
A Combined mRNA and microRNA Transcriptome Analysis of B. oleracea Response to Plasmodiophora brassicae Infection

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction and Sequencing
2.3. mRNA and miRNA Sequence Alignment and Annotation
2.4. Functional Analysis of Differentially Expressed Unigenes
2.5. Identification and Classification of NBS-LRR Gene Family in Cabbage
2.6. Co-Expression Networks
2.7. miRNA Target Prediction
2.8. Quantitative Real-Time PCR Validation
3. Results
3.1. RNA Sequencing and Differentially Expressed Genes (DEGs)
3.2. GO and KEGG Pathway Enrichment Analyses
3.3. The Expression of DEGs in Important Signaling Pathways
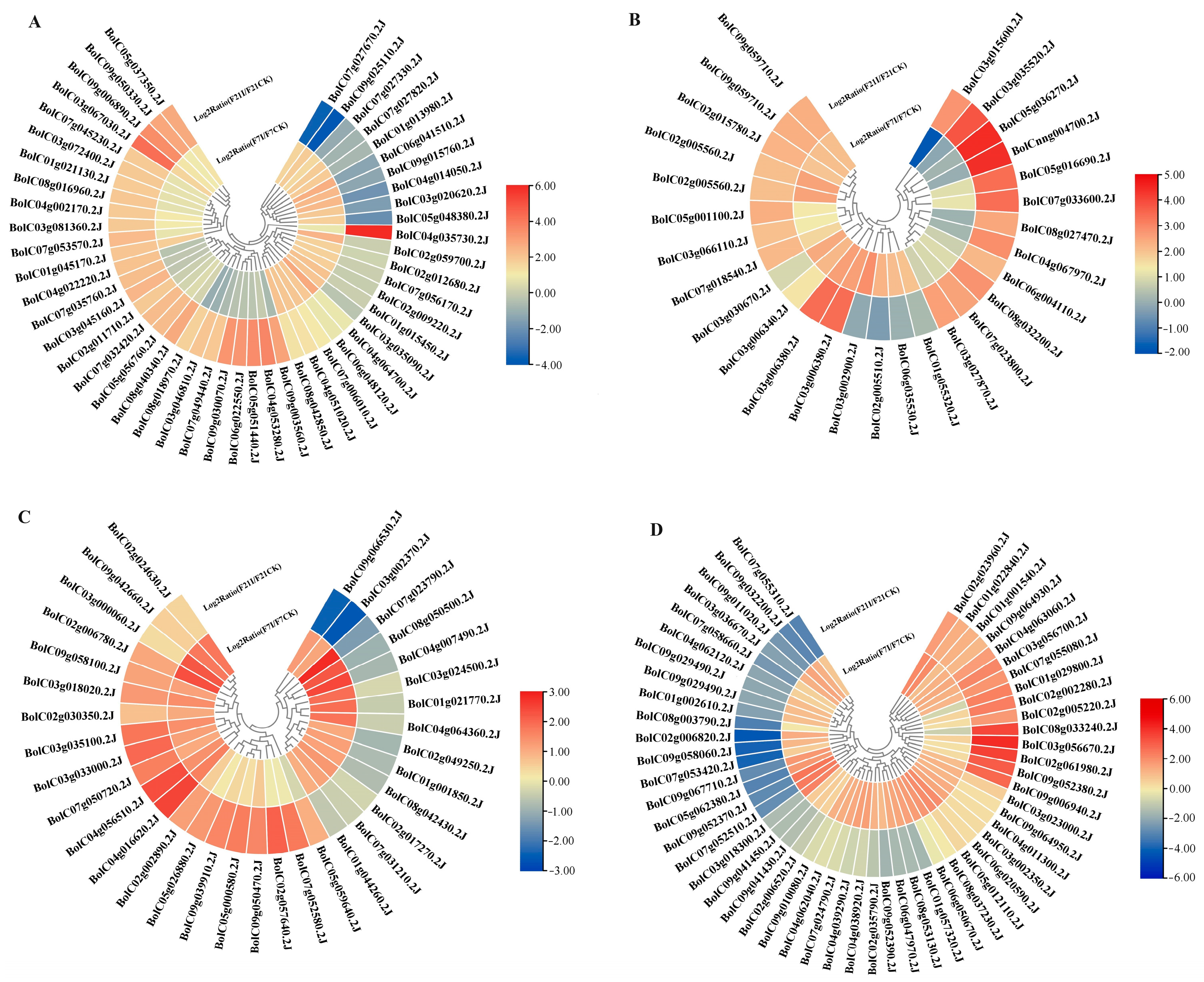
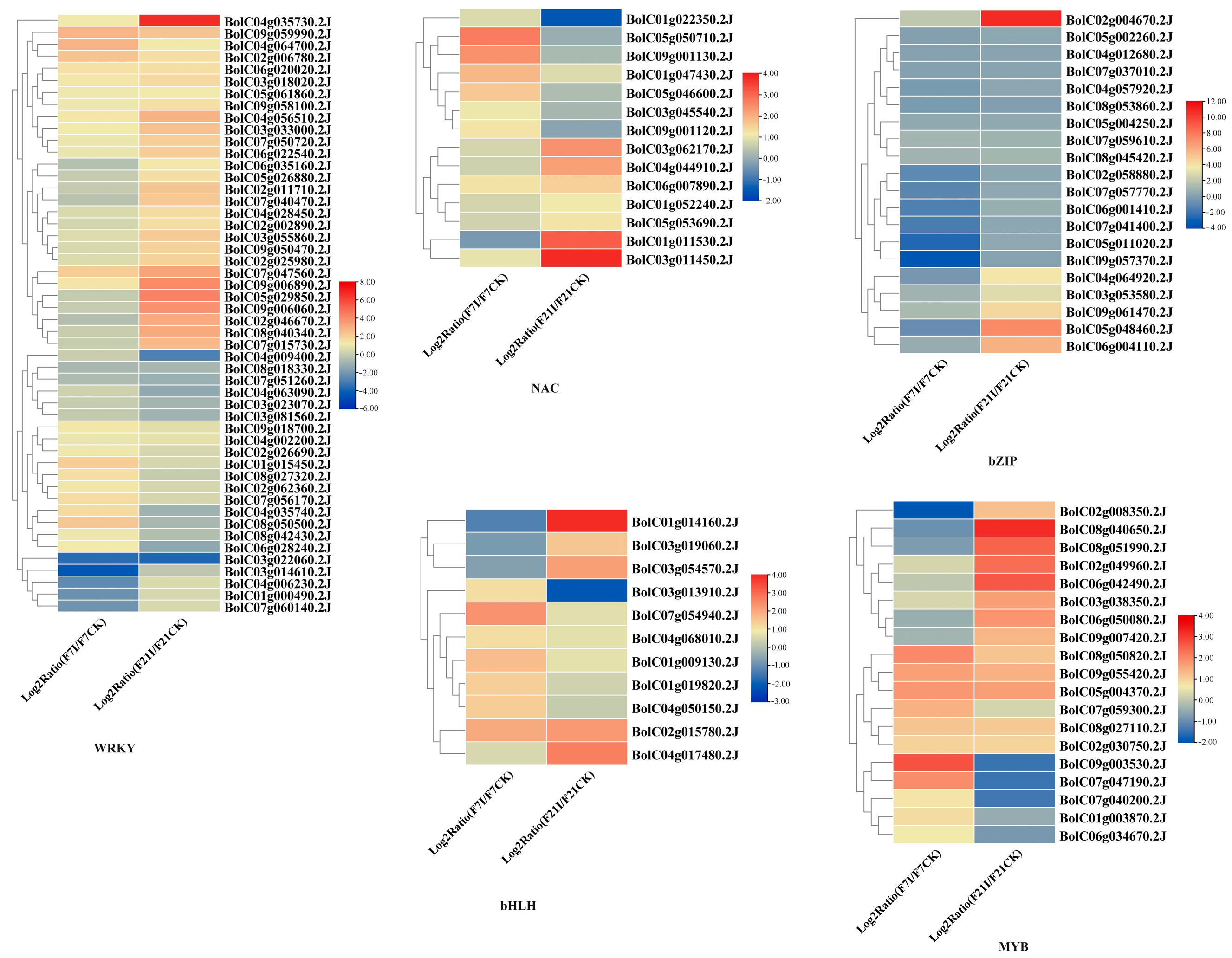
3.4. Expression of NBS-LRR Genes and Transcription Factors
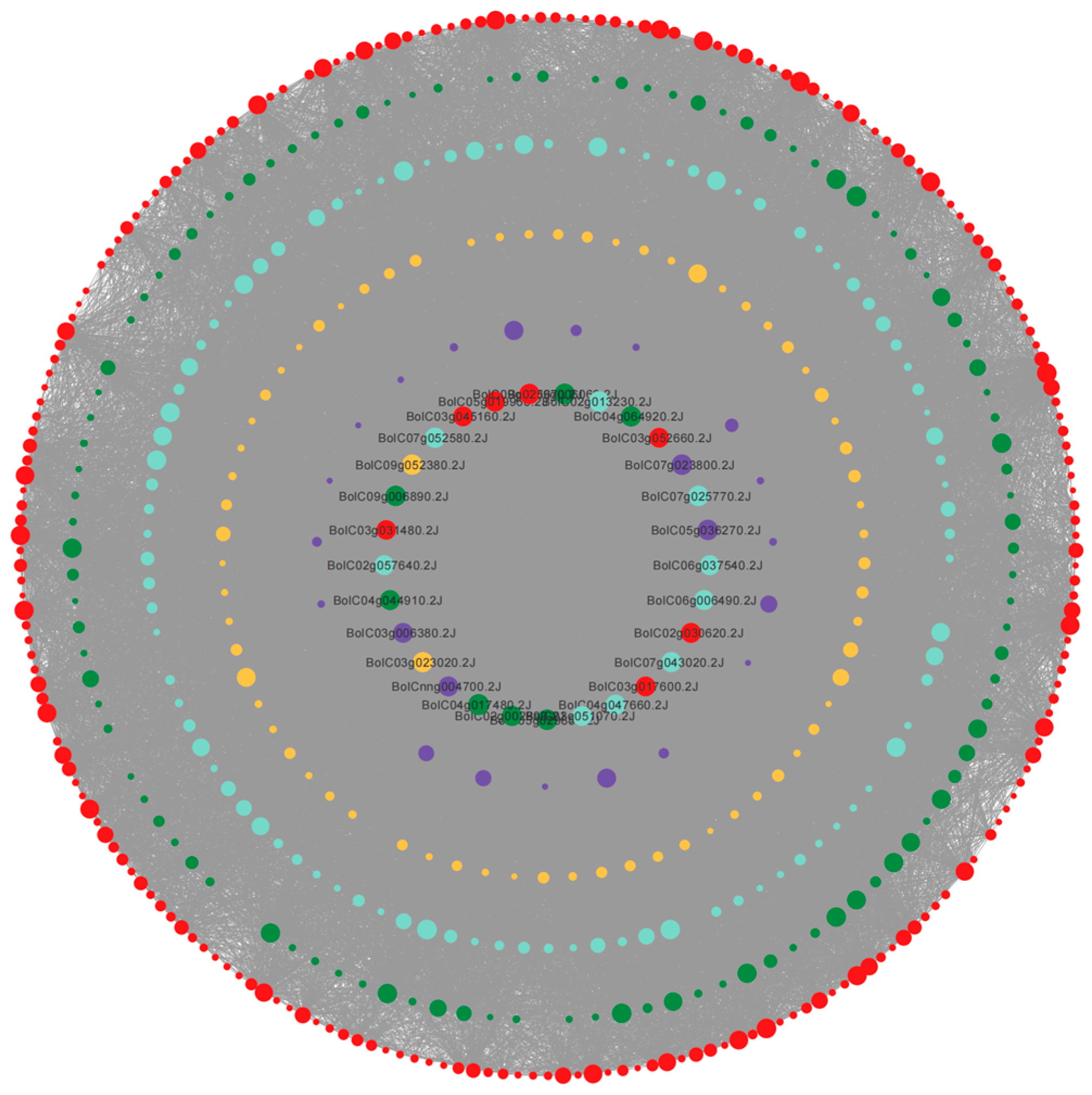
3.5. Co-Expression Network
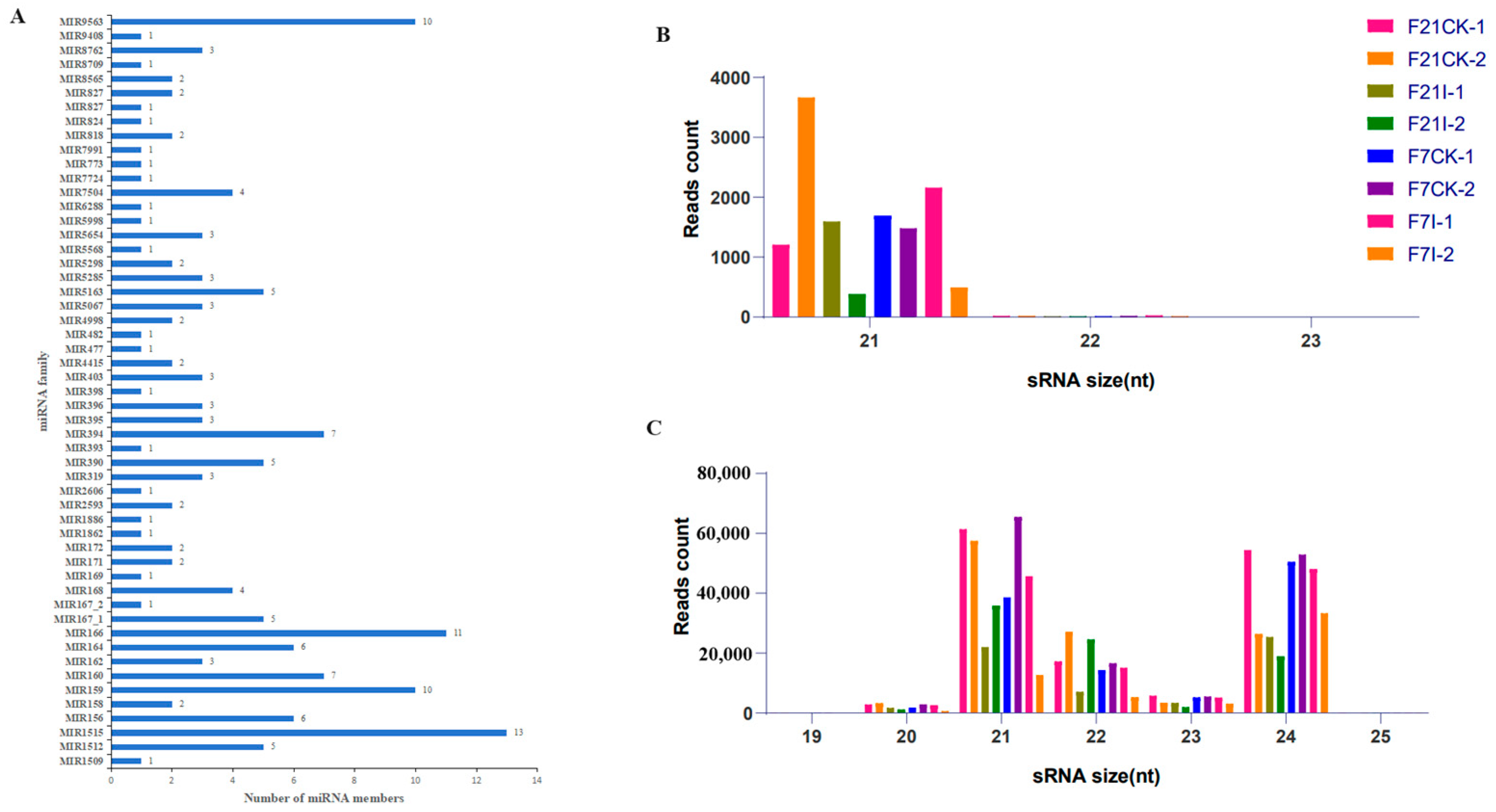
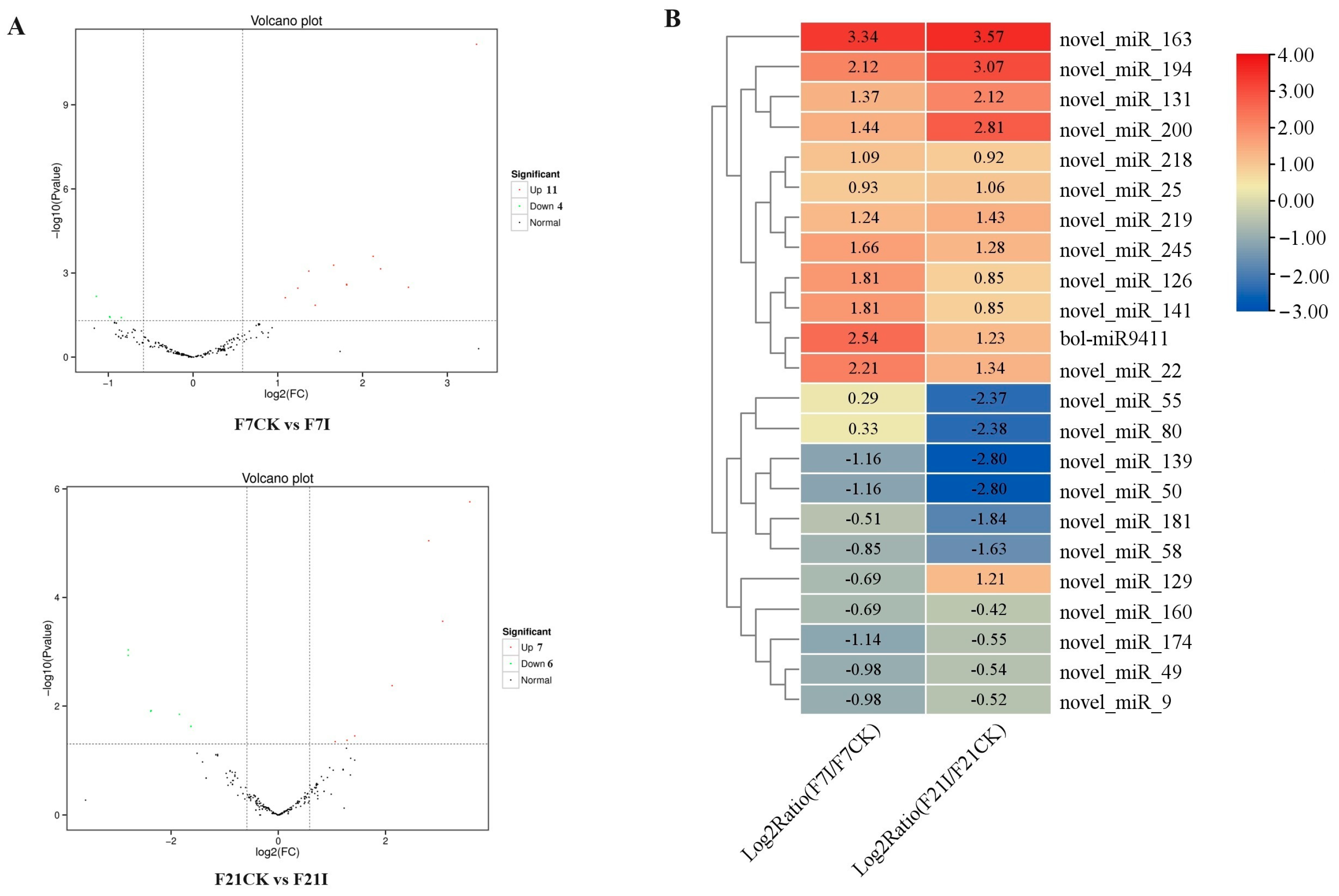
3.6. Identification of miRNAs in Cabbage Roots in Response to P. brassicae
3.7. Identification of miRNA-mRNA Pairs Associated with P. brassicae Infection
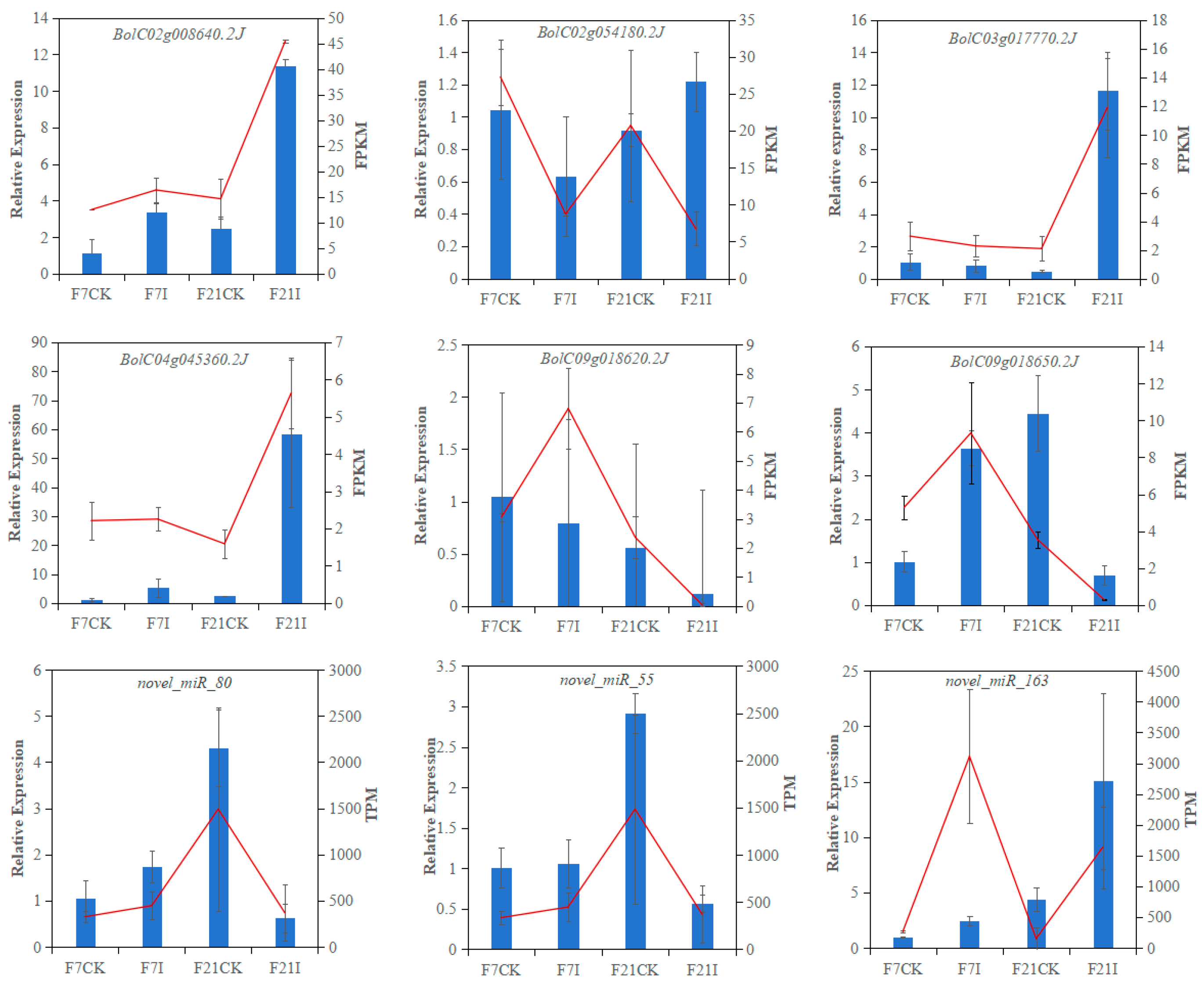
3.8. Validation of the DEGs and DEMs Data Using RT-qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Javed, M.A.; Schwelm, A.; Zamani-Noor, N.; Salih, R.; Silvestre Vañó, M.; Wu, J.; González García, M.; Heick, T.M.; Luo, C.; Prakash, P.; et al. The clubroot pathogen Plasmodiophora brassicae: A profile update. Mol. Plant Pathol. 2023, 24, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Rocherieux, J.; Glory, P.; Giboulot, A.; Boury, S.; Barbeyron, G.; Thomas, G.; Manzanares-Dauleux, M.J. Isolate-specific and broad-spectrum QTLs are involved in the control of clubroot in Brassica oleracea. Theor. Appl. Genet. 2004, 108, 1555–1563. [Google Scholar] [CrossRef]
