Abstract
Recombinantly produced enzymes are applied in many fields, ranging from medicine to food and nutrition, production of detergents, textile, leather, paper, pulp, and plastics. Thus, the cost-effectiveness of recombinant enzyme synthesis is an important issue in biotechnological industry. Isopropyl-β-D-thiogalactoside (IPTG), an analog of lactose, is currently the most widely used chemical agent for the induction of recombinant enzyme synthesis. However, the use of IPTG can lead to production of toxic elements and can introduce physiological stress to cells. Thus, this study aims to find a simpler, cheaper, and safer way to produce recombinant enzymes. In this study, production of several previously designed recombinant lipolytic enzymes (GDEst-95 esterase, GD-95RM lipase, fused GDEst-lip lipolytic enzyme, and putative cutinase Cut+SP from Streptomyces scabiei 87.22) is induced in E. coli BL21 (DE3) using 4 mM milk permeate, a type of waste of the milk manufacturing process possessing >82% lactose. The SDS-PAGE analysis clearly indicates synthesis of all target enzymes during a 2–12 h post-induction timeframe. Further investigation of GDEst-95, GD-95RM, GDEst-lip, and Cut+SP biocatalysts was carried out spectrophotometrically and using zymography method, confirming production of fully active enzymes.
1. Introduction
The demand for enzymes, both native, and recombinant, is increasing at a tremendous pace in various industries (from food, agriculture, chemical to plastic decomposition or pharmacy). Enzymes reduce the processing time and require low energy input; they are cost effective, nontoxic, and eco-friendly. It was concluded that the global market for enzymes in industrial applications should grow from USD 6.4 billion in 2021 to USD 8.7 billion by 2026, at a compound annual growth rate (CAGR) of 6.3% for the period of 2021–2026. The global recombinant protein market was estimated at USD 1.65 billion in 2021 and is predicted to reach USD 3.93 billion by 2030, representing a CAGR of 10.12% throughout the forecast period of 2021–2030. It has also been calculated that by 2003 approximately 90% of all industrial enzymes were recombinant [1]. Recombinantly produced enzymes are applied in many fields, ranging from medicine to food and nutrition or the production of detergents, textile, leather, paper, pulp, and plastics [2,3]. Thus, the cost-effectiveness of recombinant enzyme production is an important issue in the biotechnological industry.
Escherichia coli-based heterologous systems are the most frequently used hosts for the production of recombinant enzymes [4,5,6,7,8,9]. The advantages of E. coli are its relative simplicity, fast growth in chemically defined media, growth independent of oxygen, inability to form aggregates, well-known genetics, availability of expression vectors and production strains, well-developed fermentation technologies, easy manipulation, adaptation to metabolic stress, several strains being considered biosafety level 1, and cost-effectiveness in producing recombinant proteins [5,7,9,10,11,12]. Nowadays, many proteins of commercial interest are produced in E. coli. Furthermore, about 30% of therapeutic proteins are produced using these bacteria [10,13]. The organism can accumulate heterologous proteins of up to 50% of its dry cell weight [1]. For lab-scale experiments including structural and functional studies of recombinant proteins, E. coli is the host of choice for most. Thus, the improvement of cultivation conditions and target molecule production to reduce process costs remain among the common research and industry goals.
The most frequently used strain for heterologous protein production in E. coli is the strain BL21(DE3). It offers a number of advantageous features including the lack of Lon and OmpT protease genes—resulting in the extended lifetime of recombinant proteins [12,14], faster growth in minimal medium [15], and production of less acetate during growth compared to other strains, which leads to improved production of recombinant proteins by lowering acetate stress [16,17]. The strain BL21(DE3) is mostly used together with the T7 expression system. The T7 expression system is based on the T7 promoter, which features exceptionally high transcription rates—the target protein is transcribed by the T7 polymerase, which is faster compared to native E. coli polymerases, leading to a high number of transcripts and thus a higher production of the target enzyme [6,14,18,19,20]. The BL21(DE3) strain carries a copy of the phage T7 RNA polymerase (T7RNAP) gene under the control of the lacUV5 promoter. Genes of interest are cloned under control of a T7 promoter in expression plasmids, and protein production begins upon the addition of IPTG (isopropyl-β-D-thiogalactoside), a structural non-metabolizable analogue of allolactose [12,21,22].
IPTG is currently one of the most common chemical agents used for regulating this promoter’s transcriptional activity [11,23]. IPTG itself is not degradable by β-galactosidase and its concentration remains steady throughout the experiment [23,24]. However, limitations have been observed in large-scale, as well as microplate-scale, production, including the toxicity, high cost, and demanding monitoring of culture growth [3,21,25]. The use of IPTG produces more toxic elements and can introduce physiological stress to cells in comparison to natural sugars [25]. Because of its toxicity, IPTG is not used in the production of therapeutic proteins. Moreover, unwanted production of inclusion bodies is also to be expected when using this inducer [3,23,26]. Thus, a more effective, safer, and cheaper alternative to IPTG is required.
Although IPTG is not consumed by cells, it may accumulate in cells and lead to stress. To avoid the negative effects of IPTG, several alternative inducers have also been suggested. One of them is lactose. Lactose plays two roles during expression: (1) it acts as an inducer, and (2) it is a source of carbon [23,27,28]. The application of lactose as an inducer has been previously reported [28,29,30,31]. Khani and Bagheri [23] have previously suggested skimmed milk as an attractive, cheaper alternative to IPTG for recombinant enzyme production. Thus, an inexpensive waste product could potentially serve as an inducer replacing the costly IPTG.
Lipolytic enzymes (lipases, esterases, cutinases, etc.) are attractive biocatalysts in a variety of different industries. Lipases (EC 3.1.1.3) are versatile enzymes that can catalyze the hydrolysis of triglycerides, as well as esterification, interesterification, alcoholysis, and aminolysis reactions. The market for such enzymes is estimated to have been USD 425.0 million in 2018, and it is projected to reach USD 590.2 million by 2023, growing at a CAGR of 6.8% from 2018 [32]. Lipases are the third-most-used enzyme class after proteases and amylases and their production has constantly increased throughout the years [1,33,34]. They are widely used in biodiesel, food, nutraceutical production, oil degumming and detergent manufacturing, bioremediation, agriculture, cosmetics, perfumery, the flavor industry, stereospecific compound production, esters, amino acid derivatives, fine chemical production, leather and paper, biosensors, pharmaceutical and medical industries, polyester waste conversion, and wastewater treatment [32,33,35,36,37,38,39]. E. coli represents 50% of the total heterologous production of lipases by eukaryotic and prokaryotic hosts and, from an industrial viewpoint, the selection of an efficient expression system and host for recombinant lipase production is highly important [40].
Another extensively studied group of microbial lipolytic enzymes are carboxylesterases (EC 3.1.1.1) that cleave carboxylic esters; however, the lengths of the acyl chains of the substrates are much shorter than the substrates of lipases [41,42]. Esterases display stereospecificity and regioselectivity, which make them attractive biocatalysts for the production of optically pure compounds in fine-chemical synthesis [43,44,45,46,47]. Many carboxylesterases exhibit excellent activity in organic solvents and because of this they have become among the most-used enzymes in organic compound synthesis [42]. Carboxylesterases have also gained attention for their wide application in the biodegradation of pesticides (phyrethroid, organophosphate, and carbamate), as biosensors of toxic materials, and for their ability to depolymerize plastic [48,49,50,51,52,53].
Due to their ability to catalyze reactions involving many different soluble esters, insoluble triglycerides, and various polyesters, cutinases (EC 3.1.1.74) can also be defined as lipolytic enzymes, and they are often even misclassified as esterases or lipases. However, cutinases primarily are hydrolytic enzymes that degrade cutin, the cuticular polymer of higher plants, which is a polyester composed of hydroxy and epoxy fatty acids [54,55], and they differ from classical lipases as they do not display or display little interfacial activation [55]. Cutinases have already seen use in the dairy and textile industries, household detergents, the oleochemical industry, the synthesis of structured triglycerides, polymers, and surfactants, and the production of ingredients for personal-care products, pharmaceuticals, and agrochemicals containing one or more chiral centers [54,56]. Moreover, in recent years, it has been found that cutinases can biodegrade plastic and are the most promising enzymes for solving issues related to polyester plastic pollution [53,57,58,59,60,61]. Thus, a simpler and cheaper production of recombinant lipolytic enzymes can be helpful both for industry and the fundamental investigations of these enzymes.
