
Metabolic Engineering for Production of Small Molecule Drugs: Challenges and Solutions

Abstract

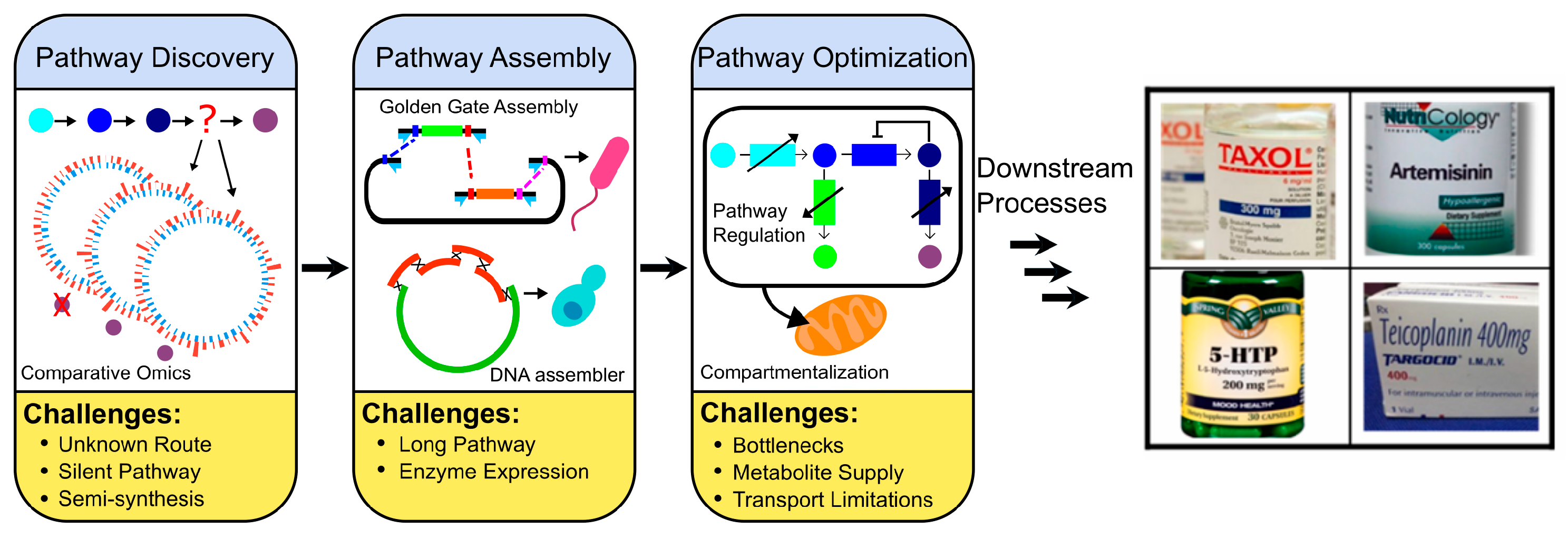
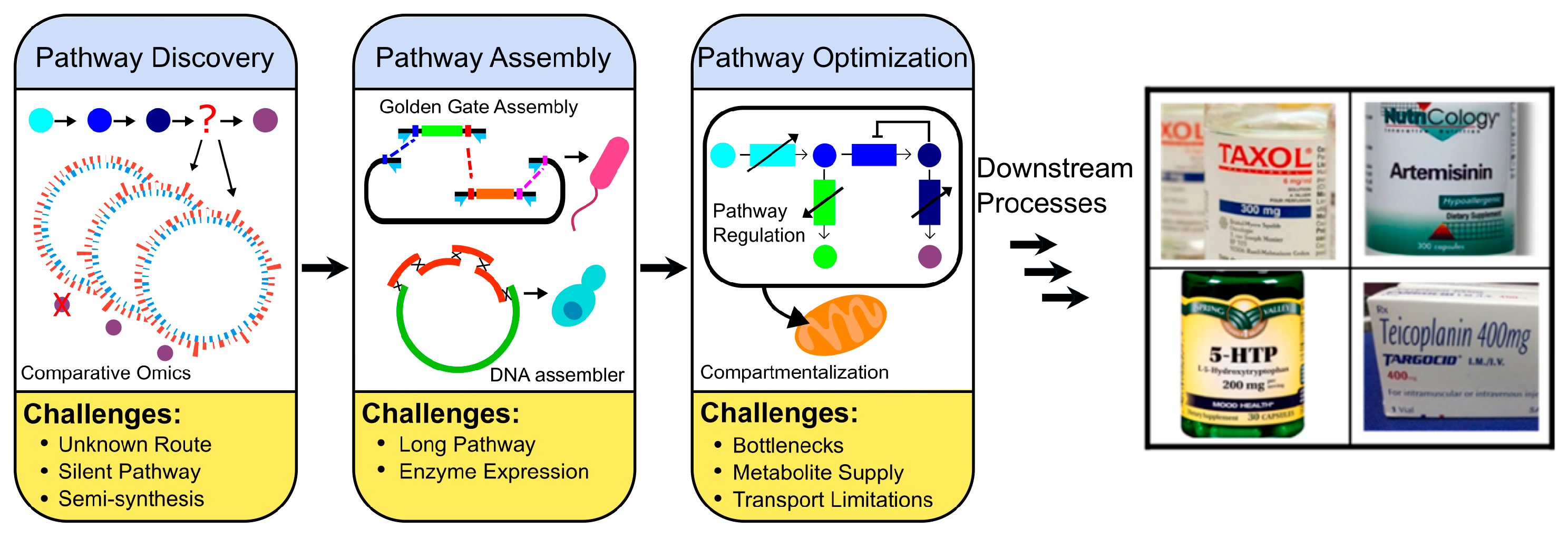
