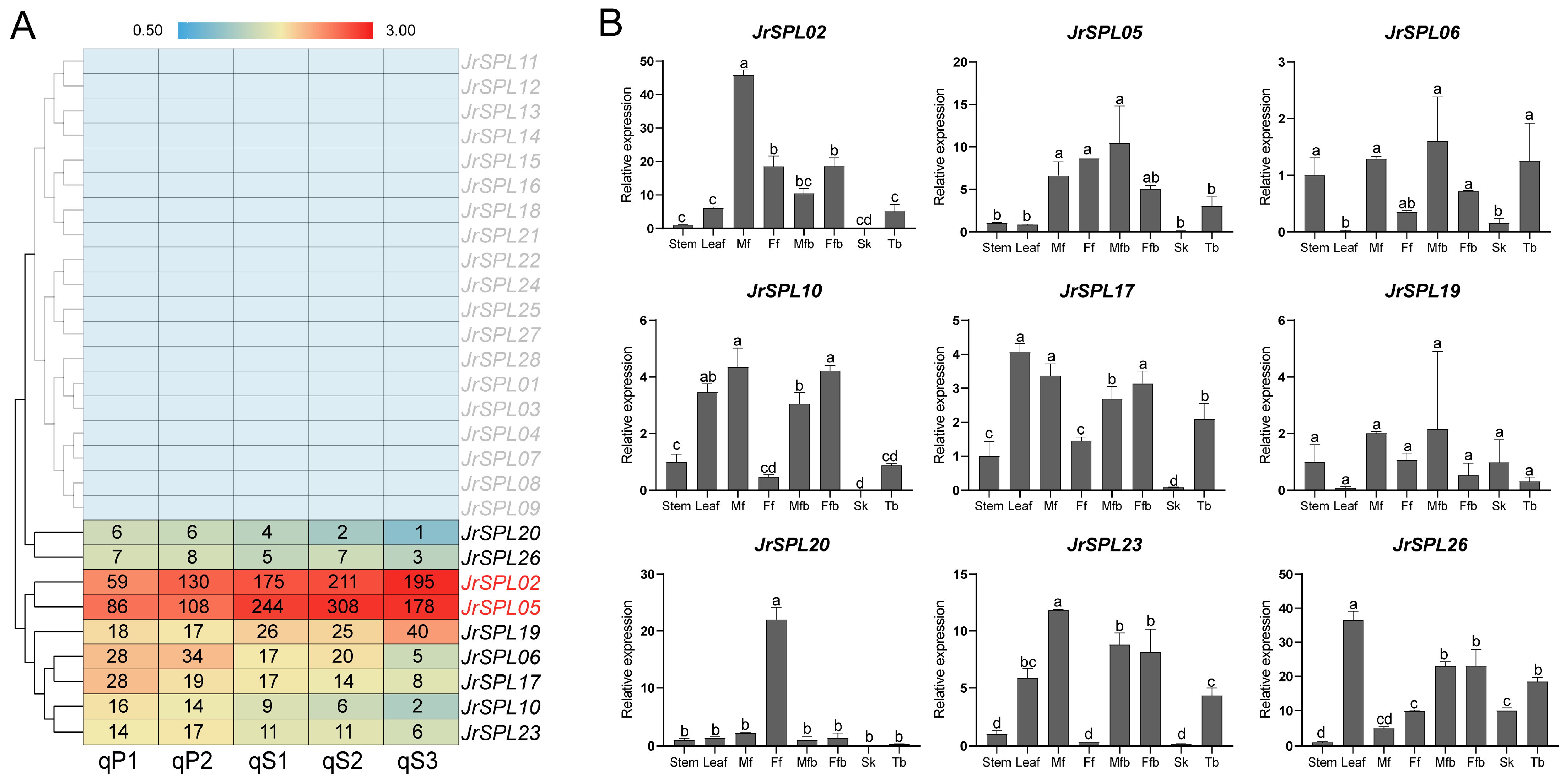
2.3. Expression Pattern Analysis of Walnut JrSPL Genes in Different Tissues
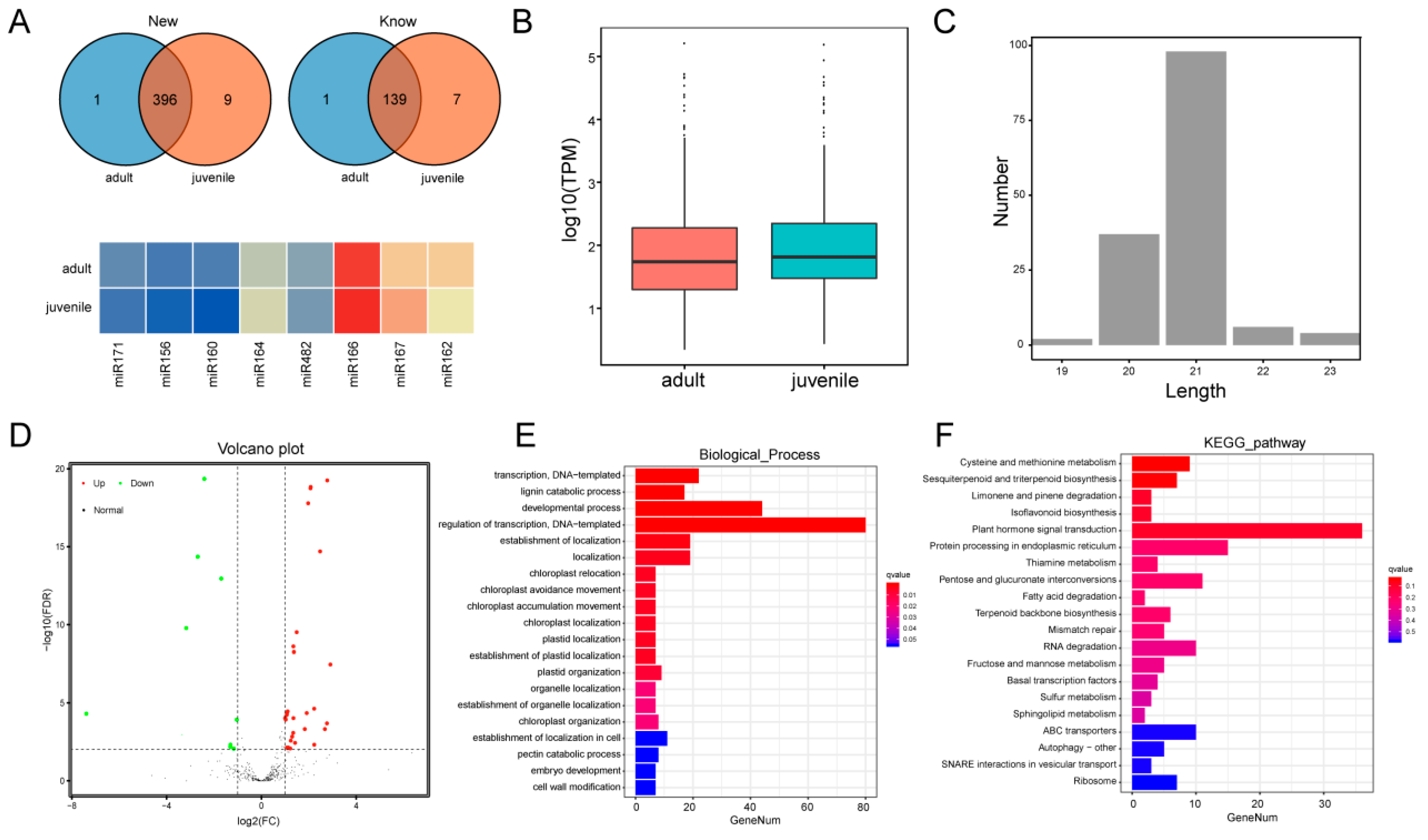
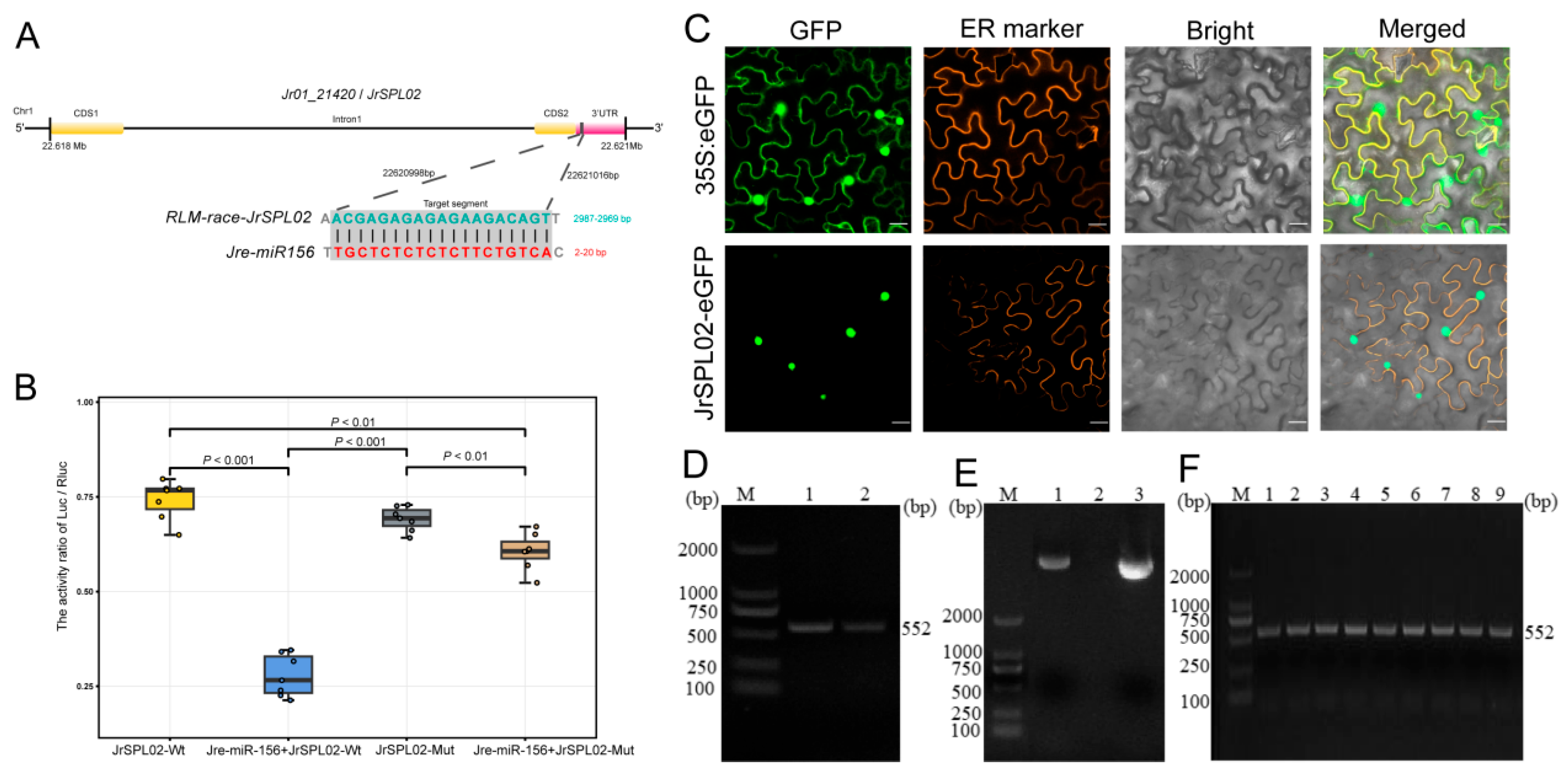
2.4. Walnut miRNA Sequencing and Target Gene Prediction
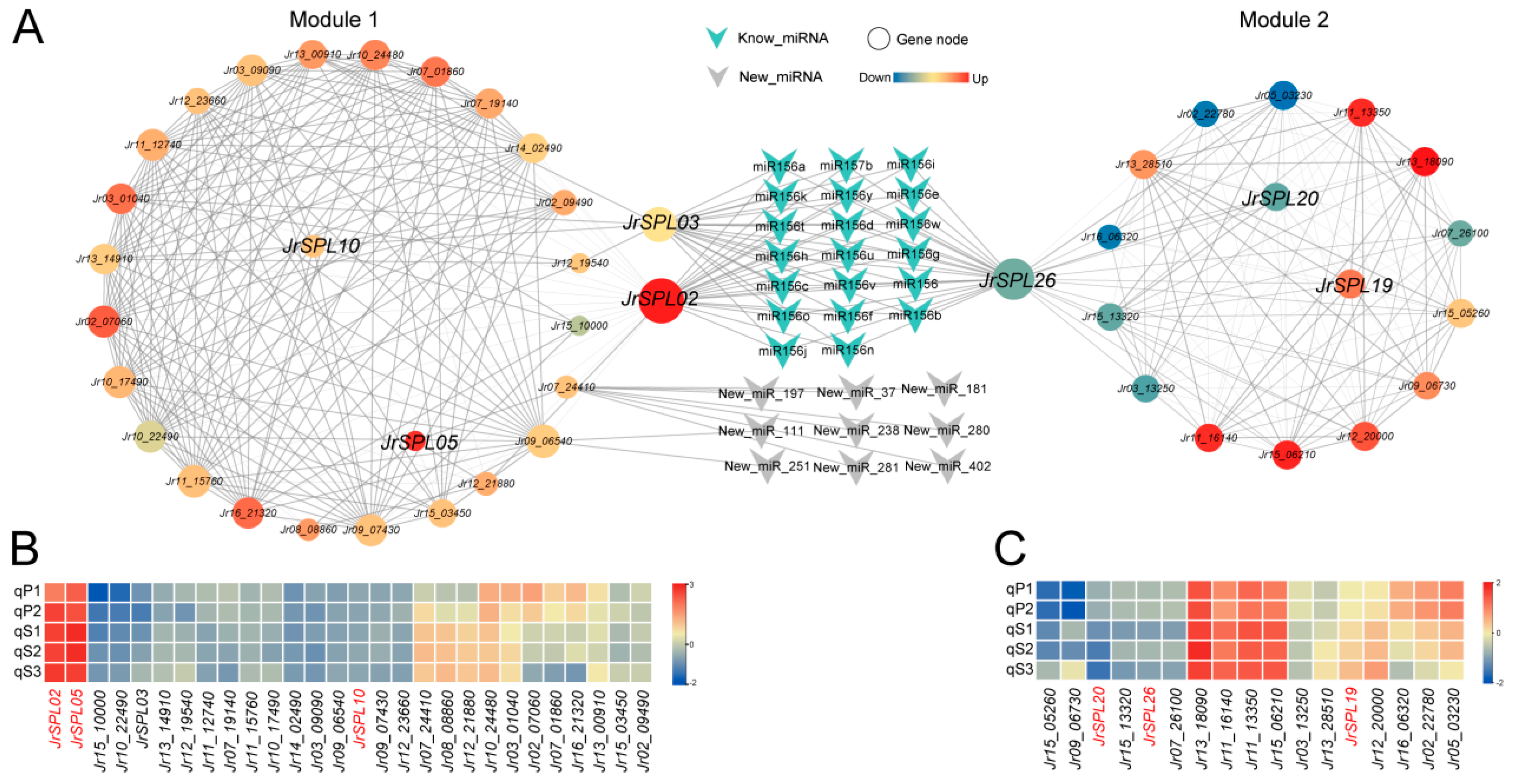
2.5. Co-Expression Network Construction
2.6. Gene Cloning and Interaction Verification
2.7. Subcellular Localization of JrSPL02 Protein in Walnut
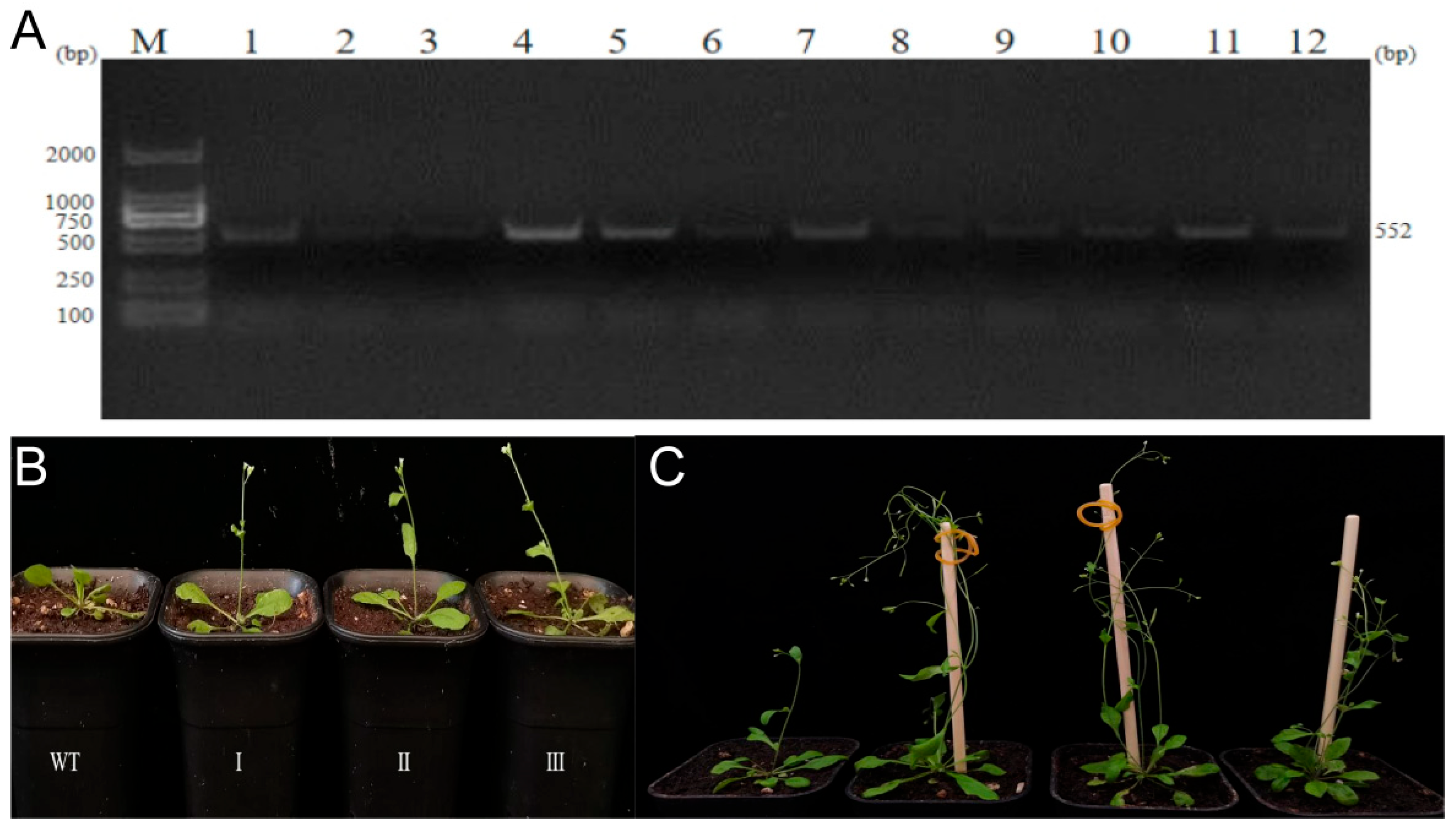
2.8. Phenotype Identification of JrSPL02-3301 Transgenic Arabidopsis
3. Discussion
4. Materials and Methods
4.1. Walnut Materials
4.2. Identification of Walnut SPL Genes
4.3. Characterization of SPL Genes
4.4. Fluorescent Real-Time Quantitative PCR
4.5. Small RNA Sequencing and Co-Expression Network Construction
4.6. PPM-RLM-5′RACE
4.7. Dual Fluorescence Reporter Gene Experiment
4.8. Subcellular Localization
4.9. Arabidopsis Genetic Transformation and Phenotype Identification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, H.; Lu, Z.; Xu, Y.; Kong, L.; Han, L. Genome-wide characterization of SPL family in Medicago truncatula reveals the novel roles of miR156/SPL module in spiky pod development. BMC Genom. 2019, 20, 552. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Poethig, R.S. Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development 2006, 133, 3539–3547. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Schwab, R.; Czech, B.; Mica, E.; Weigel, D. Dual effects of miR156-targeted SPL genes and CYP78A5/KLUH on plastochron length and organ size in Arabidopsis thaliana. Plant Cell 2008, 20, 1231–1243. [Google Scholar] [CrossRef] [PubMed]
- Gou, J.Y.; Felippes, F.F.; Liu, C.J.; Weigel, D.; Wang, J.W. Negative Regulation of Anthocyanin Biosynthesis in Arabidopsis by a miR156-Targeted SPL Transcription Factor. Plant Cell 2011, 23, 1512–1522. [Google Scholar] [CrossRef] [PubMed]