As models in this study, several previously designed recombinant lipolytic enzymes were used: GDEst-95 esterase [62], GD-95RM lipase [63], a fused GDEst-lip lipolytic enzyme [62], and Cut+SP putative cutinase from Streptomyces scabiei 87.22 [64,65]. A range of different inducers has been used in this study: IPTG (1 mM), different concentrations of lactose as control (2, 4, 6 mM), and different concentration of milk permeate (MP) (2, 4, 6 mM). Moreover, the enormous use of antibiotics worldwide has led to a widespread antibiotic resistance of bacteria and the use of antibiotics in the cultivation medium increases the overall price of recombinant enzyme production. Thus, lower antibiotic concentrations in the cultivation medium have also been tested in this work. This study aimed to find a simpler, cheaper, and safer way to produce recombinant enzymes. MP, a waste of milk manufacturing, has been revealed to be an attractive alternative to IPTG for the induction of recombinant enzyme production. This study is beneficial in the following points: 1. the production of industrially attractive biocatalysts in a cheaper way is presented; 2. MP showed great results for recombinant enzyme synthesis induction; and 3. the growth of recombinant E. coli BL21 (DE3) cells without additional supplementation with antibiotics in the cultivation medium and successful production of recombinant enzymes has been shown in this work.
2. Materials and Methods
2.1. Chemicals
Isopropyl-β-D-1-thiogalactopyranoside (IPTG), PageRuler™ Unstained Protein Ladder, and PageRuler™ Unstained Broad Range Protein Ladder were purchased from Thermo Fisher Scientific Baltics, Vilnius, Lithuania. PageBlue™ Protein Staining Solution, Pierce™ Unstained Protein MW Marker, and tributyrin were purchased from Thermo Scientific™, Waltham, MA, USA. Bis-acrylamide was purchased from AppliChem, Darmstadt, Germany. Tetramethylethylenediamine (TEMED) was purchased from Bio-Rad, Hercules, CA, USA. All other reagents and chemicals used in this study were purchased from Carl Roth Gmbh & Co. Kg, Karlsruhe, Germany, unless otherwise specified. All chemical reagents used in this study were of analytical grade. LACTOPRIMA LAC MPP85 (milk permeate powder MPP85, https://www.baltmilk.eu/en/products/mpp85, accessed on 4 June 2022), which was used as an inducer, was obtained from BaltMilk (Company “Pienas LT”, Kaunas, Lithuania).
2.2. Bacterial Strains and Plasmids
Escherichia coli strain BL21 (DE3) (Thermo Fisher Scientific, Waltham, MA, USA) was used for recombinant protein expression procedures. The gene of the cutinase family protein from Streptomyces scabiei 87.22 was synthesized by Invitrogen (part of Thermo Fisher Scientific) according to a gene sequence in the Streptomyces scabiei 87.22 complete genome (GenBank Accession no. FN554889) [66]; locus 8717205 bp–8717846 bp (NCBI Reference Sequence: NC_013929.1).
E. coli BL21 (DE3) cells harboring a pET-21c(+) vector with inserted genes encoding GDEst-95 esterase [62], GDEst-lip chimeric enzyme [62], GD-95RM lipase [63], and Cut+SP cutinase [64,65] have also been used in this study.
Electrocompetent E. coli BL21 (DE3) cells were prepared, and transformation via electroporation has been performed using protocols from Sambrook and Rusell 2001 [67]. Further E. coli BL21 (DE3) transformants were screened on LB agar plates containing 100 μg/mL of ampicillin.
2.3. Culture Conditions
For the cultivation of E. coli BL21 (DE3) transformants, two media were used in this work: (1) LB (Luria–Bertani) medium and (2) M9 mineral medium with modifications (mM9). The composition of LB medium was 1% peptone from casein, 0.5% yeasts extract, and 0.5% NaCl. Additionally, 1.5% agar-agar was added to prepare solid medium. mM9 medium was composed of 20% M9 salt solution (5×): 1.5% KH2PO4; 0.5% NH4Cl; 0.25% NaCl; 10% amino acid solution (10×): L-alanine (126 mg/L), L-arginine hydrochloride (522.5 mg/L), L-asparagine monohydrate (300 mg/L), L-cystine (120 mg/L), L-glutamic acid (294 mg/L), glycine (150 mg/L), L-histidine hydrochloride monohydrate (155 mg/L), L-isoleucine (263 mg/L), L-glutamine (294 mg/L), L-leucine (262 mg/L), L-lysine hydrochloride (363 mg/L), L-methionine (76 mg/L), L-phenylalanine (165 mg/L), L-proline (230 mg/L), L-serine (210 mg/L), L-threonine (238 mg/L), L-tryptophan (51 mg/L), L-valine (234 mg/L), L-aspartic acid (266 mg/L); 0.2% yeast extract; 0.05% 1000× vitamin solution: (D(+)-biotin (10 mg/L); meso-Inositol (100 mg/L); nicotinic acid (100 mg/L); pyridoxine hydrochloride (100 mg/L); riboflavin (100 mg/L), thiamine hydrochloride (100 mg/L)). After autoclaving mM9 medium was supplemented with 0.2% 1 M MgSO4 and 0.005% 1 M CaCl2. For the cultivation of E. coli BL21 (DE3) transformants in LB or mM9, the media were supplemented with 100 μg/mL (50 μg/mL; 25 μg/mL; 10 μg/mL; 0 μg/mL in optimization experiments) ampicillin. The cells were cultivated at 37 °C in pH ~7. All growth conditions are presented in Table 1.

Table 1.
Conditions used for recombinant E. coli BL21 (DE3) cell cultivation and Cut+SP, GD-95RM, GDEst-95, and GDEst-lip recombinant enzyme production. IPTG—isopropyl-β-D-thiogalactoside; MP—milk permeate.
2.4. Induction of Recombinant Enzyme Synthesis
IPTG (1 mM), lactose (2, 4, 6, mM), and MP (2, 4, 6 mM) were used for induction of recombinant enzyme synthesis. The inoculum for induction was prepared by growing one colony of a transformant of E. coli BL21(DE3) harboring a pET-21c(+) plasmid with one of the inserted genes (GD-95RM lipase, Cut+SP putative cutinase, GDEst-lip chimeric enzyme, and GDEst-95 esterase) at 37 °C overnight with agitation (150/180 rpm) in LB broth containing 100 μg/mL ampicillin. The culture was then transferred using 1% inoculum to 50/250/1000 and 2000 mL fresh mM9 medium containing the same or lower concentrations (50 μg/mL, 25 μg/mL; 10 μg/mL, and 0 μg/mL) of ampicillin to a final OD600 of 0.02–0.03. The culture was incubated at 37 °C with agitation (Table 1) until OD600 reached 0.4 and IPTG/ lactose or MP was added to a final tested concentration. All growth and induction conditions are presented in Table 1.
For protein detection by SDS-PAGE (Section 2.5), cell samples obtained at different times after induction (2, 4, 6, 8, 12, 24 h after adding the inducer) were adjusted so that their OD600 value was 0.4, mixed with 80 µL 4X SDS–PAGE sample loading buffer (50 mM Tris-HCl buffer (pH 6.8), 2% sodium dodecyl sulphate, 10% glycerol, and 0.02% bromphenol blue) and subjected to glycine or tricine SDS-PAGE analysis. The negative control was prepared by the same method using cells not subjected to IPTG/lactose/MP induction [68].
2.5. Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE)
To evaluate recombinant enzyme production, glycine-SDS-PAGE (for GD-95RM, GDEst-lip, GDEst-95) and tricine-SDS-PAGE (for Cut+SP) were used. Glycine-SDS-PAGE was carried out on 10% running gel using Laemmli’s discontinuous tris-glycine buffer system [69]. The conditions and gel compositions for tricine-SDS-PAGE were adapted according to Schägger [70] and Haider et al. [71], with minor modifications to the number of reagents. For SDS-PAGE, the Mini-PROTEAN system (Bio-Rad, Hercules, CA, USA) was used.
After electrophoresis, gels were stained with PageBlue™ protein-staining solution following the manufacturer’s recommendations. A broad range of protein standards PageRuler™ Unstained Broad Range Protein Ladder, Pierce™ Unstained Protein MW Marker, and PageRuler™ Unstained Protein Ladder were used as molecular mass markers.
2.6. Zymography and Lipolytic Activity Assay
Lipolytic activity was determined on SDS-PAGE gels using tributyrin (TB) as substrate. The gel renaturation procedure was carried out following Levisson et al. [72] with slight modification. The part of SDS-PAGE that was used for zymography analysis was washed twice with renaturation solution (20% isopropanol solution in Tris-HCl (50 mM, pH 8) buffer) for 15 min and three times using only Tris-HCl (50 mM, pH 8) buffer. The renatured gel was placed into a Petri dish and 50 mL TB-agar (agar 1.5% (w/v), 0.5% emulsified TB, and 50 mM Tris–HCl buffer (pH 8)) was poured over them and left to set. The Petri dish was incubated overnight at 30 °C. The molecular weight of the expressed enzymes that showed activity was determined by the comparison of stained protein gels and their zymogram counterpart.
