
Screening l-Lysine-Overproducing Escherichia coli Using Artificial Rare Codons and a Rare Codon-Rich Marker

Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains, Plasmids, and Culture Media
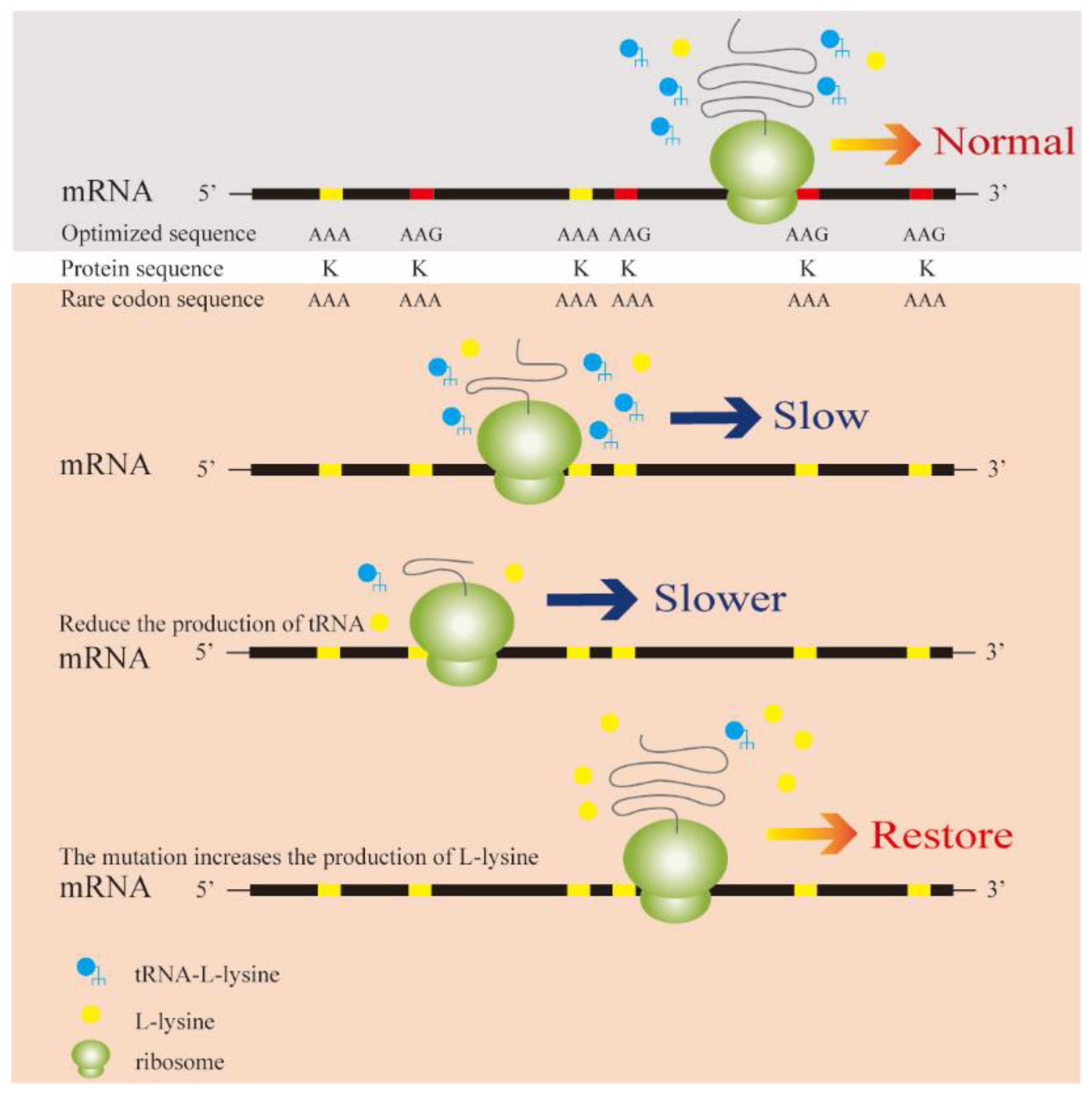
2.2. Construction of l-Lysine Codon-Rich Fluorescent Fusion Protein Screening Vectors
2.3. Construction of an Artificial Rare l-Lysine Codon in E. coli
2.4. Transformation and Expression of l-Lysine Codon-Rich Screening Vectors
2.5. Correlation Analysis of Fluorescence Intensity and Lysine Content in Recombinant Strains of E. coli QD01
2.6. Atmospheric Pressure and Room Temperature Plasma Mutagenesis
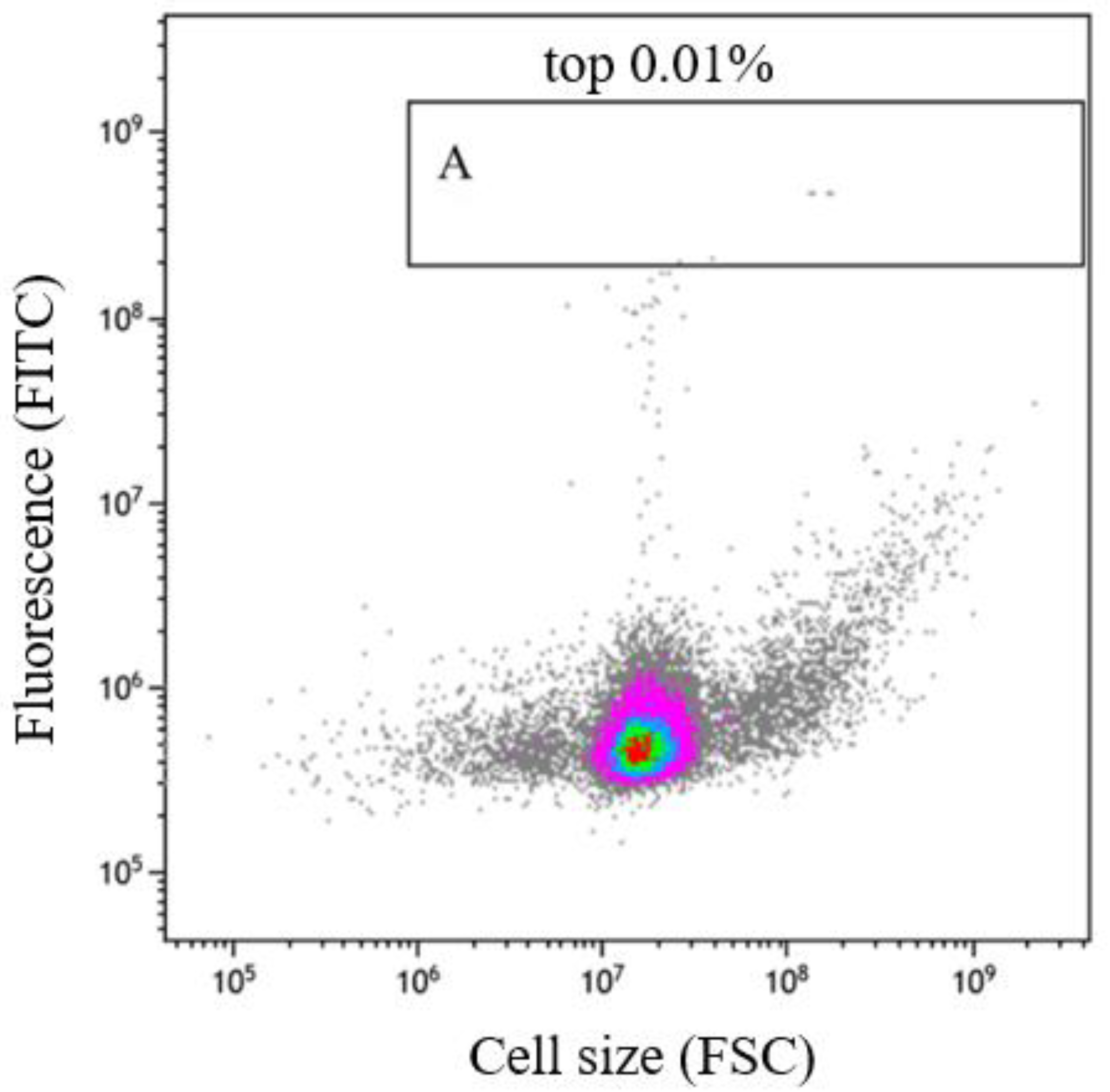
2.7. Flow Cytometric Cell Sorting of Mutant Strains
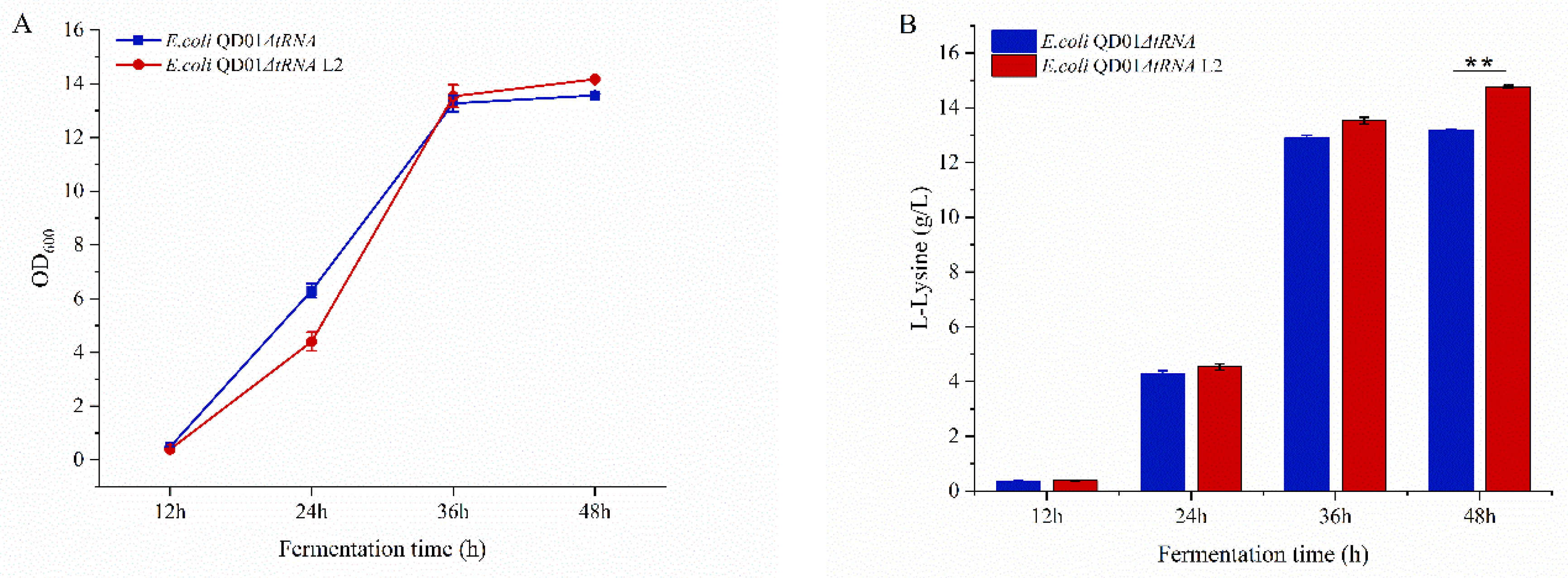
2.8. Fermentation Analysis of Mutant Strains
3. Results
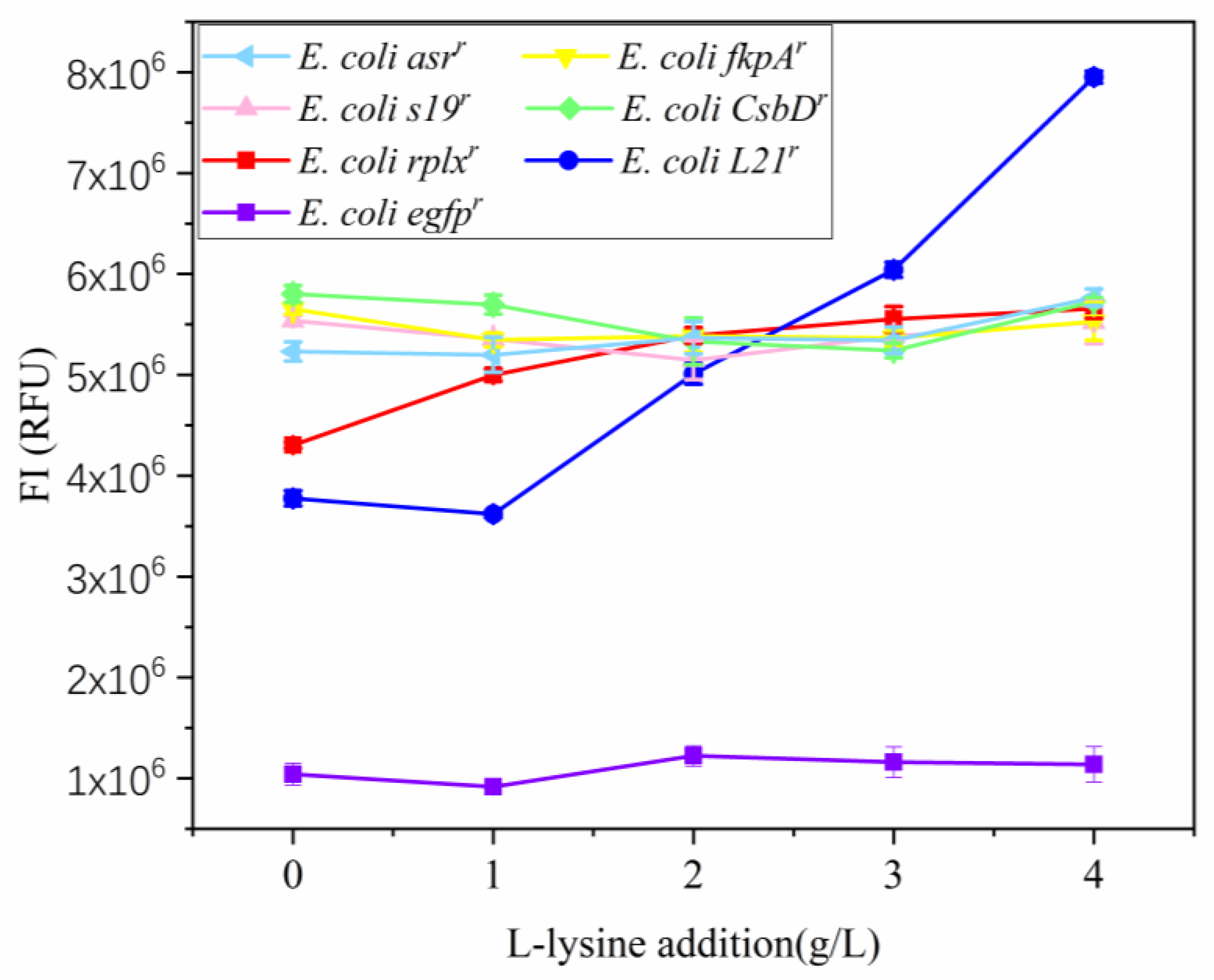
3.1. Construction and Expression of Lysine-Rich Fluorescent Fusion Protein Expression Vectors
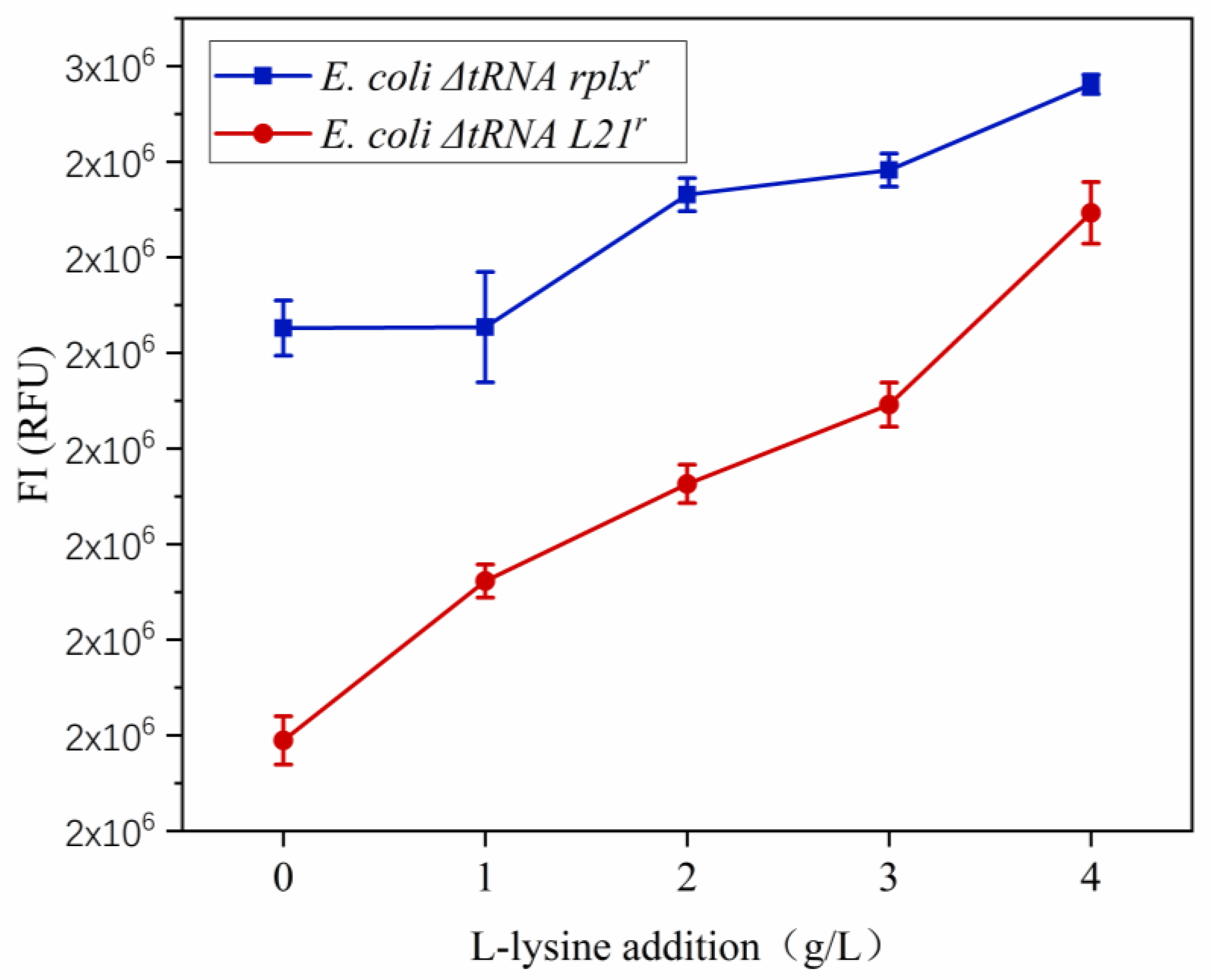
3.2. tRNA Knockout in E. coli QD01
3.3. Construction of a Rare Codon-Rich Marker Screening System for E. coli QD01ΔtRNA
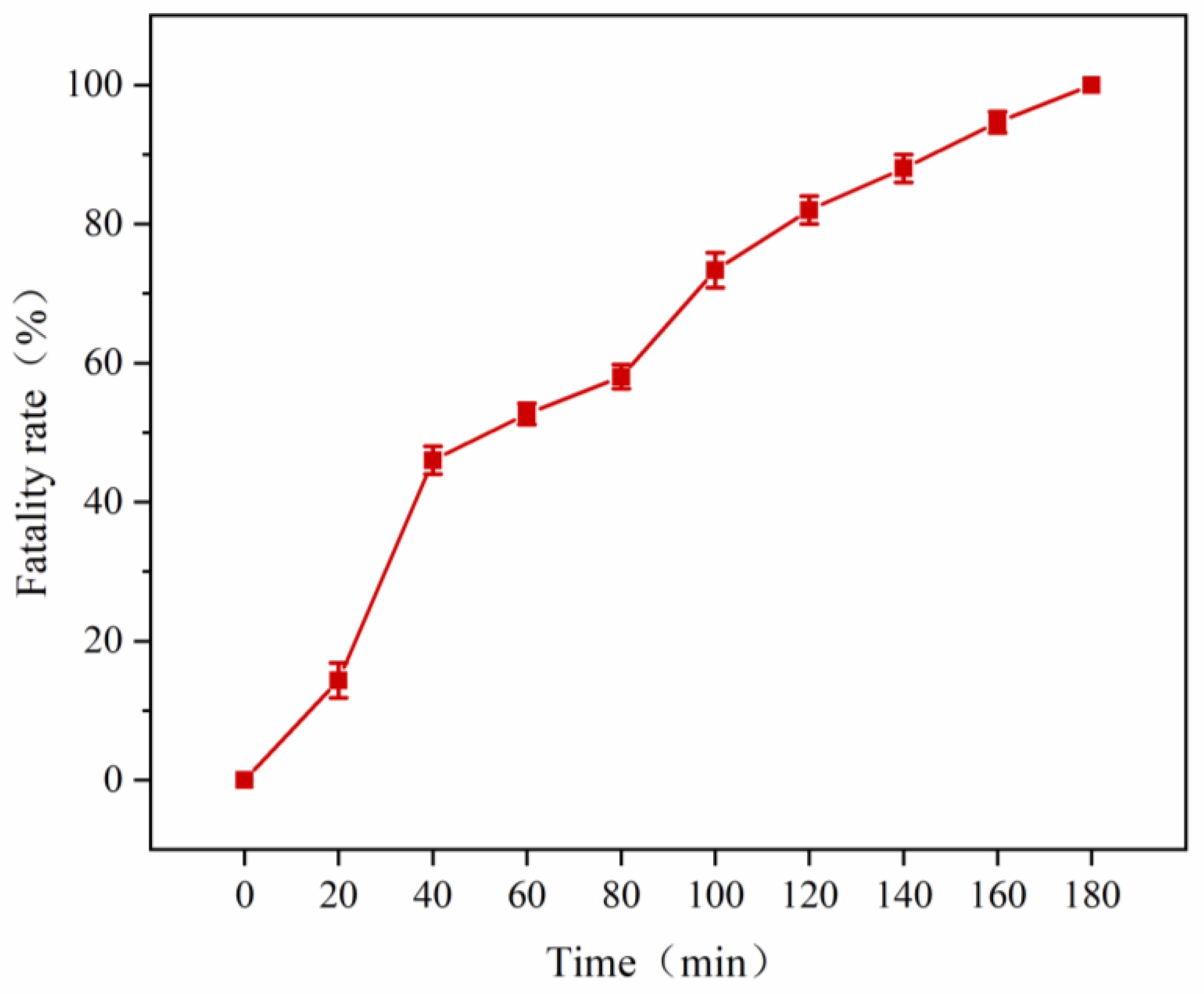
3.4. Mutant Library Construction Using ARTP
3.5. Fermentation of E. coli ΔtRNA L21r Mutants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eggeling, L.; Bott, M. A giant market and a powerful metabolism: L-lysine provided by Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 2015, 99, 3387–3394. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Davis, D.A. Comparison of crystalline lysine and intact lysine used as a supplement in practical diets of channel catfish (Ictalurus punctatus) and Nile tilapia (Oreochromis niloticus). Aquaculture 2016, 464, 331–339. [Google Scholar] [CrossRef]
