
Degradation Kinetics of Lignocellulolytic Enzymes in a Biogas Reactor Using Quantitative Mass Spectrometry

 ,
,
Abstract
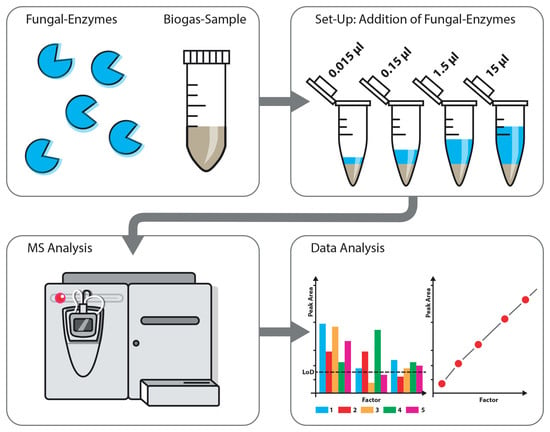
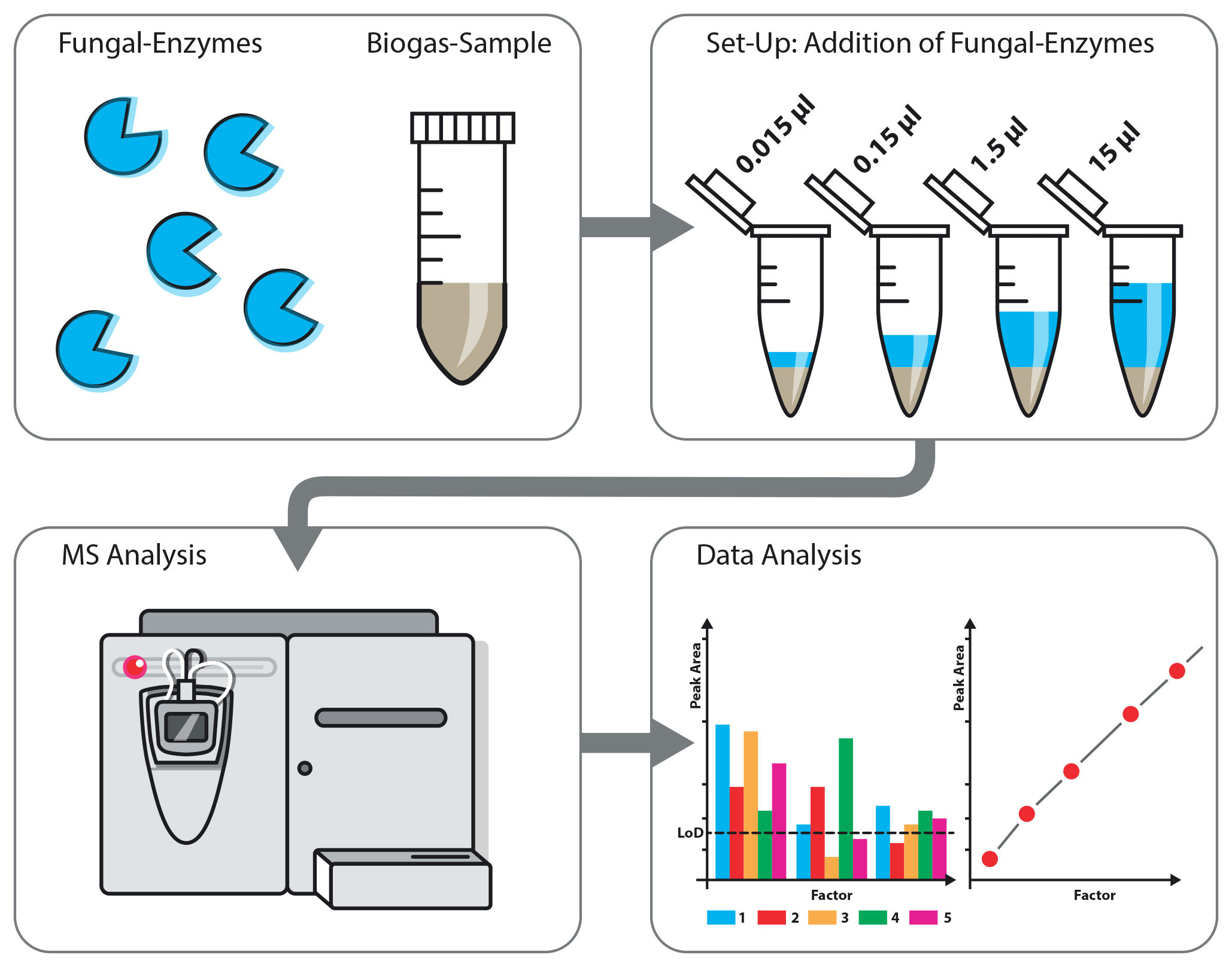
1. Introduction
2. Materials and Methods
2.1. Bioreactor Set-Up and Sampling
2.2. Enzyme Preparation
2.3. Addition of Fungal Enzymes
2.4. Sample Preparation
2.4.1. Protein Precipitation
2.4.2. Protein Quantification
2.5. Tryptic Digestion
2.6. Chromatography and Mass Spectrometry
2.7. Data Analysis
3. Results and Discussion
3.1. Detection of Enzyme Proteins
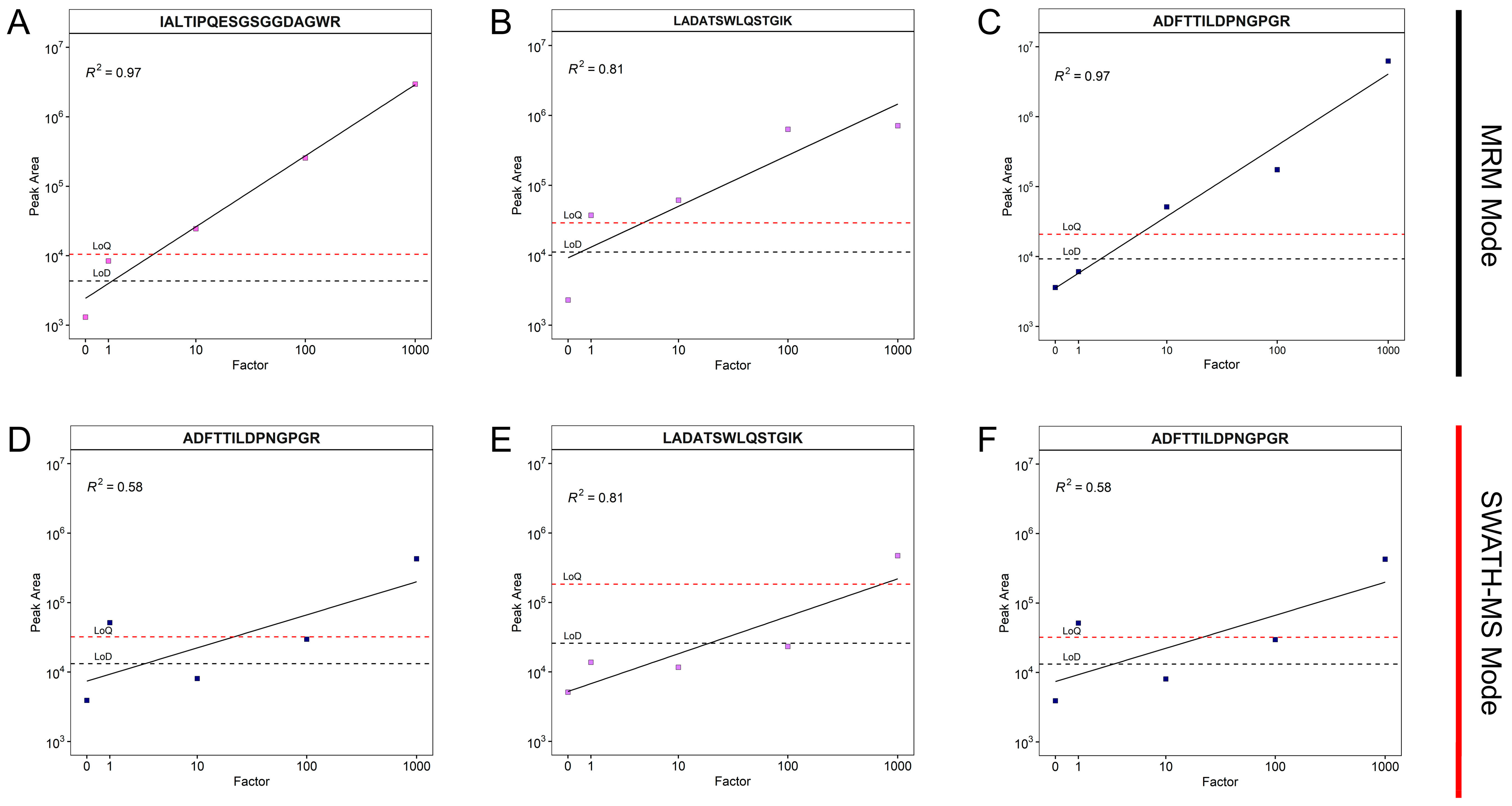
3.2. Selection of Signature Peptides and Validation of the Assay
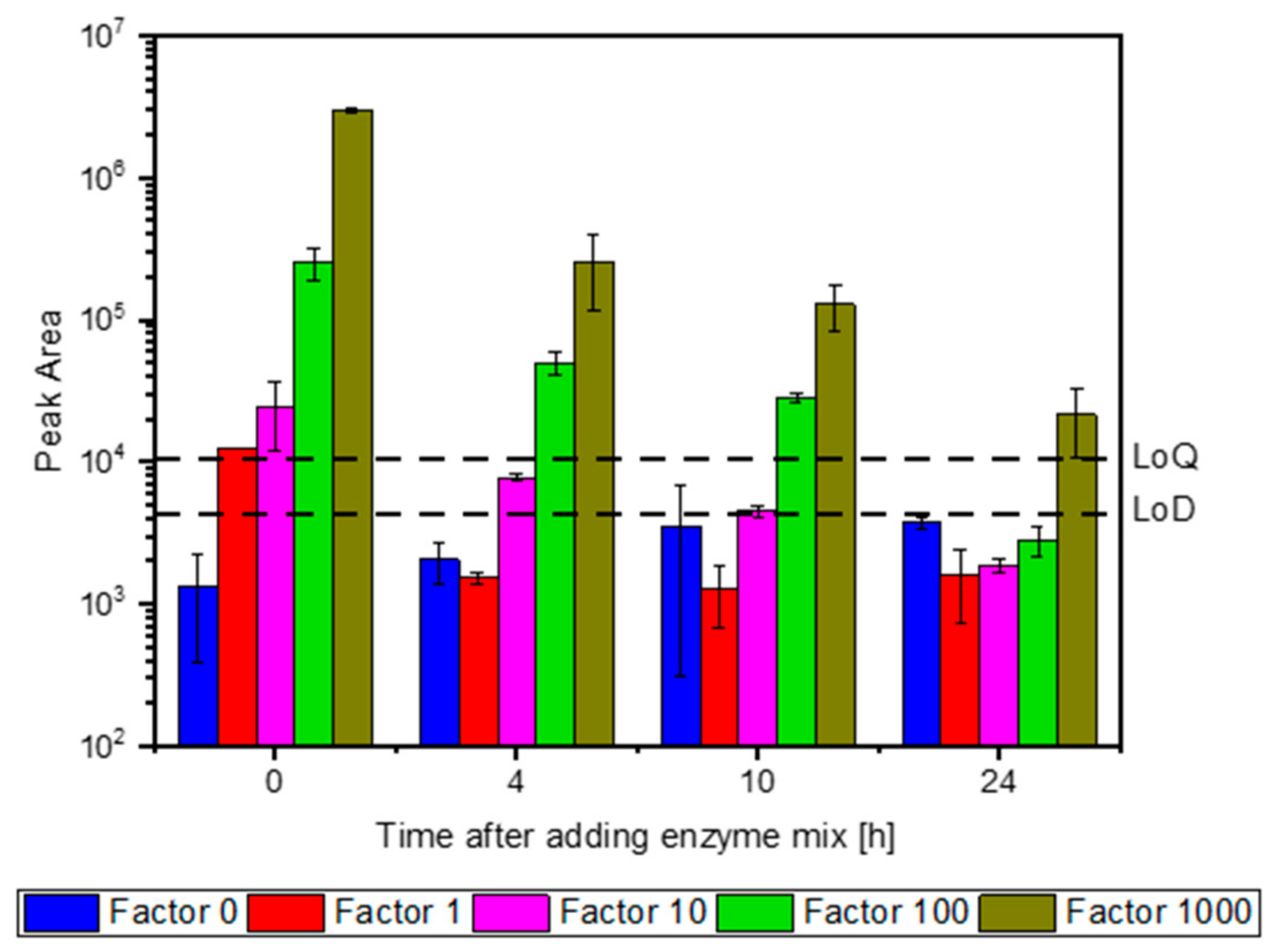
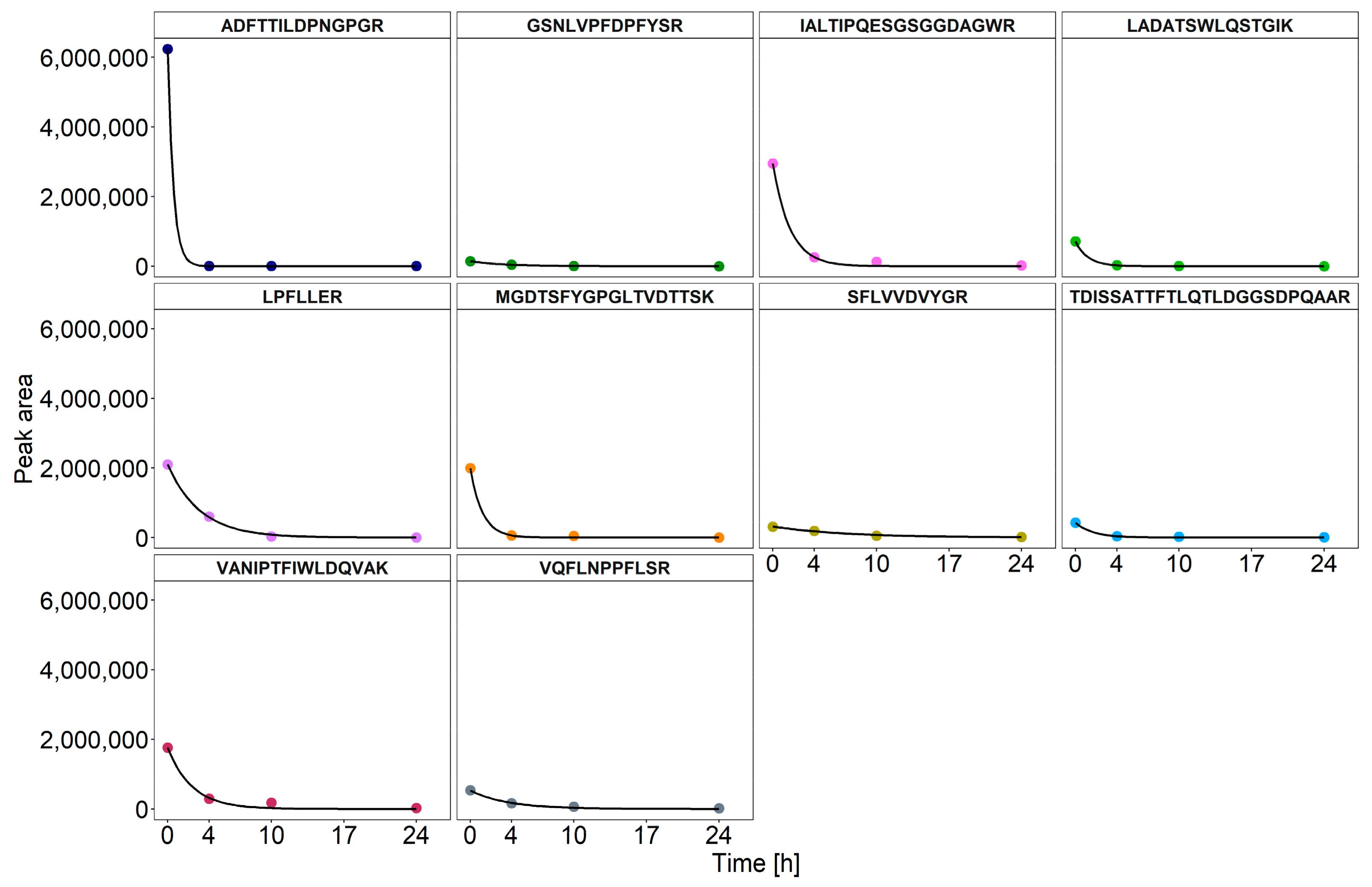
3.3. Dynamics of Enzyme Degradation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taherzadeh, M.J.; Karimi, K. Pretreatment of lignocellulosic wastes to improve ethanol and bi-ogas production: A review. Int. J. Mol. Sci. 2008, 9, 1621–1651. [Google Scholar] [CrossRef] [PubMed]
- Parawira, W. Enzyme research and applications in biotechnological intensification of biogas production. Crit. Rev. Biotechnol. 2012, 32, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Speda, J.; Johansson, M.A.; Odnell, A.; Karlsson, M. Enhanced biomethane production rate and yield from lignocellulosic ensiled forage ley by in situ anaerobic digestion treatment with endogenous cellulolytic enzymes. Biotechnol. Biofuels 2017, 10, 129. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, T.; Bos, G.K.; Zeeman, G.; Sanders, J.; van Lier, J. Effects of thermo-chemical pre-treatment on anaerobic biodegradability and hydrolysis of lignocellulosic biomass. Bioresour. Technol. 2009, 100, 2575–2579. [Google Scholar] [CrossRef] [PubMed]