- Nagaoka, T.; Doullah, M.A.; Matsumoto, S.; Kawasaki, S.; Ishikawa, T.; Hori, H.; Okazaki, K. Identification of QTLs that control clubroot resistance in Brassica oleracea and comparative analysis of clubroot resistance genes between B. rapa and B. oleracea. Theor. Appl. Genet. 2010, 120, 1335–1346. [Google Scholar] [CrossRef]
- Lee, J.; Izzah, N.K.; Choi, B.S.; Joh, H.J.; Lee, S.C.; Perumal, S.; Seo, J.; Ahn, K.; Jo, E.J.; Choi, G.J.; et al. Genotyping-by-sequencing map permits identification of clubroot resistance QTLs and revision of the reference genome assembly in cabbage (Brassica oleracea L.). DNA Res. 2016, 23, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Ce, F.; Mei, J.; He, H.; Zhao, Y.; Hu, W.; Yu, F.; Li, Q.; Ren, X.; Si, J.; Song, H.; et al. Identification of Candidate Genes for Clubroot-Resistance in Brassica oleracea Using Quantitative Trait Loci-Sequencing. Front. Plant Sci. 2021, 12, 703520. [Google Scholar] [CrossRef] [PubMed]
- Dakouri, A.; Zhang, X.; Peng, G.; Falk, K.C.; Gossen, B.D.; Strelkov, S.E.; Yu, F. Analysis of genome-wide variants through bulked segregant RNA sequencing reveals a major gene for resistance to Plasmodiophora brassicae in Brassica oleracea. Sci. Rep. 2018, 8, 17657. [Google Scholar] [CrossRef]
- Ning, Y.; Wang, Y.; Fang, Z.; Zhuang, M.; Zhang, Y.; Lv, H.; Liu, Y.; Li, Z.; Yang, L. Comparative transcriptome analysis of cabbage (Brassica oleracea var. capitata) infected by Plasmodiophora brassicae reveals drastic defense response at secondary infection stage. Plant Soil 2019, 443, 167–183. [Google Scholar]
- Zhang, X.; Liu, Y.; Fang, Z.; Li, Z.; Yang, L.; Zhuang, M.; Zhang, Y.; Lv, H. Comparative Transcriptome Analysis between Broccoli (Brassica oleracea var. italica) and Wild Cabbage (Brassica macrocarpa Guss.) in Response to Plasmodiophora brassicae during Different Infection Stages. Front. Plant Sci. 2016, 7, 1929. [Google Scholar]
- Li, H.; Sun, X.; Ma, C.; Meng, H.; Chen, Q. Identification and characterization of microRNAs during common buckwheat (Fagopyrum esculentum) seed development. Int. J. Agric. Biol. 2020, 24, 1115–1124. [Google Scholar]