- Felix, F.K.D.; Letti, L.A.; Pereira, G.V.D.; Bonfim, P.G.B.; Soccol, V.T.; Soccol, C.R. L-lysine production improvement: A review of the state of the art and patent landscape focusing on strain development and fermentation technologies. Crit. Rev. Biotechnol. 2019, 39, 1031–1055. [Google Scholar] [CrossRef]
- Xu, J.Z.; Yang, H.K.; Liu, L.M.; Wang, Y.Y.; Zhang, W.G. Rational modification of Corynebacterium glutamicum dihydrodipicolinate reductase to switch the nucleotide-cofactor specificity for increasing L-lysine production. Biotechnol. Bioeng. 2018, 115, 1764–1777. [Google Scholar] [CrossRef] [PubMed]
- Suvachan, A.; Lal, R.; Nampoothiri, K.M.; Sindhu, R.; Bhaskar, T.; Binod, P.; Pandey, A.; Yasarla, R. Valorization of paper industry rejects by combined thermo-chemical pretreatment and biological conversion to L-lysine. Environ. Technol. Innov. 2021, 24, 101882. [Google Scholar] [CrossRef]
- Wang, J.P.; Gao, C.; Chen, X.L.; Liu, L.M. Chapter one—Expanding the lysine industry: Biotechnological production of L-lysine and its derivatives. Adv. Appl. Microbiol. 2021, 115, 1–33. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, S.X.; Yang, X.N.; Qiu, L.; Gao, B.; Li, R.; Chen, J. Reactive extraction of amino acids mixture in hydrolysate from cottonseed meal with di(2-ethylhexyl) phosphoric acid. J. Chem. Technol. Biotechnol. 2016, 91, 483–489. [Google Scholar] [CrossRef]
- Xiao, J.; Wang, D.T.; Wang, L.; Jiang, Y.J.; Xue, L.; Sui, S.S.; Wang, J.; Guo, C.; Wang, R.; Wang, J.; et al. Increasingl-lysine production in Corynebacterium glutamicum by engineering amino acid transporters. Amino Acids 2020, 52, 1363–1374. [Google Scholar] [CrossRef]
- Yu, C.H.; Dang, Y.K.; Zhou, Z.P.; Wu, C.; Zhao, F.Z.; Sachs, M.S.; Liu, Y. Codon Usage Influences the Local Rate of Translation Elongation to Regulate Co-translational Protein Folding. Mol. Cell 2015, 59, 744–754. [Google Scholar] [CrossRef]