- Ferdeș, M.; Dincă, M.N.; Moiceanu, G.; Zăbavă, B.Ș.; Paraschiv, G. Microorganisms and Enzymes Used in the Biological Pretreatment of the Substrate to Enhance Biogas Production: A Review. Sustainability 2020, 12, 7205. [Google Scholar] [CrossRef]
- Ometto, F.; Quiroga, G.; Pšenička, P.; Whitton, R.; Jefferson, B.; Villa, R. Impacts of microalgae pre-treatments for improved anaerobic digestion: Thermal treatment, thermal hydrolysis, ultrasound and en-zymatic hydrolysis. Water Res. 2014, 65, 350–361. [Google Scholar] [CrossRef]
- Szűcs, C.; Kovács, E.; Bagi, Z.; Rákhely, G.; Kovács, K.L. Enhancing biogas production from agroindustrial waste pre-treated with filamentous fungi. Biol. Futur. 2021, 72, 341–346. [Google Scholar] [CrossRef]
- Schimpf, U.; Schulz, R. Industrial by-products from white-rot fungi production. Part I: Generation of enzyme preparations and chemical, protein biochemical and molecular biological characterization. Process Biochem. 2016, 51, 2034–2046. [Google Scholar] [CrossRef]
- Binner, R.; Menath, V.; Huber, H.; Thomm, M.; Bischof, F.; Schmack, D.; Reuter, M. Comparative study of stability and half-life of enzymes and enzyme aggregates implemented in anaerobic biogas processes. Biomass Conv. Bioref. 2011, 1, 1–8. [Google Scholar] [CrossRef]