For quantitative activity measurements, target enzymes were purified from the E. coli BL21 (DE3) cells collected during cultivation with MP. The purification process was carried out based on the previous reports [62,63,64,65,68]. Lipolytic activity was determined spectrophotometrically by using p-nitrophenyl (p-NP) dodecanoate as a substrate for GDEst-95, GD-95RM, and GDEst-lip enzymes, and p-NP butyrate—for Cut+SP cutinase. For the Cut+SP activity determination, the assay mixture contained 890 μL of KH2PO4 buffer (50 mM, pH 7), 100 μL of substrate solution (2.5 mM p-NP butyrate in DMSO), and 10 μL of purified enzyme solution. Before the reaction, the assay mixture was preincubated at 30 °C temperature for 10 min and then the enzyme solution was added. Other measurement conditions were used as described in previous works [62,63,68].
3. Results
3.1. Optimization of Milk Permeate (MP) Concentration
Since the milk permeate powder (LACTOPRIMA LAC MPP85) is composed of >82% lactose, <2% fat, and <4% protein, first, lactose as an inducer was tested. In this step, GD-95RM lipase and Cut+SP enzymes were used as models because of their medium (~43 kDa) and low molecular weight (~25 kDa), respectively. The E. coli BL21 (DE3) cells harboring plasmids encoding GD-95RM lipase and Cut+SP cutinase were grown in 50 mL mM9 medium, and lactose to a final concentration of 2, 4, and 6 mM was added at the point of induction. The results clearly indicated GD-95RM lipase and Cut+SP putative cutinase production after induction with 2, 4, and 6 mM lactose (Figure 1). The protein bands corresponding to the size of GD-95RM lipase were detected immediately 2 h post-induction (Figure 1c), while the accumulation of Cut+SP cutinase in the cells started 4 h post-induction (Figure 1a).
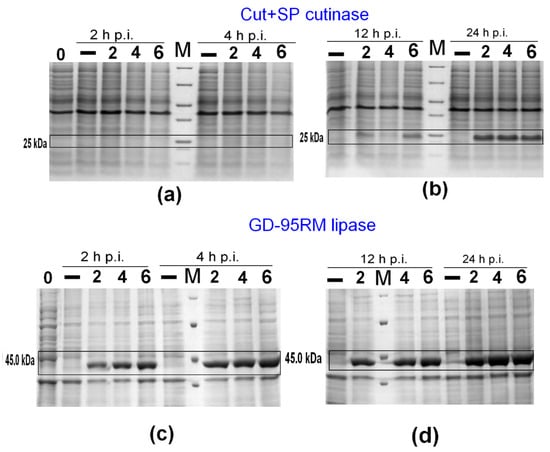
Figure 1.
The expression of recombinant Cut+SP cutinase (a,b) and GD-95RM lipase (c,d) using lactose as an inducer. M—Pierce™ Unstained Protein MW Marker; 2/4/6—concentration of lactose as an inducer (mM); 0—time point when E. coli BL21 (DE3) has been subjected for induction; –—control sample of recombinant E. coli BL21 (DE3) cells growing without induction; p.i.—post-induction time in hours. The target enzymes are marked with black boxes. Tricine-SDS-PAGE was used for Cut+SP analysis and glycine-SDS-PAGE for GD-95RM, respectively.
The obtained results revealed that 2 mM lactose is already a suitable concentration for production of target recombinant enzymes; however, for the next-stage experiments, a 4 mM concentration was chosen to ensure effective expression of recombinant enzymes.
In the next step, the volume of the media was increased to 250 mL and MP was tested as an inducer; 2, 4, and 6 mM concentrations of MP was tested for induction of synthesis of Cut+SP and GD-95RM enzymes to compare MP’s effectiveness with that of lactose. The SDS-PAGE analysis confirmed target enzyme synthesis after recombinant E. coli BL21 (DE3) treatment with 2, 4, and 6 mM MP at different post-induction time points (Figure 2). However, the accumulation of Cut+SP was not detected at a 24 h post-induction time point. It is worth noticing that production of Cut+SP, using both IPTG and lactose as inducers, also could not be detected using SDS-PAGE analysis after 24 h post-induction. Moreover, SDS-PAGE analysis of Cut+SP producing recombinant E. coli cells did not show any cellular proteins either. This suggests a possible toxic effect of cutinase to recombinant host cells using a larger production scale and this case will be discussed in further detail in the Discussion section.
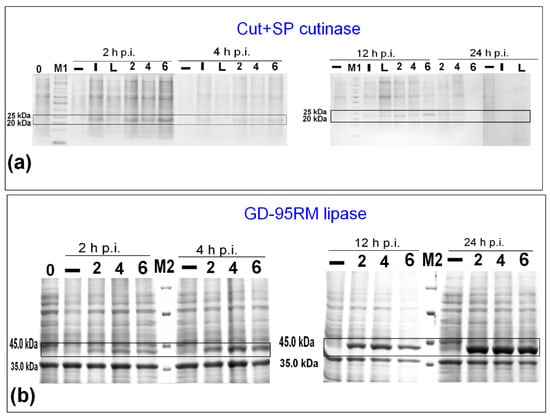
Figure 2.
The expression induction of recombinant Cut+SP cutinase (a) and GD-95RM lipase (b) using MP as an inducer. M1—PageRuler™ Unstained Protein Ladder; M2—Pierce™ Unstained Protein MW Marker; 2/4/6—concentration of MP as an inducer (mM); 0—time point when E. coli BL21 (DE3) was subjected for induction; –—control sample of recombinant E. coli BL21 (DE3) cells growing without induction; I—for induction IPTG (1 mM) was used as an inducer; L—for induction lactose (4 mM) was used as an inducer; p.i.—post-induction time in hours. The target enzymes are marked with black boxes. Tricine-SDS-PAGE was used for Cut+SP analysis and glycine-SDS-PAGE for GD-95RM.
As was the case with lactose, 2 mM MP is an appropriate concentration to induce the synthesis of recombinant enzymes. In this study, to ensure effective production of recombinant enzymes, a higher concentration (4 mM) of MP was chosen for further experiments.
3.2. Influence of Lower Antibiotic Concentrations on Recombinant Enzyme Production
The enormous use of different antibiotics leads to widespread resistance of bacteria and also increases production costs of recombinant enzymes. To decrease costs of the synthesis process, lower concentrations of ampicillin were tested in the cultivation media. The obtained results suggested that recombinant enzyme production is possible without additional supplementation of antibiotics (Figure 3). However, some antibiotics from an overnight inoculum still enter the fresh cultivation medium. It was calculated that the final concentration of ampicillin in the fresh medium can be ~0.5 µg/mL. It could be the case that such an amount of antibiotic is suitable to maintain recombinant plasmids within cells. A significant difference in recombinant enzyme production using a lower concentration of ampicillin was not detected (Figure 3). The protein bands corresponding to the target Cut+SP putative cutinase (Figure 3a,b) and GD-95RM lipase (Figure 3c), were clearly detected in mM9 medium at different time points post-induction with MP (4 mM). The different post-induction time points for Cut+SP and GD-95RM are presented to illustrate various analysis periods. The Cut+SP cutinase and GD-95RM lipase was successfully produced even 12 h and 24 h post-induction (Figure 3b,c). It is important to emphasize that a similar yield of recombinant enzymes can be achieved using MP instead IPTG as an inducer (Figure 2 and Figure 3).
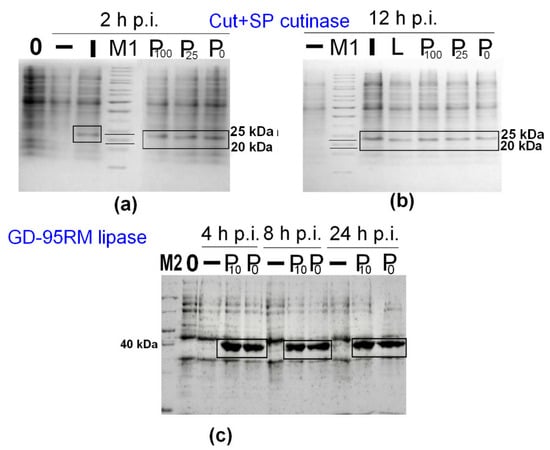
Figure 3.
Influence of ampicillin concentration in the cultivation medium for recombinant Cut+SP (a,b) and GD-95RM enzymes (c) synthesis. M1—PageRuler™ Unstained Protein Ladder; M2—PageRuler™ Unstained Broad Range Protein Ladder; P—for induction MP (4 mM) was used as an inducer; 100, 25, 10, 0—mark a concentration of ampicillin (µg/ mL) added to the medium; I—for induction IPTG (1 mM) as an inducer was used as an inducer; L—for induction lactose (4 mM) as an inducer was used as an inducer; –—control sample of recombinant E. coli BL21 (DE3) cells growing without induction; p.i.—post-induction time in hours. The target enzymes are marked with black boxes. Tricine-SDS-PAGE was used for Cut+SP analysis and glycine-SDS-PAGE for GD-95RM.