- D’Ario, M.; Griffiths-Jones, S.; Kim, M. Small RNAs: Big impact on plant development. Trends Plant Sci. 2017, 22, 1056–1068. [Google Scholar] [CrossRef]
- Chen, C.H.; Zhong, Y.D.; Yu, F.X.; Xu, M. Deep sequencing identifies miRNAs and their target genes involved in the biosynthesis of terpenoids in Cinnamomum camphora. Ind. Crop. Prod. 2020, 145, 111853. [Google Scholar] [CrossRef]
- Shivaprasad, P.V.; Chen, H.M.; Patel, K.; Bond, D.M.; Santos, B.A.; Baulcombe, D.C. A microRNA superfamily regulates nucleotide binding site-leucine-rich repeats and other mRNAs. Plant Cell 2012, 24, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Gupta, O.P.; Sharma, P.; Gupta, R.K.; Sharma, I. Current status on role of miRNAs during plant-fungus interaction. Physiol. Mol. Plant Pathol. 2014, 85, 1–7. [Google Scholar] [CrossRef]
- Mishra, R.; Mohanty, J.N.; Chand, S.K.; Joshi, R.K. Can-miRn37a mediated suppression of ethylene response factors enhances the resistance of chilli against anthracnose pathogen Colletotrichum truncatum L. Plant Sci. 2018, 267, 135–147. [Google Scholar] [CrossRef]
- Navarro, L.; Dunoyer, P.; Jay, F.; Arnold, B.; Dharmasiri, N.; Estelle, M.; Voinnet, O.; Jones, J.D. A plant miRNA contributes to antibac-terial resistance by repressing auxin signaling. Science 2006, 312, 436–439. [Google Scholar] [CrossRef]
- Robert-Seilaniantz, A.; MacLean, D.; Jikumaru, Y.; Hill, L.; Yamaguchi, S.; Kamiya, Y.; Jones, J.D. The microRNA miR393 re-directs secondary metabolite biosynthesis away from camalexin and towards glucosinolates. Plant J. 2011, 67, 218–231. [Google Scholar] [CrossRef]