- Guimaraes, J.C.; Mittal, N.; Gnann, A.; Jedlinski, D.; Riba, A.; Buczak, K.; Schmidt, A.; Zavolan, M. A rare codon-based translational program of cell proliferation. Genome Biol. 2020, 21, 44. [Google Scholar] [CrossRef]
- Dittmar, K.A.; Sorensen, M.A.; Elf, J.; Ehrenberg, M.; Pan, T. Selective charging of tRNA isoacceptors induced by amino-acid starvation. Embo Rep. 2005, 6, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, A.R.; Zid, B.M.; O’Shea, E.K. An Integrated Approach Reveals Regulatory Controls on Bacterial Translation Elongation. Cell 2014, 159, 1200–1211. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Ma, X.Y.; Wang, N.; Ding, T.T.; Guo, L.W.; Zhang, X.R.; Yang, Y.; Li, C.; Huo, Y.-X. Utilization of rare codon-rich markers for screening amino acid overproducers. Nat. Commun. 2018, 9, 3616. [Google Scholar] [CrossRef] [PubMed]
- Schrumpf, B.; Eggeling, L.; Sahm, H.J.A.M. Isolation and prominent characteristics of an L-lysine hyperproducing strain of Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 1992, 37, 566–571. [Google Scholar] [CrossRef]
- Javed, A.; Jamil, A.; Rezaei-Zarchi, S. Optimization and hyper-expressed production of lysine through chemical mutagenesis of Brevibacterium flavum by N-nitroso-N-ethylurea. Afr. J. Microbiol. Res. 2011, 5, 5230–5238. [Google Scholar] [CrossRef]
- Holatko, J.; Elisakova, V.; Prouza, M.; Sobotka, M.; Nesvera, J.; Patek, M. Metabolic engineering of the L-valine biosynthesis pathway in Corynebacterium glutamicum using promoter activity modulation. J. Biotechnol. 2009, 139, 203–210. [Google Scholar] [CrossRef]