3.3. Synthesis of GDEst-lip and GDEst-95 in Low Scale
MP as an inducer was also tested for production of GDEst-95 esterase (55 kDa) and fused GDEst-lip enzyme (98 kDa) to evaluate the suitability of optimized conditions for synthesis of higher molecular weight lipolytic enzymes. Moreover, both biocatalysts showed attractive industrial characteristics: temperature activity (5-80 °C) and thermostability, as well as high tolerance to organic solvents [62,73]. Thus, the cost-efficient production of these enzymes is crucial for their application and further testing. The growth at a lower scale was performed in 250 mL mM9 cultivation medium without additional supplementation of ampicillin. Glycine-SDS-PAGE results indicated the successful production of target enzymes after E. coli BL21 (DE3) cell treatment with 4 mM MP (Figure 4). The molecular weight of the newly produced proteins corresponded to the molecular weights of GDEst-95 (Figure 4a) and GDEst-lip enzymes (Figure 4b). The SDS-PAGE analysis did not show significant differences in recombinant enzyme synthesis using IPTG (1 mM) and MP (4 mM), confirming MP as a potential alternative inducer able to rival IPTG. The selected mM9 medium and induction conditions allowed the production and accumulation of both enzymes even after 24 h post-induction.
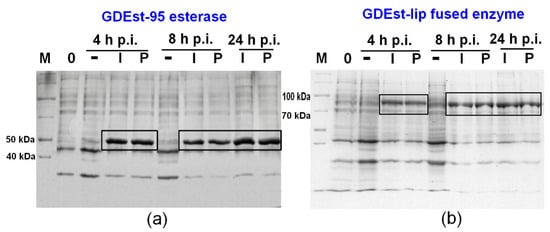
Figure 4.
Synthesis of recombinant GDEst-95 esterase (a) and GDEst-lip fused enzyme (b) in E. coli BL21 (DE3) cells using 4 mM milk permeate (P) as an inducer. Induction in mM9 medium supplemented with 100 µg/mL ampicillin and 1 mM of IPTG (I) as an inducer were used as control. M—PageRuler™ Unstained Broad Range Protein Ladder; –—control sample of recombinant E. coli BL21 (DE3) cells growing without induction; p.i.—post-induction time (in hours). The target enzymes are marked with black boxes. Glycine-SDS-PAGE was used for analysis.
3.4. Up-Scale Synthesis of Cut+SP, GD-95RM, GDEst-lip and GDEst-95 Enzymes
For the up-scaling synthesis experiments of Cut+SP, GD-95RM, GDEst-lip, and GDEst-95 enzymes, one (Figure 5) and two liters (Figure 6) of mM9 medium were used. The recombinant E. coli BL21 (DE3) cells were cultivated 24 h post-induction without additional supplementation of ampicillin, and MP (4 mM) was used as an inducer. Data analysis clearly indicated GD-95RM, GDEst-lip, and GDEst-95 enzyme synthesis 2, 4, 12, and 24 h post-induction in 1 L (Figure 5) and 2 L (Figure 6) media. Because the synthesis of the Cut+SP putative cutinase was not detected in small-scale experiments at a 24 h post-induction time point (Figure 2), the large-scale cultivation experiments were stopped at a final time point of 12 h post-induction (Figure 5a and Figure 6a).
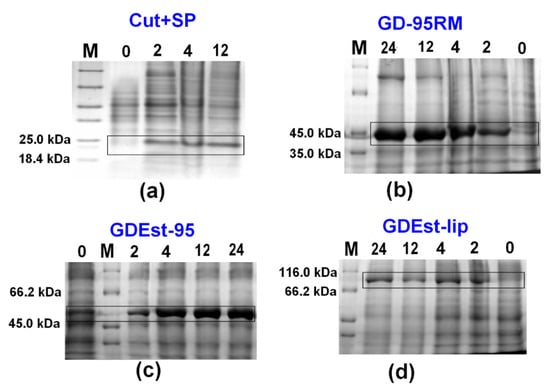
Figure 5.
Analysis of production of recombinant Cut+SP (a), GD-95RM (b), GDEst-95 (c) and GDEst-lip (d) enzymes in 1 L mM9 cultivation medium without additional supplementation with ampicillin and using MP (4 mM) as an inducer. M—Pierce™ Unstained Protein MW Marker; 0/2/4/12/24—time point after induction with MP (in hours). The target enzymes are marked with black boxes. Tricine-SDS-PAGE was used for Cut+SP analysis and glycine-SDS-PAGE for GD-95RM, GDEst-95, and GDEst-lip.
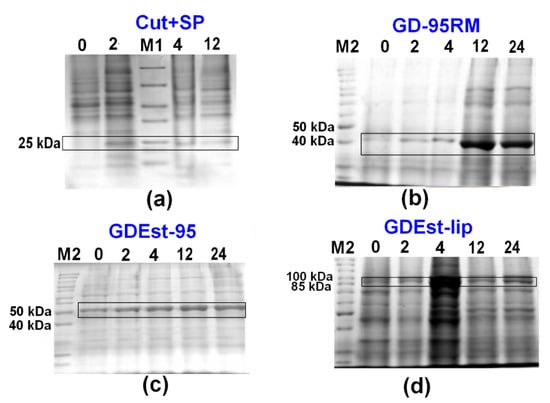
Figure 6.
Analysis of production of recombinant Cut+SP (a), GD-95RM (b), GDEst-95 (c), and GDEst-lip (d) enzymes in 2 L mM9 cultivation medium without additional supplementation with ampicillin and using MP (4 mM) as an inducer. M1—Pierce™ Unstained Protein MW Marker; M2—PageRuler™ Unstained Protein Ladder; 0/2/4/12/24—time point after induction with MP (in hours). The target enzymes are marked with black boxes. Tricine-SDS-PAGE was used for Cut+SP analysis and glycine-SDS-PAGE for GD-95RM, GDEst-95, and GDEst-lip.
Zymography analysis confirmed production of active target recombinant enzymes in 2 L of cultivation medium at different time points after induction with MP (4 mM) (Figure 7).
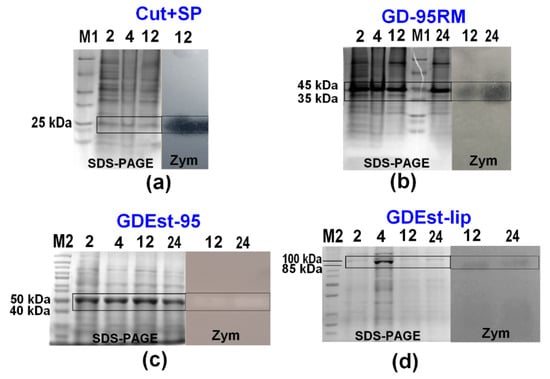
Figure 7.
Activity analysis of recombinant Cut+SP (a), GD-95RM (b), GDEst-95 (c), and GDEst-lip (d) enzymes in zymograms (Zym) during production in 2 L mM9 cultivation medium without additional supplementation with ampicillin and using MP (4 mM) as an inducer. M1—Pierce™ Unstained Protein MW Marker; M2—PageRuler™ Unstained Protein Ladder; 2/4/12/24—time point after induction with MP (in hours). The target enzymes are marked with black boxes. Tricine-SDS-PAGE was used for Cut+SP analysis, and glycine-SDS-PAGE for GD-95RM, GDEst-95, and GDEst-lip.
Additionally, the recombinant E. coli BL21 (DE3) cells produced Cut+SP, GDEst-95, GD-95RM, and GDEst-lip lipolytic enzymes were collected at the end of expression (Cut+SP: 12 h post-induction, and other enzymes: 24 h post-induction), stored at –20 °C and further used for target biocatalysts purification and application experiments (not shown). Confirmation of fully active enzyme production was done by using p-NP fatty acid esters as substrates. The activity of purified recombinant GDEst-95 esterase, GD-95RM lipase, and GDEst-lip enzymes was 120 U/mg, 1800 U/mg, and 580 U/mg, respectively, and these values were very similar to previous detected activities using p-NP dodecanoate as a substrate [62,63]. The activity of Cut+SP was detected using p-PN butyrate as substrate and was 540 U/mg.
4. Discussion
The cheaper production of recombinant proteins is a goal of both scientists and industry. In this study, it was shown for the first time that milk permeate is a suitable inducer for target enzyme production, while lactose has already been previously shown to be usable as an inducer [28,29,30,31,74,75,76,77]. There have already been strategies proposed to replace IPTG. One of them is the use of an autoinduction medium consisting of at least two carbon sources: glucose and lactose (glycerol can also be added to increase yields) [78,79,80,81]. The induction starts once all the available glucose has been consumed. The glucose metabolism prevents uptake of lactose until glucose is depleted, usually in mid to late log phase. Consumption of glycerol and lactose follows, the latter being also the inducer of lac-controlled protein expression [21,82]. The researchers also proposed a new acetate containing a medium for the production of a recombinant protein in E. coli BL21(DE3) [83] together with 1 mM lactose as inducer. The authors [83] took advantage of E. coli BL21(DE3) as a high-acetate-tolerant host, which allows us to reach high-density cell cultures with controlled feeding. It was observed that acetate metabolism can be employed to efficiently produce recombinant proteins in E. coli BL21 by growth in a culture medium with acetate as a carbon source [83].