- Wellmer, F.; Riechmann, J.L. Gene networks controlling the initiation of flower development. Trends Genet. 2010, 26, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Gandikota, M.; Birkenbihl, R.P.; Höhmann, S.; Cardon, G.H.; Saedler, H.; Huijser, P. The miRNA156/157 recognition element in the 3′ UTR of the Arabidopsis SBP box gene SPL3 prevents early flowering by translational inhibition in seedlings. Plant J. 2007, 49, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Seo, P.J.; Kang, S.K.; Park, C.M. miR172 signals are incorporated into the miR156 signaling pathway at the SPL3/4/5 genes in Arabidopsis developmental transitions. Plant Mol. Biol. 2011, 6, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hou, H.; Li, X.; Xiang, J.; Yin, X.J.; Gao, H.; Zheng, Y.; Bassett, C.L.; Wang, X.P. Genome-wide identification and analysis of the SBP-box family genes in apple (Malus × domestica Borkh.). Plant Physiol. Biochem. 2013, 70, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Ni, J.; Niu, Q.; Bai, S.; Bao, L.; Li, J.; Sun, Y.; Dong, Z.; Teng, Y. Response of miR156-SPL module during the red peel coloration of bagging-treated Chinese sand pear (Pyrus pyrifolia Nakai). Front. Physiol. 2017, 8, 550. [Google Scholar] [CrossRef]
- Wu, X.M.; Liu, M.Y.; Ge, X.X.; Xu, Q.; Guo, W.W. Stage and tissue-specific modulation of ten conserved miRNAs and their targets during somatic embryogenesis of Valencia sweet orange. Planta 2011, 233, 495–505. [Google Scholar] [CrossRef]
- Jiang, Y.; Peng, J.; Wang, M.; Su, W.; Gao, Y. The role of EjSPL3, EjSPL4, EjSPL5, and EjSPL9 in regulating flowering in loquat (Eriobotrya japonica Lindl.). Int. J. Mol. Sci. 2019, 21, 248. [Google Scholar] [CrossRef]