- Xu, H.Y.; Andi, B.; Qian, J.H.; West, A.H.; Cook, P.F. The alpha-aminoadipate pathway for lysine biosynthesis in fungi. Cell Biochem. Biophys. 2006, 46, 43–64. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, T.T.; Rao, Z.M.; Zhang, W.G.; Xu, J.Z. Reconstruction of the Diaminopimelic Acid Pathway to Promote L-lysine Production in Corynebacterium glutamicum. Int. J. Mol. Sci. 2021, 22, 9065. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, S.; Jiang, P.X.; Yao, J.; He, Y.Z.; Chen, L.C.; Gui, X.; Dong, Z.; Tang, S.-Y. Design of an ectoine-responsive AraC mutant and its application in metabolic engineering of ectoine biosynthesis. Metab. Eng. 2015, 30, 149–155. [Google Scholar] [CrossRef]
- Cress, B.F.; Trantas, E.A.; Ververidis, F.; Linhardt, R.J.; Koffas, M.A.G. Sensitive cells: Enabling tools for static and dynamic control of microbial metabolic pathways. Curr. Opin. Biotechnol. 2015, 36, 205–214. [Google Scholar] [CrossRef]
- Binder, S.; Schendzielorz, G.; Stabler, N.; Krumbach, K.; Hoffmann, K.; Bott, M.; Eggeling, L. A high-throughput approach to identify genomic variants of bacterial metabolite producers at the single-cell level. Genome Biol. 2012, 13, R40. [Google Scholar] [CrossRef] [PubMed]
- Cazier, A.P.; Blazeck, J. Advances in promoter engineering: Novel applications and predefined transcriptional control. Biotechnol. J. 2021, 16, 2100239. [Google Scholar] [CrossRef]
- Della Corte, D.; van Beek, H.L.; Syberg, F.; Schallmey, M.; Tobola, F.; Cormann, K.U.; Schlicker, C.; Baumann, P.T.; Krumbach, K.; Sokolowsky, S.; et al. Engineering and application of a biosensor with focused ligand specificity. Nat. Commun. 2020, 11, 4851. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.P.; Peng, Z.H.; Yang, L.; Du, B.W.; Guo, C.Z.; Sui, S.S.; Wang, J.; Li, J.; Wang, J.; Li, N. Design and application of artificial rare L-lysine codons in Corynebacterium glutamicum. Front. Bioeng. Biotechnol. 2023, 11, 1194511. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Ando, R.; Shimozono, S.; Sugiyama, M.; Takeda, N.; Kurokawa, H.; Deguchi, R.; Endo, K.; Haga, K.; Takai-Todaka, R.; et al. A highly photostable and bright green fluorescent protein. Nat. Biotechnol. 2022, 40, 1132. [Google Scholar] [CrossRef]