A few years ago, Briand and co-authors [21] suggested a SILEX (self-inducible expression) system as a convenient, cost-effective alternative that did not required IPTG induction. The SILEX system is presented as compatible with classical culture media temperatures and allows protein expression modulation. This system is based on the engineered BL21(DE3) strain containing the SILEX plasmid encoding the human heat shock protein 70 (hHsp70) and a second plasmid encoding for the protein of interest [21]. The system allows recombinant protein overexpression using a lac-inducible plasmid by autoinduction. hHsp70 is a stress protein that plays an anti-aggregation function and demonstrates a high affinity between the two partners and an autoinduction phenomenon. However, in this strategy, two plasmids and co-expression are required.
Regarding the toxicity and cost of IPTG, milk manufacturing wastes that are rich in lactose such as skimmed milk [23] or milk permeate can be used instead of IPTG, helping overcome problems arising from the use of IPTG and making the overall production of recombinant biocatalysts more cost-efficient. During milk processing, lactose is separated from the fluid milk or other fluid dairy products to improve its functionality and storage stability, and to make reduced lactose products for consumers [84]. Ultrafiltration itself is a very useful process for producing reduced lactose dairy products. Ultrafiltration membranes retain all the fat globules and milk proteins and allow lactose to permeate. Permeate contains lactose, vitamins, and minerals, while skimmed milk is also known as fat-free or non-fat milk. Permeate is a very rich in lactose (>82%), a dairy manufacturing waste product obtained after milk or skim-milk processing. Two forms of permeates are known: milk and whey permeates. Milk permeate has a similar composition to whey permeate, but because of the fewer processing steps, its organoleptic profile may be different. The permeates usually have a minimum of 76% lactose, a maximum of 14% ash, and typically between 2–7% protein. MP used in this study also had fat (<2%), protein (<4%), and ash (<7.5%). Ash can consist of different chemical components. However, the main chemical component of ash is carbon, with varying amounts of other elements including calcium, magnesium, potassium, manganese, and phosphorus [85]. The minerals and fatty acids included in MP composition can have a positive effect on microorganism growth, and Ca2+ and Mg2+ can improve the activity of recombinant and native enzymes.
In this study, mM9 medium has been used instead of the classical LB medium. The mM9 medium has been chosen because of the possible high basic induction level in LB medium. The lower amount of yeast extract (0.2%) and amino acid solution instead of peptone was used in the composition of the mM9 medium. It was shown that the use of plant-derived peptones (soy peptone and malt extract) in a culture medium causes the T7-lac expression system to leak [86]. The authors detected that the presence of raffinose and stachyose (galactoside derivatives) in such peptones cause a premature and uncontrolled induction of a gene expression controlled by a promoter. In soy peptones, there is a large amount of saccharides and other compounds that in some expression systems can act as gene-inducing factors [86]. Defined media are also preferred in industrial applications because of the possibility of easy scale-up and careful control of all nutrient concentrations.
Another important aspect that could be discussed is the difference in lipases/esterases and Cut+SP production levels. It was observed that the yield of recombinant Cut+SP cutinase was lower than other enzymes produced, and Cut+SP was not synthesized in E. coli after 24 h post-induction. This may be related to the fact that the GD-95RM lipase/GDEst-95 esterase/GDEst-lip fused enzyme do not have a signal peptide and are accumulated in high levels using heterologous systems [62,63], while the Cut+SP putative cutinase possesses an N-terminal signal peptide for secretion via the Sec secretion system. Additional experiments showed that the secreted form of Cut+SP cutinase can be successfully concentrated from culture medium and purified through IMAC (results not shown in this manuscript). Moreover, it is known that T. fusca cutinase possesses hydrolytic activity toward phospholipids [87]. Our preliminary analysis of Cut+SP cutinase also proposed the ability of Cut+SP cutinase to negatively affect phospholipidic membranes, but this currently requires further experimental evidence. This hypothesis could explain why after induction with IPTG, lactose, and MP, no protein bands were observed in SDS-PAGE 24 h post-induction. It is possible that Cut+SP production results in the disturbance of the cell envelope of recombinant E. coli BL21 (DE3) for long-term cultivation in large volumes of culture media. Another important aspect is that additional compounds like ash, which are included in MP composition, can lead to activation of Cut+SP cutinase, resulting in higher phospholipase activity. It is well-documented that calcium ions can play structural and thermal stabilization roles for microbial cutinases [88,89,90,91,92]. It can thus be hypothesized that Ca2+, which can be included in the composition of MP, can improve the phospholipase activity of the Cut+SP enzyme; however, a deeper characterization of Cut+SP as a cutinase with industrial applications, as well as a phospholipase, is a large subject of our further research. Currently, only the cutinase/suberinase Sub1 from S. scabiei EF-35 has been characterized [93,94].
In our study, we decided to investigate the possibility of using lower concentrations of antibiotics in culture medium as well. Antibiotics are used during gene engineering experiments and protein expression processes as both selection markers and pressure to ensure that only bacteria with plasmids coding resistance genes are allowed to grow [95]. However, at larger scales, the use of antibiotics results in higher production costs, environmental pollution, and regulatory restrictions [12]. Thus, the optimization of recombinant enzyme production included not only testing a cheaper and safer inducer but also the possibility of using a lower concentration of antibiotics. It is known that cultures growing without selection pressure tend to be outcompeted by descendants that have lost the plasmid. Using low concentrations of antibiotic could not maintain a sufficient selection pressure to ensure only cells with the plasmids grow. It was reported that for recombinant E. coli cells growing in the absence of antibiotics only 5% of the population retains the plasmid [12]. Several authors investigated effects of ampicillin concentration on recombinant enzyme production and plasmid copy numbers [95,96]; however, the tested concentrations range was 50–300 µg/mL. Our results suggest that only an ~0.5 µg/mL concentration of ampicillin can support production of recombinant enzymes. The qualitative analysis of plasmid maintenance (plasmid DNA purification from the same recombinant cell amounts at the start and end points of induction; restriction analysis) suggested that target genetic constructs encoded Cut+SP, GD-95RM, GDEst-95, and GDEst-lip enzymes can be successfully purified from recombinant E. coli BL21 (DE3) cells after 24 h post-induction (in case of Cut+SP cutinase, the plasmid DNA has been purified after 12 h) (results not included in the report). Although additional quantitative experiments are required to count plasmid copy numbers, there is no doubt that we can reach target enzyme-production levels with lower concentrations of antibiotics. The development and use of an antibiotic-free selection system would be a high breakthrough in the future.
This report highlights production of four different recombinant lipolytic enzymes (GD-95RM lipase, GDEst-95 esterase, Cut+SP putative cutinase, and GDEst-lip-fused enzyme) from low laboratory scale-up to a 2 L medium using a cheaper inducer than IPTG and a lower antibiotic concentration. It can be hypothesized that the selected cultivation and induction conditions could be successfully applied in even higher-scale experiments or industrial production. The obtained results clearly indicate that milk permeate can be successfully used for production of value-added products like recombinant biocatalysts.
5. Conclusions
The obtained results suggested that use of milk permeate would be a cost-effective alternative to produce recombinant proteins and could solve the problems related to induction with expensive materials like IPTG. Moreover, it was shown that recombinant enzyme production can be achieved using only ~0.5 µg/mL concentration of antibiotic in a culture medium. This may be useful to reduce not only the price of recombinant protein production but also the huge worldwide crisis of bacterial antibiotic resistance.
Author Contributions
Conceptualization, A.G. and R.G.; methodology, A.G. and R.G.; investigation, A.G., T.B.; resources, E.L.; writing—original draft preparation, R.G.; writing—review and editing, A.G and R.G; visualization, A.G. and R.G.; supervision, R.G.; funding acquisition, A.G and R.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the “Development of scientific competence of scientists, other researchers, students through practical scientific activities” measure, Grant No. P-SV-22-160, under grant agreements with the Research Council of Lithuania (LMTLT).