- Wong, J.; Gao, L.; Yang, Y.; Zhai, J.; Arikit, S.; Yu, Y.; Duan, S.; Chan, V.; Xiong, Q.; Yan, J.; et al. Roles of small RNAs in soybean defense against Phytophthora sojae infection. Plant J. 2014, 79, 928–940. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Q.; Zhang, J.; Wu, L.; Qi, Y.; Zhou, J.M. Identification of microRNAs involved in pathogen-associated molecular pattern-triggered plant innate immunity. Plant Physiol. 2020, 152, 2222–2231. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, Y.G.; Shi, Y.; Wu, L.; Xu, Y.J.; Huang, F.; Guo, X.Y.; Zhang, Y.; Fan, J.; Zhao, J.Q.; et al. Multiple rice microRNAs are involved in immunity against the blast fungus Magnaporthe oryzae. Plant Physiol. 2014, 164, 1077–1092. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, S.L.; Li, J.L.; Hu, X.H.; Wang, H.; Cao, X.L.; Xu, Y.J.; Zhao, Z.X.; Xiao, Z.Y.; Yang, N.; et al. Osa-miR169 Negatively Regulates Rice Immunity against the Blast Fungus Magnaporthe oryzae. Front. Plant Sci. 2017, 8, 2. [Google Scholar] [CrossRef]
- Wu, J.; Yang, R.; Yang, Z.; Yao, S.; Zhao, S.; Wang, Y.; Li, P.; Song, X.; Jin, L.; Zhou, T.; et al. ROS accumulation and antiviral defence control by microRNA528 in rice. Nat. Plants 2017, 3, 16203. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Hou, Y.; Chen, W.; Wang, S.; Wang, P.; Qu, S. Malus hupehensis miR168 Targets to ARGONAUTE1 and Contributes to the Resistance against Botryosphaeria dothidea Infection by Altering Defense Responses. Plant Cell Physiol. 2017, 58, 1541–1557. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Liao, R.; Zhang, X.; Zhao, Y.; Xie, Z.; Yang, S.; Su, H.; Wang, Z.; Zhang, L.; Tian, B.; et al. Integrative Transcriptome, miRNAs, Degradome, and Phytohormone Analysis of Brassica rapa L. in Response to Plasmodiophora brassicae. Int. J. Mol. Sci. 2023, 24, 2414. [Google Scholar] [CrossRef]
- Yuan, Y.; Qin, L.; Su, H.; Yang, S.; Wei, X.; Wang, Z.; Zhao, Y.; Li, L.; Liu, H.; Tian, B.; et al. Transcriptome and Coexpression Network Analyses Reveal Hub Genes in Chinese Cabbage (Brassica rapa L. ssp. pekinensis) During Different Stages of Plasmodiophora brassicae Infection. Front Plant Sci. 2021, 12, 650252. [Google Scholar]