- Yang, Y.Q.; Yu, Q.L.; Wang, M.; Zhao, R.; Liu, H.W.; Xun, L.Y.; Xia, Y. Escherichia coli BW25113 Competent Cells Prepared Using a Simple Chemical Method Have Unmatched Transformation and Cloning Efficiencies. Front. Microbiol. 2022, 13, 838698. [Google Scholar] [CrossRef]
- East, A.K.; Mauchline, T.H.; Poole, P.S. Chapter 5 Biosensors for Ligand Detection. Adv. Appl. Microbiol. 2008, 64, 137–166. [Google Scholar] [CrossRef]
- Schallmey, M.; Frunzke, J.; Eggeling, L.; Marienhagen, J. Looking for the pick of the bunch: High-throughput screening of producing microorganisms with biosensors. Curr. Opin. Biotechnol. 2014, 26, 148–154. [Google Scholar] [CrossRef]
- Niu, F.X.; He, X.; Huang, Y.B.; Liu, J.Z. Biosensor-Guided Atmospheric and Room-Temperature Plasma Mutagenesis and Shuffling for High-Level Production of Shikimic Acid from Sucrose in Escherichia coli. J. Agric. Food Chem. 2020, 68, 11765–11773. [Google Scholar] [CrossRef]
- Shen, Y.P.; Pan, Y.Y.; Niu, F.X.; Liao, Y.L.; Huang, M.T.; Liu, J.Z. Biosensor-assisted evolution for high-level production of 4-hydroxyphe- nylacetic acid in Escherichia coli. Metab. Eng. 2022, 70, 1–11. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, Y.; Xu, Y.; Zhang, J.; Ma, L.L.; Qi, Q.S.; Wang, Q. Development of bifunctional biosensors for sensing and dynamic control of glycolysis flux in metabolic engineering. Metab. Eng. 2021, 68, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, J.A.; Shis, D.L.; Alikhani, A.; Keasling, J.D. Transcription Factor-Based Screens and Synthetic Selections for Microbial Small-Molecule Biosynthesis. Acs Synth. Biol. 