- Willenbücher, K.; Wibberg, D.; Huang, L.; Conrady, M. Phage Genome Diversity in a Biogas-Producing Microbiome Analyzed by Illumina and Nanopore GridION Sequencing. Microorganisms 2022, 10, 368. [Google Scholar] [CrossRef]
- Heyer, R.; Schallert, K.; Büdel, A.; Zoun, R.; Dorl, S.; Behne, A.; Kohrs, F.; Püttker, S.; Siewert, C.; Muth, T.; et al. A Robust and Universal Metaproteomics Workflow for Research Studies and Routine Diagnostics Within 24 h Using Phenol Extraction, FASP Digest, and the MetaProteomeAnalyzer. Front. Microbiol. 2019, 10, 1883. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and ac-curate proteome analysis. Mol. Cell. Proteom. MCP 2012, 11, O111.016717. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef]
- Currie, L.A. Limits for qualitative detection and quantitative determination. In Application to Radiochemistry; Analytical Chemistry Division, National Bureau of Standards: Washington, DC, USA, 1968. [Google Scholar]
- Mani, D.R.; Abbatiello, S.E.; Carr, S.A. Statistical characterization of multiple-reaction monitoring mass spectrometry (MRM-MS) assays for quantitative proteomics. BMC Bioinform. 2012, 13 (Suppl. 16), S9. [Google Scholar] [CrossRef]
- Odnell, A.; Recktenwald, M.; Stensén, K.; Jonsson, B.-H.; Karlsson, M. Activity, life time and effect of hydrolytic enzymes for enhanced biogas production from sludge anaerobic digestion. Water Res. 2016, 103, 462–471. [Google Scholar] [CrossRef]
- Davidsson, Å.; Wawrzynczyk, J.; Norrlöw, O.; Jansen, J.L.C. Strategies for Enzyme Dosing to Enhance Anaerobic Digestion of Sewage Sludge. J. Residuals Sci. Technol. 2007, 4, 1–7. [Google Scholar]