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors are very grateful to BaltMilk (Company “Pienas LT”) for the milk permeate powder and to L. Kalediene (Life Sciences Center, Vilnius University) for allowing to use the Wisd23 ViseStir MSH-D hotplate magnetic stirrers.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Demain, A.L.; Vaishnav, P. Production of recombinant enzymes. In Reference Module in Food Science; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar] [CrossRef]
- Liu, L.; Yang, H.; Shin, H.-D.; Chen, R.R.; Li, J.; Du, G.; Chen, J. How to achieve high-level expression of microbial enzymes. Bioengineered 2013, 4, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Hausjell, J.; Weissensteiner, J.; Molitor, C.; Halbwirth, H.; Spadiut, O. E. coli HMS174(DE3) is a sustainable alternative to BL21(DE3). Microb. Cell Factories 2018, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Makino, T.; Skretas, G.; Georgiou, G. Strain engineering for improved expression of recombinant proteins in bacteria. Microb. Cell Factories 2011, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Fakruddin, M.; Mazumdar, R.M.; Mannan, K.S.; Chowdhury, A.; Hossain, M.N. Critical Factors Affecting the Success of Cloning, Expression, and Mass Production of Enzymes by Recombinant E. coli. ISRN Biotechnol. 2013, 2013, 590587. [Google Scholar] [CrossRef] [PubMed]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef] [PubMed]
- Idalia, V.-M.N.; Bernardo, F. Escherichia coli as a model organism and its application in biotechnology. In Escherichia coli-Recent Advances on Physiology, Pathogenesis and Biotechnological Applications; Amidou, S., Ed.; IntchOpen: London, UK, 2017; pp. 253–274. [Google Scholar] [CrossRef]
- Castiñeiras, T.S.; Williams, S.G.; Hitchcock, A.G.; Smith, D.C. E. coli strain engineering for the production of advanced biopharmaceutical products. FEMS Microbiol. Lett. 2018, 365, fny162. [Google Scholar] [CrossRef]
- Terol, G.L.; Gallego-Jara, J.; Martínez, R.A.S.; Vivancos, A.M.; Díaz, M.C.; Puente, T.D.D. Impact of the Expression System on Recombinant Protein Production in Escherichia coli BL21. Front. Microbiol. 2021, 12, 682001. [Google Scholar] [CrossRef]
- Huang, C.-J.; Lin, H.; Yang, X. Industrial production of recombinant therapeutics in Escherichia coli and its recent advancements. J. Ind. Microbiol. Biotechnol. 2012, 39, 383–399. [Google Scholar] [CrossRef]
- Theisen, M.; Liao, J.C. Industrial biotechnology: Escherichia coli as a host. In Industrial Biotechnology: Microorganisms, 1st ed.; Wittmann, C., Liao, J.C., Eds.; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2017; Volume 1, pp. 149–181. [Google Scholar] [CrossRef]
- Rosano, G.L.; Morales, E.S.; Ceccarelli, E.A. New tools for recombinant protein production in Escherichia coli: A 5-year update. Protein Sci. 2019, 28, 1412–1422. [Google Scholar] [CrossRef]
- Baeshen, M.N.; Al-Hejin, A.M.; Bora, R.S.; Ahmed, M.M.M.; Ramadan, H.A.I.; Saini, K.S.; Baeshen, N.A.; Redwan, E.M. Production of Biopharmaceuticals in E. coli: Current Scenario and Future Perspectives. J. Microbiol. Biotechnol. 2015, 25, 953–962. [Google Scholar] [CrossRef]
- Ratelade, J.; Miot, M.-C.; Johnson, E.; Betton, J.-M.; Mazodier, P.; Benaroudj, N. Production of Recombinant Proteins in the lon -Deficient BL21(DE3) Strain of Escherichia coli in the Absence of the DnaK Chaperone. Appl. Environ. Microbiol. 2009, 75, 3803–3807. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Jeong, H.; Kwon, S.-K.; Kim, J.F. Genomics, biological features, and biotechnological applications of Escherichia coli B: “is B for better?!”. In Systems Biology and Biotechnology of Escherichia coli; Lee, S.Y., Ed.; Springer: Dordrecht, The Netherlands, 2009; pp. 1–17. [Google Scholar] [CrossRef]
- Son, Y.-J.; Phue, J.-N.; Trinh, L.B.; Lee, S.J.; Shiloach, J. The role of Cra in regulating acetate excretion and osmotic tolerance in E. coli K-12 and E. coli B at high density growth. Microb. Cell Factories 2011, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, F.; Wang, W.; Yao, X.; Wei, D.; Cheng, H.; Deng, Z. Improving the Expression of Recombinant Proteins in E. coli BL21 (DE3) under Acetate Stress: An Alkaline pH Shift Approach. PLoS ONE 2014, 9, e112777. [Google Scholar] [CrossRef] [PubMed]
- Studier, F.W.; Moffatt, B.A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 1986, 189, 113–130. [Google Scholar] [CrossRef]
- Tegel, H.; Ottosson, J.; Hober, S. Enhancing the protein production levels in Escherichia coli with a strong promoter. FEBS J. 2010, 278, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Durani, V.; Sullivan, B.J.; Magliery, T.J. Simplifying protein expression with ligation-free, traceless and tag-switching plasmids. Protein Expr. Purif. 2012, 85, 9–17. [Google Scholar] [CrossRef]
- Briand, L.; Marcion, G.; Kriznik, A.; Heydel, J.-M.; Artur, Y.; Garrido, C.; Seigneuric, R.; Neiers, F. A self-inducible heterologous protein expression system in Escherichia coli. Sci. Rep. 2016, 6, 33037. [Google Scholar] [CrossRef] [PubMed]
- Marbach, A.; Bettenbrock, K. lac operon induction in Escherichia coli: Systematic comparison of IPTG and TMG induction and influence of the transacetylase LacA. J. Biotechnol. 2012, 157, 82–88. [Google Scholar] [CrossRef]
- Khani, M.-H.; Bagheri, M. Skimmed milk as an alternative for IPTG in induction of recombinant protein expression. Protein Expr. Purif. 2020, 170, 105593. [Google Scholar] [CrossRef]
- Donovan, R.S.; Robinson, C.W.; Glick, B.R. Review: Optimizing inducer and culture conditions for expression of foreign proteins under the control of thelac promoter. J. Ind. Microbiol. Biotechnol. 1996, 16, 145–154. [Google Scholar] [CrossRef]
- Dvorak, P.; Chrast, L.; Nikel, P.I.; Fedr, R.; Soucek, K.; Sedlackova, M.; Chaloupkova, R.; de Lorenzo, V.; Prokop, Z.; Damborsky, J. Exacerbation of substrate toxicity by IPTG in Escherichia coli BL21(DE3) carrying a synthetic metabolic pathway. Microb. Cell Factories 2015, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Haddadin, F.T.; Harcum, S.W. Transcriptome profiles for high-cell-density recombinant and wild-type Escherichia coli. Biotechnol. Bioeng. 2005, 90, 127–153. [Google Scholar] [CrossRef] [PubMed]
- Ko, K.-S.; Kruse, J.; Pohl, N.L. Synthesis of Isobutyl-C-galactoside (IBCG) as an Isopropylthiogalactoside (IPTG) Substitute for Increased Induction of Protein Expression. Org. Lett. 2003, 5, 1781–1783. [Google Scholar] [CrossRef] [PubMed]
- Menzella, H.G.; Ceccarelli, E.A.; Gramajo, H.C. Novel escherichia coli strain allows efficient recombinant protein production using lactose as inducer. Biotechnol. Bioeng. 2003, 82, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Woyski, D.; Cupp-Vickery, J.R. Enhanced Expression of Cytochrome P450s from lac-Based Plasmids Using Lactose as the Inducer. Arch. Biochem. Biophys. 2001, 388, 276–280. [Google Scholar] [CrossRef]
- Striedner, G.; Cserjan-Puschmann, M.; Pötschacher, F.; Bayer, K. Tuning the Transcription Rate of Recombinant Protein in Strong Escherichia coli Expression Systems through Repressor Titration. Biotechnol. Prog. 2003, 19, 1427–1432. [Google Scholar] [CrossRef]
- Kotik, M.; Kočanová, M.; Marešová, H.; Kyslík, P. High-level expression of a fungal pyranose oxidase in high cell-density fed-batch cultivations of Escherichia coli using lactose as inducer. Protein Expr. Purif. 2004, 36, 61–69. [Google Scholar] [CrossRef]
- Chandra, P.; Enespa; Singh, R.; Arora, P.K. Microbial lipases and their industrial applications: A comprehensive review. Microb. Cell Fact. 2020, 19, 169. [Google Scholar] [CrossRef]