- Cui, M.; Wang, C.; Zhang, W.; Pervaiz, T.; Haider, M.S.; Tang, W.; Fang, J.G. Characterization of Vv-miR156: Vv-SPL pairs involved in the modulation of grape berry development and ripening. Mol. Genet. Genom. 2018, 293, 1333–1354. [Google Scholar] [CrossRef]
- Li, H.; Ma, B.; Luo, Y.; Wei, W.; Yuan, J.; Zhai, C.; He, N. The Morus SPL gene family and the response of MnSPL7 to silkworm herbivory through activating the transcription of MnTT2L2 in the catechin biosynthesis pathway. Int. J. Mol. Sci. 2022, 23, 1141. [Google Scholar] [CrossRef]
- Klein, J.; Saedler, H.; Huijser, P. A new family of DNA binding proteins includes putative transcriptional regulators of the Antirrhinum majus floral meristem identity gene SQUAMOSA. Mol. Gen. Genet. 1996, 250, 7–16. [Google Scholar]
- Cardon, G.H.; Höhmann, S.; Nettesheim, K.; Saedler, H.; Huijser, P. Functional analysis of the Arabidopsis thaliana SBP-box gene SPL3: A novel gene involved in the floral transition. Plant J. 1997, 12, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Lännenpää, M.; Jänonen, I.; Hölttä-Vuori, M.; Gardemeister, M.M.; Sopanen, T. A new SBP-box gene BpSPL1 in silver birch (Betula pendula). Physiol. Plant. 2010, 120, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Wu, M.F.; Yang, L.; Wu, G.; Poethig, R.S.; Wagner, D. The microRNA-regulated SBP-box transcription factor SPL3 is a direct upstream activator of LEAFY, FRUITFULL, and APETALA1. Dev. Cell 2009, 17, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.W.; Song, X.M.; Duan, W.K.; Wang, Y.; Hou, X.L. Genome-wide analysis of the SBP-box gene family in Chinese cabbage (Brassica rapa subsp. pekinensis). Genome 2015, 58, 463–477. [Google Scholar] [CrossRef]