- Garcia, N.H.; Mattioli, A.; Gil, A.; Frison, N.; Battista, F.; Bolzonella, D. David Evaluation of the methane potential of different agricultural and food processing substrates for improved biogas produc-tion in rural areas. Renew. Sustain. Energy Rev. 2019, 112, 1–10. [Google Scholar] [CrossRef]
- Heyer, R.; Schallert, K.; Siewert, C.; Kohrs, F.; Greve, J.; Maus, I.; Klang, J.; Klocke, M.; Heiermann, M.; Hoffmann, M.; et al. Metaproteome analysis reveals that syntrophy, competition, and phage-host interaction shape microbial communities in biogas plants. Microbiome 2019, 7, 69. [Google Scholar] [CrossRef]
- Colgrave, M.L.; Byrne, K.; Blundell, M.; Heidelberger, S.; Lane, C.S.; Tanner, G.J.; Howitt, C.A. Comparing Multiple Reaction Monitoring and Sequential Window Acquisition of All Theoret-ical Mass Spectra for the Relative Quantification of Barley Gluten in Selectively Bred Barley Lines. Anal. Chem. 2016, 88, 9127–9135. [Google Scholar] [CrossRef]
- Montemurro, N.; Orfanioti, A.; Manasfi, R.; Thomaidis, N.S.; Pérez, S. Comparison of high resolution mrm and sequential window acquisition of all theoretical fragment-ion acquisition modes for the quantitation of 48 wastewater-borne pollutants in lettuce. J. Chromatogr. A 2020, 1631, 461566. [Google Scholar] [CrossRef] [PubMed]
- Schievano, A.; Scaglia, B.; D’Imporzano, G.; Malagutti, L.; Gozzi, A.; Adani, F. Prediction of biogas potentials using quick laboratory analyses: Upgrading previous models for application to heterogeneous organic matrices. Bioresour. Technol. 2009, 100, 5777–5782. [Google Scholar] [CrossRef] [PubMed]