2013, 2, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Chen, Z.Y.; Guo, S.Y.; Zhang, C.Y.; Huo, Y.X. Engineering Transcription Factor BmoR Mutants for Constructing Multifunctional Alcohol Biosensors. Acs Synth. Biol. 2022, 11, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.H.; Liu, P.R.; Chen, J.; Du, G.C.; Li, H.Z.; Zhou, J.W. Characterization of mutants of a tyrosine ammonia-lyase from Rhodotorula glutinis. Appl. Microbiol. Biotechnol. 2016, 100, 10443–10452. [Google Scholar] [CrossRef]
- Zhang, Y.; Ptacin, J.L.; Fischer, E.C.; Aerni, H.R.; Caffaro, C.E.; Jose, K.S.; Feldman, A.W.; Turner, C.R.; Romesberg, F.E. A semi-synthetic organism that stores and retrieves increased genetic information. Nature 2017, 551, 644. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.X.; Zheng, B.; Wang, N.; Yang, Y.P.; Liang, X.X.; Ma, X.Y. Identifying Amino Acid Overproducers Using Rare-Codon-Rich Markers. Jove-J. Vis. Exp. 2019, 148, e59331. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Correlated Characteristic | Source |
|---|---|---|
| Strains | ||
| E. coli QD01 | l-lysine producing bacteria | This laboratory constructs and preserves |
| E. coli BL21 | High levels of recombinant protein expression | Vazyme |
| E. coli DH5α | Transformation of recombinant plasmids | Vazyme |
| E. coli QD01ΔtRNA | Five l-lysine tRNAs were knocked out in the E. coli QD01 genome | Construction of this study |
| E. coli ΔtRNA rplxr | E.coli QD01ΔtRNA was transferred into the plasmid pET-22b(+)-staygold-rplxr | Construction of this study |
| E. coli ΔtRNA L21r | E.coli QD01ΔtRNA was transferred into the plasmid pET-22b(+)-staygold-L21r | Construction of this study |
| E. coli ΔtRNA egfpr | E.coli QD01ΔtRNA was transferred into the plasmid pET-22b(+)-egfpr | Construction of this study |