- Borrelli, G.M.; Trono, D. Recombinant Lipases and Phospholipases and Their Use as Biocatalysts for Industrial Applications. Int. J. Mol. Sci. 2015, 16, 20774–20840. [Google Scholar] [CrossRef]
- Yao, W.; Liu, K.; Liu, H.; Jiang, Y.; Wang, R.; Wang, W.; Wang, T. A Valuable Product of Microbial Cell Factories: Microbial Lipase. Front. Microbiol. 2021, 12, 743377. [Google Scholar] [CrossRef]
- Hasan, F.; Shah, A.A.; Hameed, A. Industrial applications of microbial lipases. Enzym. Microb. Technol. 2006, 39, 235–251. [Google Scholar] [CrossRef]
- Ramnath, L.; Sithole, B.; Govinden, R. Classification of lipolytic enzymes and their biotechnological applications in the pulping industry. Can. J. Microbiol. 2017, 63, 179–192. [Google Scholar] [CrossRef]
- Vanleeuw, E.; Winderickx, S.; Thevissen, K.; Lagrain, B.; Dusselier, M.; Cammue, B.P.A.; Sels, B.F. Substrate-Specificity of Candida rugosa Lipase and Its Industrial Application. ACS Sustain. Chem. Eng. 2019, 7, 15828–15844. [Google Scholar] [CrossRef]
- Kumar, A.; Gudiukaite, R.; Gricajeva, A.; Sadauskas, M.; Malunavicius, V.; Kamyab, H.; Sharma, S.; Sharma, T.; Pant, D. Microbial lipolytic enzymes—promising energy-efficient biocatalysts in bioremediation. Energy 2019, 192, 116674. [Google Scholar] [CrossRef]
- Vishnoi, N.; Dixit, S.; Mishra, J. Microbial Lipases and Their Versatile Applications. In Microbial Enzymes: Roles and Applications in Industries. Microorganisms for Sustainability; Arora, N., Mishra, J., Mishra, V., Eds.; Springer: Singapore, 2020; Volume 11, pp. 207–230. [Google Scholar] [CrossRef]
- Contesini, F.J.; Davanço, M.G.; Borin, G.P.; Vanegas, K.G.; Cirino, J.P.G.; De Melo, R.R.; Mortensen, U.H.; Hildén, K.; Campos, D.R.; Carvalho, P.D.O. Advances in Recombinant Lipases: Production, Engineering, Immobilization and Application in the Pharmaceutical Industry. Catalysts 2020, 10, 1032. [Google Scholar] [CrossRef]
- Oh, C.; Kim, T.D.; Kim, K.K. Carboxylic Ester Hydrolases in Bacteria: Active Site, Structure, Function and Application. Crystals 2019, 9, 597. [Google Scholar] [CrossRef]
- Johan, U.U.M.; Rahman, R.N.Z.R.A.; Kamarudin, N.H.A.; Ali, M.S.M. An integrated overview of bacterial carboxylesterase: Structure, function and biocatalytic applications. Colloids Surfaces B Biointerfaces 2021, 205, 111882. [Google Scholar] [CrossRef] [PubMed]
- Baba, A.; Yoshioka, T. Characterization of chemo- and regioselectivity in enzyme-catalyzed consecutive hydrolytic deprotection of methyl acetyl derivatives of 1-β-O-acyl glucuronides. J. Mol. Catal. B Enzym. 2011, 69, 74–82. [Google Scholar] [CrossRef]
- Jeon, J.H.; Kim, S.-J.; Lee, H.S.; Cha, S.-S.; Lee, J.H.; Yoon, S.-H.; Koo, B.-S.; Lee, C.-M.; Choi, S.H.; Lee, S.H.; et al. Novel Metagenome-Derived Carboxylesterase That Hydrolyzes β-Lactam Antibiotics. Appl. Environ. Microbiol. 2011, 77, 7830–7836. [Google Scholar] [CrossRef]
- Romano, D.; Bonomi, F.; de Mattos, M.C.; Fonseca, T.D.S.; Oliveira, M.D.C.F.D.; Molinari, F. Esterases as stereoselective biocatalysts. Biotechnol. Adv. 2015, 33, 547–565. [Google Scholar] [CrossRef]
- Bollinger, A.; Molitor, R.; Thies, S.; Koch, R.; Coscolín, C.; Ferrer, M.; Jaeger, K.-E. Organic-Solvent-Tolerant Carboxylic Ester Hydrolases for Organic Synthesis. Appl. Environ. Microbiol. 2020, 86, e00106-20. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Jeong, G.S.; Lee, H.W.; Kim, H. Biochemical characterization of a family IV esterase with R-form enantioselectivity from a compost metagenomic library. Appl. Biol. Chem. 2021, 64, 1–16. [Google Scholar] [CrossRef]
- Hajighasemi, M.; Nocek, B.P.; Tchigvintsev, A.; Brown, G.; Flick, R.; Xu, X.; Cui, H.; Hai, T.; Joachimiak, A.; Golyshin, P.N.; et al. Biochemical and Structural Insights into Enzymatic Depolymerization of Polylactic Acid and Other Polyesters by Microbial Carboxylesterases. Biomacromolecules 2016, 17, 2027–2039. [Google Scholar] [CrossRef] [PubMed]
- Singh, B. Review on Microbial Carboxylesterase: General Properties and Role in Organophosphate Pesticides Degradation. Biochem. Mol. Biol. 2014, 2, 1. [Google Scholar] [CrossRef]
- Mustafa, S.A. The Development of Bacterial Carboxylesterase Biological Recognition Elements for Cocaine Detection. Mol. Biotechnol. 2018, 60, 601–607. [Google Scholar] [CrossRef]
- Kaushal, J.; Khatri, M.; Arya, S.K. Recent insight into enzymatic degradation of plastics prevalent in the environment: A mini—review. Clean. Eng. Technol. 2021, 2, 100083. [Google Scholar] [CrossRef]
- Ghodke, V.M.; Punekar, N.S. Environmental role of aromatic carboxylesterases. Environ. Microbiol. 2021, 24, 2657–2668. [Google Scholar] [CrossRef]
- Gricajeva, A.; Nadda, A.K.; Gudiukaite, R. Insights into polyester plastic biodegradation by carboxyl ester hydrolases. J. Chem. Technol. Biotechnol. 2021, 97, 359–380. [Google Scholar] [CrossRef]
- Carvalho, C.M.L.; Aires-Barros, M.R.; Cabral, J.M.S. Cutinase structure, function and biocatalytic applications. Electron. J. Biotechnol. 1998, 1, 160–173. [Google Scholar] [CrossRef]
- Dutta, K.; Sen, S.; Veeranki, V.D. Production, characterization and applications of microbial cutinases. Process. Biochem. 2009, 44, 127–134. [Google Scholar] [CrossRef]
- Martínez, A.; Maicas, S. Cutinases: Characteristics and Insights in Industrial Production. Catalysts 2021, 11, 1194. [Google Scholar] [CrossRef]
- Danso, D.; Schmeisser, C.; Chow, J.; Zimmermann, W.; Wei, R.; Leggewie, C.; Li, X.; Hazen, T.; Streit, W.R. New Insights into the Function and Global Distribution of Polyethylene Terephthalate (PET)-Degrading Bacteria and Enzymes in Marine and Terrestrial Metagenomes. Appl. Environ. Microbiol. 2018, 84, e02773-17. [Google Scholar] [CrossRef] [PubMed]
- Nikolaivits, E.; Kanelli, M.; Dimarogona, M.; Topakas, E. A Middle-Aged Enzyme Still in Its Prime: Recent Advances in the Field of Cutinases. Catalysts 2018, 8, 612. [Google Scholar] [CrossRef]
- Kawai, F.; Kawabata, T.; Oda, M. Current knowledge on enzymatic PET degradation and its possible application to waste stream management and other fields. Appl. Microbiol. Biotechnol. 2019, 103, 4253–4268. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Alcántara, L.; Oliart-Ros, R.M.; García-Bórquez, A.; Peña-Montes, C. Expression of a Cutinase of Moniliophthora roreri with Polyester and PET-Plastic Residues Degradation Activity. Microbiol. Spectr. 2021, 9, e00976-21. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Zou, H. Biotechnological Application of Cutinase: A Powerful Tool in Synthetic Biology. SynBio 2022, 1, 54–64. [Google Scholar] [CrossRef]
- Gudiukaite, R.; Sadauskas, M.; Gegeckas, A.; Malunavicius, V.; Citavicius, D. Construction of a novel lipolytic fusion biocatalyst GDEst-lip for industrial application. J. Ind. Microbiol. Biotechnol. 2017, 44, 799–815. [Google Scholar] [CrossRef] [PubMed]
- Druteika, G.; Sadauskas, M.; Malunavicius, V.; Lastauskiene, E.; Statkeviciute, R.; Savickaite, A.; Gudiukaite, R. New engineered Geobacillus lipase GD-95RM for industry focusing on the cleaner production of fatty esters and household washing product formulations. World J. Microbiol. Biotechnol. 2020, 36, 1–15. [Google Scholar] [CrossRef]
- Savickaitė, A.; Gudiukaitė, R. In silico analysis of cutinase from Streptomyces scabiei 87.22. In Proceedings of the 64th International Conference for Students of Physics and Natural Sciences Open Readings 2021, Vilnius, Lithuania, 16–19 March 2021. [Google Scholar]
- Greičius, A.; Savickaitė, A.; Gudiukaitė, R. Analysis of site-directed mutant (Asp94Ala) of Streptomyces scabiei 87.22 cutinase. In Proceedings of the 65th International Conference for Students of Physics and Natural Sciences Open Readings 2022, Vilnius, Lithuania, 15–18 March 2022. [Google Scholar]