- Badieyan, S.; Bevan, D.R.; Zhang, C. Study and design of stability in GH5 cellulases. Biotechnol. Bioeng. 2012, 109, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Trötschel, C.; Poetsch, A. Current approaches and challenges in targeted absolute quantification of membrane proteins. Proteomics 2015, 15, 915–929. [Google Scholar] [CrossRef]
- Liew, Y.X.; Chan, Y.J.; Manickam, S.; Chong, M.F.; Chong, S.; Tiong, T.J.; Lim, J.W.; Pan, G.-T. Enzymatic pretreatment to enhance anaerobic bioconversion of high strength wastewater to biogas: A review. Sci. Total Environ. 2020, 713, 136373. [Google Scholar] [CrossRef]
- Aghtaei, H.K.; Püttker, S.; Maus, I.; Heyer, R.; Huang, L.; Sczyrba, A.; Reichl, U.; Benndorf, D. Adaptation of a microbial community to demand-oriented biological methanation. Biotechnol. Biofuels Bioprod. 2022, 15, 125. [Google Scholar] [CrossRef]
- Küchler, J.; Püttker, S.; Lahmann, P.; Genzel, Y.; Kupke, S.; Benndorf, D.; Reichl, U. Absolute quantification of viral proteins during single-round replication of MDCK suspension cells. J. Proteom. 2022, 259, 104544. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acc | Description | Score | Mass | Matches | Sequences |
|---|---|---|---|---|---|
| A0A1Q3EJ89 | A0A1Q3EJ89_LENED Glucanase OS = Lentinula edodes OX = 5353 GN = LENED_009211 PE = 3 SV = 1 | 1028 | 55,356 | 33 | 4 |
| A0A1Q3EU06 | A0A1Q3EU06_LENED Glucanase OS = Lentinula edodes OX = 5353 GN = LENED_012880 PE = 3 SV = 1 | 636 | 62,086 | 21 | 6 |
| A0A1Q3E856 | A0A1Q3E856_LENED Subtilisin-like protein OS = Lentinula edodes OX = 5353 GN = LENED_004953 PE = 4 SV = 1 | 620 | 63,032 | 26 | 6 |