- Kageyama, K.; Asano, T. Life Cycle of Plasmodiophora brassicae. J. Plant Growth Regul. 2009, 28, 203–211. [Google Scholar] [CrossRef]
- Liu, L.; Qin, L.; Zhou, Z.; Hendriks, W.G.H.M.; Liu, S.; Wei, Y. Refining the Life Cycle of Plasmodiophora brassicae. Phytopathology 2020, 110, 1704–1712. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, S.; Yu, F.; Tang, J.; Shan, X.; Bao, K.; Yu, L.; Wang, H.; Fei, Z.; Li, J.; et al. Genome-wide characterization and expression profiling of SWEET genes in cabbage (Brassica oleracea var. capitata L.) reveal their roles in chilling and clubroot disease responses. BMC Genom. 2019, 20, 93. [Google Scholar]
- Livak, K.J.; Schmittgen, T. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Bari, R.; Jones, J.D. Role of plant hormones in plant defence responses. Plant Mol. Biol. 2009, 69, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhao, C.; Yang, L.; Zhuang, M.; Zhang, Y.; Wang, Y.; Fang, Z.; Lv, H. A time-resolved dual transcriptome analysis reveals the molecular regulating network underlying the compatible/incompatible interactions between cabbage (Brassica oleracea) and Fusarium oxysporum f. sp. conglutinans. Plant Soil 2020, 448, 455–478. [Google Scholar] [CrossRef]
- Wang, H.; Zheng, Y.; Xiao, D.; Li, Y.; Liu, T.; Hou, X. BcWRKY33A Enhances Resistance to Botrytis cinerea via Activating BcMYB51-3 in Non-Heading Chinese Cabbage. Int. J. Mol. Sci. 2022, 23, 8222. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Fang, H.; Chen, Y.; Chen, K.; Li, G.; Gu, S.; Tan, X. Overexpression of BnWRKY33 in oilseed rape enhances resistance to Sclerotinia sclerotiorum. Mol. Plant Pathol. 2014, 15, 677–689. [Google Scholar] [CrossRef]
- Hu, Y.; Dong, Q.; Yu, D. Arabidopsis WRKY46 coordinates with WRKY70 and WRKY53 in basal resistance against pathogen Pseudomonas syringae. Plant Sci. 2012, 185–186, 288–297. [Google Scholar] [CrossRef]