| E. coli QD01ΔtRNA L2 | l-lysine-producing bacteria | Construction of this study |
| Plasmids | ||
| pET-22b(+) | Expression of E.coli protein | Laboratory preservation |
| pET-22b(+)-asrr-staygold | pET-22b(+) is inserted into the rare codon fluorescent fusion protein asrr-staygold gene | Construction of this study |
| pET-22b(+)-fkpAr-staygold | pET-22b(+) is inserted into the rare codon fluorescent fusion protein fkpAr-staygold gene | Construction of this study |
| pET-22b(+)-s19r-staygold | pET-22b(+) is inserted into the rare codon fluorescent fusion protein s19r-staygold gene | Construction of this study |
| pET-22b(+)-CsbDr-staygold | pET-22b(+) is inserted into the rare codon fluorescent fusion protein CsbDr-staygold gene | Construction of this study |
| pET-22b(+)-rplxr-staygold | pET-22b(+) is inserted into the rare codon fluorescent fusion protein rplxr-staygold gene | Construction of this study |
| pET-22b(+)-L21r-staygold | pET-22b(+) is inserted into the rare codon fluorescent fusion protein L21r-staygold gene | Construction of this study |
| pET-28a(+) | Expression of Escherichia coli protein | Laboratory storage |
| pET-28a(+)-tRNA | pET-28a(+) is inserted into the L-Kanr-R segment | Construction of this study |
| pKD13 | As a template for amplification of Kanr fragments | Laboratory storage |
| pKD46 | Exo, Beta, and Gam proteins were expressed | Laboratory storage |
| pCP20 | Coding for flipping recombinant enzyme FLP | Laboratory storage |
| Strain | Screening Technique | Selection Marker | Target Material | Screening Effect | References |
|---|---|---|---|---|---|
| E. coli SA30 | PSenSA biosensor | RFP | shikimic acid | The yield increased by 2.7 times, and the growth increased by 2.0 times | [29] |
| E. coli 4HPAA-1 | pSenSA-4HPAA biosensor | RFP | 4-hydroxyphenylacetic acid | The output is 25.42 g/L | [30] |