- Bergonzi, S.; Al, E. Mechanisms of Age-Dependent Response to Winter Temperature in Perennial Flowering of Arabis alpina. Science 2013, 340, 1094–1097. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Xu, Y.; Guo, C.; Zheng, J.R.; Zhou, B.Y.; Zhang, Y.; Lu, D.Y.; Zhu, Z.J.; Wang, H.S.; Wu, G. Modulation of miR156 to identify traits associated with vegetative phase change in tobacco (Nicotiana tabacum). J. Exp. Bot. 2016, 67, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Solar, A.; Ivani, A.; Stampar, F.; Hudina, M. Genetic resources for walnut (Juglans regia L.) improvement in Slovenia: Evaluation of the largest collection of local genotypes. Genet. Resour. Crop Evol. 2002, 49, 491–501. [Google Scholar] [CrossRef]
- Jin, Q.; Mo, R.; Chen, W.; Zhang, Q.; Sheng, F.; Wu, C.; Zhang, R.; Luo, Z. Identification and Comparative Analysis of Genes and MicroRNAs Involved in the Floral Transition of the Xinjiang Early-Flowering Walnut (Juglans regia L.). Horticulturae 2022, 8, 136. [Google Scholar] [CrossRef]
- Song, M.; Li, A.; Sun, L.; Mei, Y.; Wang, Z.; Wang, R.; Wang, R.; Li, D.; Song, J.; Zhang, C.; et al. The miR156x + p/SPL13-6 module responds to ABA, IAA, and ethylene, and SPL13-6 participates in the juvenile–adult phase transition in Pyrus. Hortic. Environ. Biotechnol. 2023, 64, 437–448. [Google Scholar] [CrossRef]
- Ahsan, M.U.; Hayward, A.; Irihimovitch, V.; Fletcher, S.; Tanurdzic, M.; Pocock, A.; Beveridge, C.A.; Mitter, N. Juvenility and Vegetative Phase Transition in Tropical/Subtropical Tree Crops. Front. Plant Sci. 2019, 10, 729. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Pan, H.; Wang, J.; Yang, W.; Cheng, T.; Zhang, Q. Identification and profiling of novel and conserved microRNAs during the flower opening process in Prunus mume via deep sequencing. Mol. Genet. Genom. 2014, 289, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Pan, X.; Jing, Y.; Zhao, Y.; Duan, Y.; Yang, J.; Wang, B.; Liu, Y.; Shen, R.; Cao, Y.; et al. ZmSPL10/14/26 are required for epidermal hair cell fate specification on maize leaf. New Phytol. 2021, 230, 1533–1549. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Liu, W.; Li, W.; Zhao, L.; Chen, G.; Bai, Y.; Ma, D.; Fu, C.; Wang, Y.; Zhang, X. Downregulation of miR156-targeted PvSPL6 in switchgrass delays flowering and increases biomass yield. Front. Plant Sci. 2022, 13, 834431. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Zhou, J.; Liu, S.; Gan, Z.; Zhang, J.; Hu, C. Genome-wide identification and characterization of SQUAMOSA-promoter binding protein (SBP) genes involved in the floral development of Citrus Clementina. Biomolecules 2019, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Zhao, Y.; Xie, Y.; Wang, H. Exploiting SPL genes to improve maize plant architecture tailored for high-density planting. J. Exp. Bot. 2018, 69, 4675–4688. [Google Scholar] [CrossRef]
- Gou, J.; Tang, C.; Chen, N.; Hui, W.; Wang, Z.Y. SPL7 and SPL8 represent a novel flowering regulation mechanism in switchgrass. New Phytol. 2019, 222, 1610–1623. [Google Scholar] [CrossRef]
- Ma, Z.B.; Li, W.; Wang, H.P.; Yu, D.Q. WRKY transcription factors WRKY12 and WRKY13 interact with SPL10 to modulate age-mediated flowering. J. Integr. Plant Biol. 2020, 62, 21–35. [Google Scholar] [CrossRef]
- Xie, Y.R.; Zhou, Q.; Zhao, Y.P.; Li, Q.Q.; Liu, Y.; Ma, M.D.; Wang, B.B.; Shen, R.X.; Zheng, Z.G.; Wang, H.Y. FHY3 and FAR1 Integrate Light Signals with the miR156-SPL Module-Mediated Aging Pathway to Regulate Arabidopsis Flowering. Mol. Plant 2020, 13, 483–498. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Zhang, S.F.; Chen, B.; Liu, L.; Wu, F.; Li, J.; Zhang, M.; Liu, B.G.; Liu, G.F. Genome-wide identification and characterization of the SBP-box gene family in Petunia. BMC Genom. 2018, 19, 193. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Lee, H.J.; Ryu, J.Y.; Park, C.M. SPL3/4/5 Integrate Developmental Aging and Photoperiodic Signals into the FT-FD Module in Arabidopsis Flowering. Mol. Plant 2016, 9, 1647–1659. [Google Scholar] [CrossRef]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.W.; Weigel, D.; Poethig, R.S. The Sequential Action of miR156 and miR172 Regulates Developmental Timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef]
- Yue, E.; Tao, H.; Jianhong, X.U. Genome-wide analysis of microRNA156 and its targets, the genes encoding SQUAMOSA promoter-binding protein-like (SPL) transcription factors, in the grass family Poaceae. J. Zhejiang Univ.-Sci. B 2021, 22, 366–382. [Google Scholar] [CrossRef]
- Wang, H.; Wang, H. The miR156/SPL module, a regulatory hub and versatile toolbox, gears up crops for enhanced agronomic traits. Mol. Plant 2015, 8, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jin, J.; He, Y.; Lu, B.; Li, D.; Chai, W.; Khan, A.; Gong, Z. Genome-wide identification and analysis of the SBP-box family genes under Phytophthora capsici stress in pepper (Capsicum annuum L.). Front. Plant Sci. 2016, 7, 504. [Google Scholar] [CrossRef]
- Li, J.; Gao, X.; Sang, S.; Liu, C. Genome-wide identification, phylogeny, and expression analysis of the SBP-box gene family in Euphorbiaceae. BMC Genom. 2019, 20, 912. [Google Scholar] [CrossRef]
- Liu, N.; Sun, W.; Ma, Z.; Huang, L.; Chen, H. Genome-wide identification of the SPL gene family in Tartary buckwheat (Fagopyrum tataricum) and expression analysis during fruit development stages. BMC Plant Biol. 2019, 19, 299. [Google Scholar] [CrossRef]
- Zuo, X.; Zhang, D.; Wang, S.; Xing, L.; Li, Y.; Fan, S. Expression of genes in the potential regulatory pathways controlling alternate bearing in ‘fuji’ (Malus domestica borkh.) apple trees during flower induction. Plant Physiol. Biochem. 2018, 132, 579–589. [Google Scholar] [CrossRef]