- Yang, X.; Fu, T.; Yu, R.; Zhang, L.; Yang, Y.; Xiao, D.; Wang, Y.; Wang, Y.; Wang, Y. miR159a modulates poplar resistance against different fungi and bacteria. Plant Physiol. Biochem. 2023, 201, 107899. [Google Scholar] [CrossRef]
- Zhang, T.; Zhao, Y.L.; Zhao, J.H.; Wang, S.; Jin, Y.; Chen, Z.Q.; Fang, Y.Y.; Hua, C.L.; Ding, S.W.; Guo, H.S. Cotton plants export microRNAs to inhibit virulence gene expression in a fungal pathogen. Nat. Plants 2016, 2, 16153. [Google Scholar] [CrossRef]
- Caruana, J.C.; Dhar, N.; Raina, R. Overexpressio of Arabidopsis microRNA167 induces salicylic acid-dependent defense against Pseudomonas syringae through the regulation of its targets ARF6 and ARF8. Plant Direct 2020, 4, e00270. [Google Scholar] [CrossRef]
- Hu, G.; Ge, X.; Wang, P.; Chen, A.; Li, F.; Wu, J. The cotton miR171a-SCL6 module mediates plant resistance through regulating GhPR1 expression. Plant Physiol. Biochem. 2023, 202, 107995. [Google Scholar] [CrossRef]
- Masri, R.; Kiss, E. The role of NAC genes in response to biotic stresses in plants. Physiol. Mol. Plant Pathol. 2023, 126, 102034. [Google Scholar] [CrossRef]
- Lee, M.H.; Jeon, H.S.; Kim, H.G.; Park, O.K. An Arabidopsis NAC transcription factor NAC4 promotes pathogen-induced cell death under negative regulation by microRNA164. New Phytol. 2017, 214, 343–360. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Lei, Y.; Liu, J.; Hao, M.; Zhang, Z.; Tang, Y.; Chen, A.; Wu, J. The ghr-miR164 and GhNAC100 modulate cotton plant resistance against Verticillium dahlia. Plant Sci. 2020, 293, 110438. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xia, Y.; Lin, S.; Wang, Y.; Guo, B.; Song, X.; Ding, S.; Zheng, L.; Feng, R.; Chen, S.; et al. Osa-miR164a targets OsNAC60 and negatively regulates rice immunity against the blast fungus Magnaporthe oryzae. Plant J. 2018, 95, 584–597. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Tong, T.; Li, X.; Chen, Q.; Dai, M.; Niu, F.; Yang, M.; Deyholos, M.K.; Yang, B.; Jiang, Y.Q. A Novel NAC-Type Transcription Factor, NAC87, from Oilseed Rape Modulates Reactive Oxygen Species Accumulation and Cell Death. Plant Cell Physiol. 2018, 59, 290–303. [Google Scholar] [CrossRef]