| E. coli Bw25113 | bifunctionalglycolysis flux biosensor | GFP | Methylvaleric acid | The output is 111.3 g/L | [31] |
| E. coli DH1 | TetA-based whole-cell biosensor | GFP | 1-butanol | Production increased by 35% | [32] |
| E. coli XL10-Gold | transcription factor-based biosensors (TFBs) | GFP | alcohol | Detection sensitivity increased by 107 times | [33] |
| E. coli BL21 (DE3) | direct detection of the visible light spectrum of the product | OD315 | p-Coumaric acid | Production increased by 65.9% | [34] |
| C. glutamicum 23604 | FASC | eGFP | L-lysine | Production increased by 9.7% | [24] |
| E. coli QD01 | FASC | l-lysine-rich staygold | L-lysine | Production increased by 12.1% | this research |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Yang, C.; Yang, L.; Wang, R.; Li, P.; Du, B.; Li, N.; Wang, J. Screening l-Lysine-Overproducing Escherichia coli Using Artificial Rare Codons and a Rare Codon-Rich Marker. Fermentation 2023, 9, 899. https://doi.org/10.3390/fermentation9100899
Liu H, Yang C, Yang L, Wang R, Li P, Du B, Li N, Wang J. Screening l-Lysine-Overproducing Escherichia coli Using Artificial Rare Codons and a Rare Codon-Rich Marker. Fermentation. 2023; 9(10):899. https://doi.org/10.3390/fermentation9100899
Chicago/Turabian StyleLiu, Hui, Cuiping Yang, Lu Yang, Ruiming Wang, Piwu Li, Bowen Du, Nan Li, and Junqing Wang. 2023. "Screening l-Lysine-Overproducing Escherichia coli Using Artificial Rare Codons and a Rare Codon-Rich Marker" Fermentation 9, no. 10: 899. https://doi.org/10.3390/fermentation9100899
APA StyleLiu, H., Yang, C., Yang, L., Wang, R., Li, P., Du, B., Li, N., & Wang, J. (2023). Screening l-Lysine-Overproducing Escherichia coli Using Artificial Rare Codons and a Rare Codon-Rich Marker. Fermentation, 9(10), 899. https://doi.org/10.3390/fermentation9100899