| A0A1Q3ENW5 | A0A1Q3ENW5_LENED Beta-xylanase OS = Lentinula edodes OX = 5353 GN = LENED_010993 PE = 3 SV = 1 | 389 | 120,358 | 13 | 4 |
| A0A1Q3EGI8 | A0A1Q3EGI8_LENED Glycoside hydrolase family 55 protein OS = Lentinula edodes OX = 5353 GN = LENED_008227 PE = 4 SV = 1 | 385 | 67,565 | 9 | 4 |
| Q9C1R4 | Q9C1R4_LENED Glucanase OS = Lentinula edodes OX = 5353 GN = cbhII-1 PE = 2 SV = 1 | 324 | 46,832 | 13 | 3 |
| A0A1Q3EBY2 | A0A1Q3EBY2_LENED Glycoside hydrolase family 5 protein OS = Lentinula edodes OX = 5353 GN = LENED_006487 PE = 3 SV = 1 | 318 | 43,559 | 8 | 2 |
| A0A0A1I5X1 | A0A0A1I5X1_LENED Glycoside hydrolase family 5 endoglucanase (Fragment) OS = Lentinula edodes OX = 5353 GN = glu PE = 3 SV = 1 | 231 | 8509 | 5 | 1 |
| A0A1Q3DWF6 | A0A1Q3DWF6_LENED Cu-oxidase-domain-containing protein OS = Lentinula edodes OX = 5353 GN = LENED_000509 PE = 3 SV = 1 | 273 | 57,232 | 14 | 3 |
| A0A1Q3EAX5 | A0A1Q3EAX5_LENED Glycoside hydrolase family 12 protein OS = Lentinula edodes OX = 5353 GN = LENED_006131 PE = 3 SV = 1 | 237 | 26,422 | 7 | 3 |
| Protein | ID | Peptide Sequence | Abbreviation | m/z | Collision Energy [eV] | LoD MRM | LoQ MRM | LoD SWATH-MS | LoQ SWATH-MS |
|---|---|---|---|---|---|---|---|---|---|
| Glycoside Hydrolase Family 16 Protein | A0A1Q3DXQ1 | ADFTTILDPNGPGR | ADF | 737.3703 | 42 | 9265.24 | 20,818.55 | 13,190.26 | 32,121.56 |
| Carbohydrate Esterase Family 15 Protein | A0A1Q3EF14 | IALTIPQESGSGGDAGWR | IAL | 907.9552 | 48 | 4319.99 | 10,469.10 | 10,078.47 | 26,025.36 |