- Hou, H.; Li, J.; Gao, M.; Singer, S.D.; Wang, H.; Mao, L.; Fei, Z.; Wang, X. Genomic organization, phylogenetic comparison and differential expression of the SBP-box family genes in grape. PLoS ONE 2013, 8, e59358. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Wang, Q.; Ma, J.; He, Q.; Zhang, B. Differentiated expression of microRNAs may regulate genotype-dependent traits in cotton. Gene 2014, 547, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, J.; Wang, C.; Shen, W.; Jia, H.; Zhu, X.; Li, X. Study on expression modes and cleavage role of miR156b/c/d and its target gene Vv-SPL9 during the whole growth stage of grapevine. J. Hered. 2016, 107, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Unte, U.S.; Sorensen, A.M.; Pesaresi, P.; Gandikota, M.; Leister, D.; Saedler, H.; Huijser, P. SPL8, an SBP-box gene that affects pollen sac development in Arabidopsis. Plant Cell 2003, 15, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Chuck, G.; Whipple, C.; Jackson, D.; Hake, S. The maize SBP-box transcription factor encoded by tasselsheath4 regulates bract development and the establishment of meristem boundaries. Development 2010, 137, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Luo, X.; Han, L.; Zhao, Y.; Mamat, A.; Li, N.; Mei, C.; Yan, P.; Zhang, R.; Hu, J.; et al. Transcriptome profiling based on Illumina- and SMRT-based RNA-seq reveals circadian regulation of key pathways in flower bud development in walnut. PLoS ONE 2021, 16, e0260017. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Quan, S.; Ma, L.; Xu, H.; Yang, J.; Niu, J. Molecular characterization of SBP-box gene family during floral induction in walnut (Juglans regia L.). Tree Genet. Genomes 2020, 16, 12. [Google Scholar] [CrossRef]
- Ma, K.; Xu, R.; Zhao, Y.; Han, L.; Xu, Y.; Li, L.; Wang, J.; Li, N. Walnut N-acetylserotonin methyltransferase gene family genome-wide identification and diverse functions characterization during flower bud development. Front. Plant Sci. 2022, 13, 861043. [Google Scholar] [CrossRef]