- Bignell, D.R.D.; Seipke, R.F.; Huguet-Tapia, J.C.; Chambers, A.H.; Parry, R.J.; Loria, R. Streptomyces scabies 87-22 Contains a Coronafacic Acid-Like Biosynthetic Cluster That Contributes to Plant–Microbe Interactions. Mol. Plant-Microbe Interact. 2010, 23, 161–175. [Google Scholar] [CrossRef]
- Sambrook, J.; Rusell, D.W. Molecular Cloning: A Laboratory Manual, 4th ed.; Cold Spring Harbor Lab Press: New York, NY, USA, 2001. [Google Scholar]
- Cigno, E.; Magagnoli, C.; Pierce, M.; Iglesias, P. Lubricating ability of two phosphonium-based ionic liquids as additives of a bio-oil for use in wind turbines gearboxes. Wear 2017, 376–377, 756–765. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Schägger, H. Tricine–SDS-PAGE. Nat. Protoc. 2006, 1, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Haider, S.R.; Reid, H.J.; Sharp, B.L. Tricine-SDS-PAGE. In Electrophoretic Separation of Proteins; Methods in Molecular Biology, 1855; Kurien, B., Scofield, R., Eds.; Humana Press: New York, NY, USA, 2019; pp. 151–161. [Google Scholar] [CrossRef]
- Levisson, M.; van der Oost, J.; Kengen, S.W.M. Characterization and structural modeling of a new type of thermostable esterase from Thermotoga maritima. FEBS J. 2007, 274, 2832–2842. [Google Scholar] [CrossRef] [PubMed]
- Savickaite, A.; Sadauskas, M.; Gudiukaite, R. Immobilized GDEst-95, GDEst-lip and GD-95RM lipolytic enzymes for continuous flow hydrolysis and transesterification reactions. Int. J. Biol. Macromol. 2021, 173, 421–434. [Google Scholar] [CrossRef]
- Hannig, G.; Makrides, S.C. Strategies for optimizing heterologous protein expression in Escherichia coli. Trends Biotechnol. 1998, 16, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Gombert, A.; Kilikian, B. Recombinant gene expression in Escherichia coli cultivation using lactose as inducer. J. Biotechnol. 1998, 60, 47–54. [Google Scholar] [CrossRef]
- Lim, H.K.; Lee, S.U.; Chung, S.I.; Jung, K.H. Induction of the T7 promoter using lactose for production of recombinant plasminogen kringle 1-3 in Escherichia coli. J. Microb. Biotechnol. 2004, 14, 225–230. [Google Scholar]
- Aucoin, M.G.; McMurray-Beaulieu, V.; Poulin, F.; Boivin, E.B.; Chen, J.; Ardelean, F.M.; Cloutier, M.; Choi, Y.J.; Miguez, C.B.; Jolicoeur, M. Identifying conditions for inducible protein production in E. coli: Combining a fed-batch and multiple induction approach. Microb. Cell Factories 2006, 5, 27. [Google Scholar] [CrossRef]
- Studier, F.W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef]
- Li, Z.; Kessler, W.; Heuvel, J.V.D.; Rinas, U. Simple defined autoinduction medium for high-level recombinant protein production using T7-based Escherichia coli expression systems. Appl. Microbiol. Biotechnol. 2011, 91, 1203–1213. [Google Scholar] [CrossRef]
- Tahara, N.; Tachibana, I.; Takeo, K.; Yamashita, S.; Shimada, A.; Hashimoto, M.; Ohno, S.; Yokogawa, T.; Nakagawa, T.; Suzuki, F.; et al. Boosting Auto-Induction of Recombinant Proteins in Escherichia coli with Glucose and Lactose Additives. Protein Pept. Lett. 2021, 28, 1180–1190. [Google Scholar] [CrossRef] [PubMed]
- Menacho-Melgar, R.; Ye, Z.; Moreb, E.A.; Yang, T.; Efromson, J.P.; Decker, J.S.; Wang, R.; Lynch, M.D. Scalable, two-stage, autoinduction of recombinant protein expression in E. coli utilizing phosphate depletion. Biotechnol. Bioeng. 2020, 117, 2715–2727. [Google Scholar] [CrossRef] [PubMed]
- Studier, F.W. Stable Expression Clones and Auto-Induction for Protein Production in E. coli. Methods Mol. Biol. 2013, 1091, 17–32. [Google Scholar] [CrossRef]
- Leone, S.; Sannino, F.; Tutino, M.L.; Parrilli, E.; Picone, D. Acetate: Friend or foe? Efficient production of a sweet protein in Escherichia coli BL21 using acetate as a carbon source. Microb. Cell Factories 2015, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vyas, H.; Tong, P. Process for Calcium Retention During Skim Milk Ultrafiltration. J. Dairy Sci. 2003, 86, 2761–2766. [Google Scholar] [CrossRef]
- Menchik, P.; Zuber, T.; Zuber, A.; Moraru, C.I. Short communication: Composition of coproduct streams from dairy processing: Acid whey and milk permeate. J. Dairy Sci. 2019, 102, 3978–3984. [Google Scholar] [CrossRef]
- Krefft, D.; Prusinowski, M.; Maciszka, P.; Skokowska, A.; Zebrowska, J.; Skowron, P.M. T7-lac promoter vectors spontaneous derepression caused by plant-derived growth media may lead to serious expression problems: A systematic evaluation. Microb. Cell Factories 2022, 21, 1–13. [Google Scholar] [CrossRef]
- Su, L.; Woodard, R.; Chen, J.; Wu, J. Extracellular Location of Thermobifida fusca Cutinase Expressed in Escherichia coli BL21(DE3) without Mediation of a Signal Peptide. Appl. Environ. Microbiol. 2013, 79, 4192–4198. [Google Scholar] [CrossRef]
- Kawai, F.; Oda, M.; Tamashiro, T.; Waku, T.; Tanaka, N.; Yamamoto, M.; Mizushima, H.; Miyakawa, T.; Tanokura, M. A novel Ca2+-activated, thermostabilized polyesterase capable of hydrolyzing polyethylene terephthalate from Saccharomonospora viridis AHK190. Appl. Microbiol. Biotechnol. 2014, 98, 10053–10064. [Google Scholar] [CrossRef]
- Miyakawa, T.; Mizushima, H.; Ohtsuka, J.; Oda, M.; Kawai, F.; Tanokura, M. Structural basis for the Ca2+-enhanced thermostability and activity of PET-degrading cutinase-like enzyme from Saccharomonospora viridis AHK190. Appl. Microbiol. Biotechnol. 2014, 99, 4297–4307. [Google Scholar] [CrossRef]
- Numoto, N.; Kamiya, N.; Bekker, G.-J.; Yamagami, Y.; Inaba, S.; Ishii, K.; Uchiyama, S.; Kawai, F.; Ito, N.; Oda, M. Structural Dynamics of the PET-Degrading Cutinase-like Enzyme from Saccharomonospora viridis AHK190 in Substrate-Bound States Elucidates the Ca2+-Driven Catalytic Cycle. Biochemistry 2018, 57, 5289–5300. [Google Scholar] [CrossRef]
- Oda, M.; Yamagami, Y.; Inaba, S.; Oida, T.; Yamamoto, M.; Kitajima, S.; Kawai, F. Enzymatic hydrolysis of PET: Functional roles of three Ca2+ ions bound to a cutinase-like enzyme, Cut190*, and its engineering for improved activity. Appl. Microbiol. Biotechnol. 2018, 102, 10067–10077. [Google Scholar] [CrossRef] [PubMed]
- Oda, M. Structural basis for Ca2+-dependent catalysis of a cutinase-like enzyme and its engineering: Application to enzymatic PET depolymerization. Biophys. Psysicobiology 2021, 18, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Komeil, D.; Simao-Beaunoir, A.-M.; Beaulieu, C. Detection of potential suberinase-encoding genes in Streptomyces scabiei strains and other actinobacteria. Can. J. Microbiol. 2013, 59, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Jabloune, R.; Khalil, M.; Ben Moussa, I.E.; Simao-Beaunoir, A.-M.; Lerat, S.; Brzezinski, R.; Beaulieu, C. Enzymatic Degradation of p-Nitrophenyl Esters, Polyethylene Terephthalate, Cutin, and Suberin by Sub1, a Suberinase Encoded by the Plant Pathogen Streptomyces scabies. Microbes Environ. 2020, 35, ME19086. [Google Scholar] [CrossRef]
- Feizollahzadeh, S.; Kouhpayeh, S.; Rahimmansh, I.; Khanahmad, H.; Sabzehei, F.; Ganjalikhani-Hakemi, M.; Andalib, A.; Hejazi, Z.; Rezaei, A. The Increase in Protein and Plasmid Yields of E. coli with Optimized Concentration of Ampicillin as Selection Marker. Iran. J. Biotechnol. 2017, 15, 128–134. [Google Scholar] [CrossRef]
- Bahreini, E.; Aghaiypour, K.; Abbasalipourkabir, R.; Goodarzi, M.T.; Saidijam, M.; Safavieh, S.S. An optimized protocol for overproduction of recombinant protein expression in Escherichia coli. Prep. Biochem. Biotechnol. 2014, 44, 510–528. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).