| Glycoside Hydrolase Family 5 endoglucanase (EC 3.2.1.4) (fragment) | A0A0A1I5X1 | LADATSWLQSTGIK | LAD | 745.8961 | 42 | 11,100.85 | 29,059.49 | 25,864.80 | 183,437.42 |
| Glucanase (EC 3.2.1.-) | Q96VU3 | MGDTSFYGPGLTVDTTSK | MGD | 938.9353 | 48 | 5227.55 | 11,224.83 | 12,511.12 | 32,676.04 |
| Q9C1R6 | VANIPTFIWLDQVAK | VAN | 857.9800 | 45 | 7526.52 | 19,345.53 | 19,870.98 | 49,686.59 | |
| Glycoside Hydrolase Family 5 Protein | A0A1Q3EBY2 | LPFLLER | LPF | 444.2711 | 27 | 37,104.11 | 92,768.77 | 39,710.62 | 105,827.83 |
| Carbohydrate Esterase Family 16 Protein | A0A1Q3DZ58 | SFLVVDVYGR | SFL | 577.8139 | 31 | 24,580.91 | 60,929.95 | 83,059.27 | 221,305.20 |
| Family S53 Protease (Kinesin-like Protein) | A0A1Q3DXE6 | TDISSATTFTLQTLDGGSDPQAA | TDI | 1227.093 | 48 | 3451.58 | 8628.03 | 25,740.59 | 65,377.70 |
| Cupper Radical Oxidase | A0A1Q3E003 | VQFLNPPFLSR | VQL | 659.3693 | 39 | 17,981.82 | 37,553.65 | 30,637.34 | 80,912.03 |
| Beta-Mannosidase (EC 3.2.1.25) | A0A1Q3E4F1 | GSNLVPFDPFYSR | GSN | 749.8699 | 42 | 9253.63 | 22,135.45 | 48,250.42 | 127,927.94 |
| Protein | Peptide | Half-Life [h] | ||
|---|---|---|---|---|
| Replicate A | Replicate B | Average | ||
| Carbohydrate Esterase Family 15 Protein | IAL | 1.17 | 1.07 | 1.12 |
| Glycoside Hydrolase Family 16 Protein | ADF | 0.44 | 0.44 | 0.44 |
| Glycoside Hydrolase Family 5 endoglucanase (EC 3.2.1.4) (fragment) | LAD | 22.84 | 22.51 | 22.68 |
| Glucanase (EC 3.2.1.) | MGD | 0.82 | 1.50 | 1.16 |
| VAN | 20.62 | 25.66 | 23.14 | |
| Glycoside Hydrolase Family 5 Protein | LPF | 1.69 | 3.10 | 2.40 |