- Jhala, A.J.; Chahal, P.S. Herbicide programs for control of glyphosate-resistant volunteer corn in glufosinate-resistant soybean. Weed Technol. A J. Weed Sci. Soc. Am. 2015, 29, 431–443. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Line | Plant Number | Height/cm | Rosette Leaf Number | Flower Day | Number of Branches | Carob Number |
|---|---|---|---|---|---|---|
| WT | 20 | 17.96 | 12.10 ± 1.34 | 23.13 ± 0.62 | 1.30 ± 0.36 | 1.62 ± 0.01 |
| L1 | 20 | 23.32 | 8.01 ± 1.02 * | 20.61 ± 0.35 * | 2.5 ± 0.65 * | 16 ± 0.27 |
| L2 | 20 | 22.5 | 9.52 ± 0.92 | 20.57 ± 0.19 | 3.57 ± 0.29 | 18 ± 0.31 |
| L3 | 20 | 21.56 | 8.39 ± 1.37 | 20.43 ± 0.38 | 3.29 ± 0.76 | 15 ± 0.18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, K.; Zhao, Y.; Han, L.; Gao, C.; Hu, J.; Guan, P. Genome-Wide Analysis of SPL Gene Family and Functional Identification of JrSPL02 Gene in the Early Flowering of Walnut. Horticulturae 2024, 10, 158. https://doi.org/10.3390/horticulturae10020158
Ma K, Zhao Y, Han L, Gao C, Hu J, Guan P. Genome-Wide Analysis of SPL Gene Family and Functional Identification of JrSPL02 Gene in the Early Flowering of Walnut. Horticulturae. 2024; 10(2):158. https://doi.org/10.3390/horticulturae10020158
Chicago/Turabian StyleMa, Kai, Yu Zhao, Liqun Han, Chaoyuan Gao, Jianfang Hu, and Pingyin Guan. 2024. "Genome-Wide Analysis of SPL Gene Family and Functional Identification of JrSPL02 Gene in the Early Flowering of Walnut" Horticulturae 10, no. 2: 158. https://doi.org/10.3390/horticulturae10020158
APA StyleMa, K., Zhao, Y., Han, L., Gao, C., Hu, J., & Guan, P. (2024). Genome-Wide Analysis of SPL Gene Family and Functional Identification of JrSPL02 Gene in the Early Flowering of Walnut. Horticulturae, 10(2), 158. https://doi.org/10.3390/horticulturae10020158