- Dong, W.; Ren, W.; Wang, X.; Mao, Y.; He, Y. MicroRNA319a regulates plant resistance to Sclerotinia stem rot. J. Exp. Bot. 2021, 72, 3540–3553. [Google Scholar] [CrossRef]
- Cho, H.; Lee, J.; Oh, E. Leucine-Rich Repeat Receptor-Like Proteins in Plants: Structure, Function, and Signaling. J. Plant Biol. 2023, 66, 99–107. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Family | Gene ID | Gene Annotation | F7CK VS F7I | F21CK VS F21I | ||
|---|---|---|---|---|---|---|---|
| Log2FC of | Log2FC of | Log2FC of | Log2FC of | ||||
| miRNA | mRNA | miRNA | mRNA | ||||
| novel_miR_58 | MIR171_1 | BolC08g051460.2J | SacI homology domain protein | −0.846 | 1.444 | −1.629 | 1.349 |
| novel_miR_80 | MIR167 | BolC05g028200.2J | Auxin response factor | 0.325 | −0.526 | −2.379 | −0.792 |
| novel_miR_160 MIR164 | BolC04g044910.2J | NAC domain protein | −0.687 | 0.678 | −0.418 | 2.205 | |
| BolC05g053690.2J | NAC domain protein | −0.687 | 0.693 | −0.418 | 1.256 | ||
| BolC02g003130.2J | NAC domain protein | −0.687 | 0.207 | −0.418 | −1.162 | ||
| BolC02g060420.2J | NAC domain protein | −0.687 | −0.326 | −0.418 | −3.979 | ||
| BolC03g086720.2J | NAC domain protein | −0.687 | 0.696 | −0.418 | −0.902 | ||
| novel_miR_129 MIR319 | BolC04g018790.2J | TCP transcription factor | −0.687 | 0.616 | 1.209 | −2.123 | |
| BolC03g071340.2J | TCP transcription factor | −0.687 | 0.338 | 1.209 | −1.537 | ||
| BolC05g027880.2J | TCP transcription factor | −0.687 | 0.025 | 1.209 | −2.025 | ||
| novel_miR_245 | MIR818 | BolC09g018620.2J | Leucine-rich receptor protein kinase | 1.659 | 1.242 | 1.282 | −4.861 |
| BolC09g018650.2J | Leucine-rich receptor kinase | 1.659 | 0.931 | 1.282 | −3.723 | ||