| Carbohydrate Esterase Family 16 Protein | SFL | 1.01 | 1.57 | 1.29 |
| Family S53 Protease (Kinesin-like Protein) | TDI | 1.28 | 1.54 | 1.41 |
| Copper Radical Oxidase | VQL | 1.38 | 1.79 | 1.59 |
| Beta-Mannosidase (EC 3.2.1.25) | GSN | 5.27 | 7.27 | 6.27 |
| Average | 1.17 | 1.78 | 1.48 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Küchler, J.; Willenbücher, K.; Reiß, E.; Nuß, L.; Conrady, M.; Ramm, P.; Schimpf, U.; Reichl, U.; Szewzyk, U.; Benndorf, D. Degradation Kinetics of Lignocellulolytic Enzymes in a Biogas Reactor Using Quantitative Mass Spectrometry. Fermentation 2023, 9, 67. https://doi.org/10.3390/fermentation9010067
Küchler J, Willenbücher K, Reiß E, Nuß L, Conrady M, Ramm P, Schimpf U, Reichl U, Szewzyk U, Benndorf D. Degradation Kinetics of Lignocellulolytic Enzymes in a Biogas Reactor Using Quantitative Mass Spectrometry. Fermentation. 2023; 9(1):67. https://doi.org/10.3390/fermentation9010067
Chicago/Turabian StyleKüchler, Jan, Katharina Willenbücher, Elisabeth Reiß, Lea Nuß, Marius Conrady, Patrice Ramm, Ulrike Schimpf, Udo Reichl, Ulrich Szewzyk, and Dirk Benndorf. 2023. "Degradation Kinetics of Lignocellulolytic Enzymes in a Biogas Reactor Using Quantitative Mass Spectrometry" Fermentation 9, no. 1: 67. https://doi.org/10.3390/fermentation9010067
APA StyleKüchler, J., Willenbücher, K., Reiß, E., Nuß, L., Conrady, M., Ramm, P., Schimpf, U., Reichl, U., Szewzyk, U., & Benndorf, D. (2023). Degradation Kinetics of Lignocellulolytic Enzymes in a Biogas Reactor Using Quantitative Mass Spectrometry. Fermentation, 9(1), 67. https://doi.org/10.3390/fermentation9010067