| novel_miR_181 | BolC02g054180.2J | SANT/Myb-like domain protein | −0.506 | −1.395 | −1.844 | −1.78 | |
| BolC03g017770.2J | SBP domain protein | −0.506 | −0.154 | −1.844 | 2.11 | ||
| BolC03g074680.2J | NB-ARC domain protein | −0.506 | 0.253 | −1.844 | −1.433 | ||
| BolC05g014870.2J | CASP-like protein | −0.506 | 0.702 | −1.844 | 2.144 | ||
| BolC07g025060.2J | SBP domain protein | −0.506 | −0.289 | −1.844 | −1.274 | ||
| BolC08g008660.2J | Leucine-rich repeats protein | −0.506 | 0.147 | −1.844 | −1.179 | ||
| BolC09g009060.2J | Ribosomal protein S28e | −0.506 | 0.031 | −1.844 | 1.076 | ||
| novel_miR_139 | BolC02g008640.2J | NAC domain protein | −1.165 | 0.536 | −2.8 | 1.278 | |
| BolC03g017770.2J | SBP domain protein | −1.165 | −0.154 | −2.8 | 2.11 | ||
| BolC04g045360.2J | Cation efflux family | −1.165 | 0.195 | −2.8 | 1.455 | ||
| BolC05g024810.2J | SBP domain protein | −1.165 | −1.369 | −2.8 | 0.436 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Zhu, X.; Tai, X.; Chen, J.; Bo, T. A Combined mRNA and microRNA Transcriptome Analysis of B. oleracea Response to Plasmodiophora brassicae Infection. Horticulturae 2024, 10, 1013. https://doi.org/10.3390/horticulturae10101013
Wang M, Zhu X, Tai X, Chen J, Bo T. A Combined mRNA and microRNA Transcriptome Analysis of B. oleracea Response to Plasmodiophora brassicae Infection. Horticulturae. 2024; 10(10):1013. https://doi.org/10.3390/horticulturae10101013
Chicago/Turabian StyleWang, Min, Xiaowei Zhu, Xiang Tai, Jinxiu Chen, and Tianyue Bo. 2024. "A Combined mRNA and microRNA Transcriptome Analysis of B. oleracea Response to Plasmodiophora brassicae Infection" Horticulturae 10, no. 10: 1013. https://doi.org/10.3390/horticulturae10101013
APA StyleWang, M., Zhu, X., Tai, X., Chen, J., & Bo, T. (2024). A Combined mRNA and microRNA Transcriptome Analysis of B. oleracea Response to Plasmodiophora brassicae Infection. Horticulturae, 10(10), 1013. https://doi.org/10.3390/horticulturae10101013