1. Introduction
2. Pathway Discovery
2.1. Unknown Route
2.2. Silent Pathway
2.3. Semi-Synthesis
3. Pathway Expression
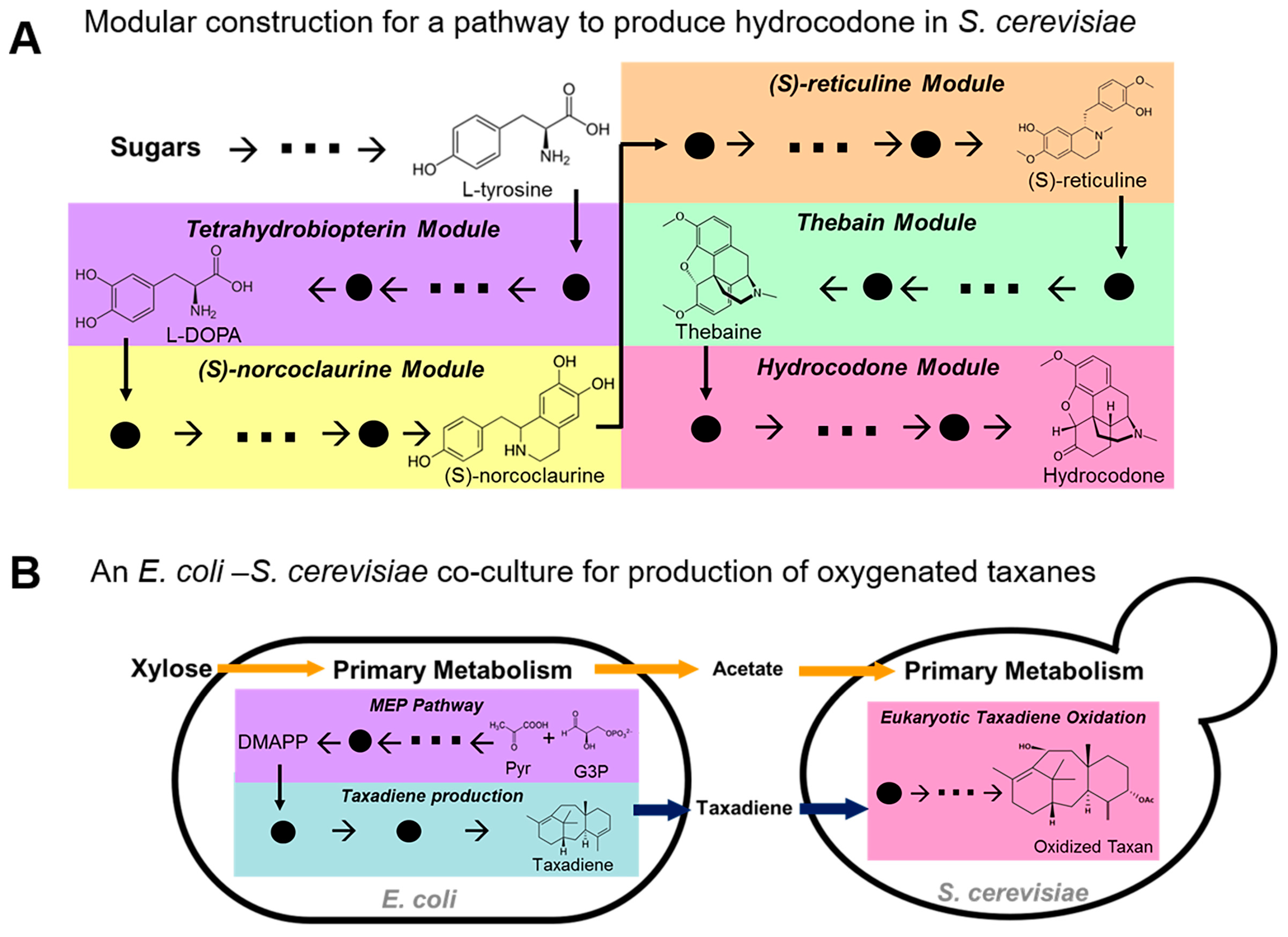
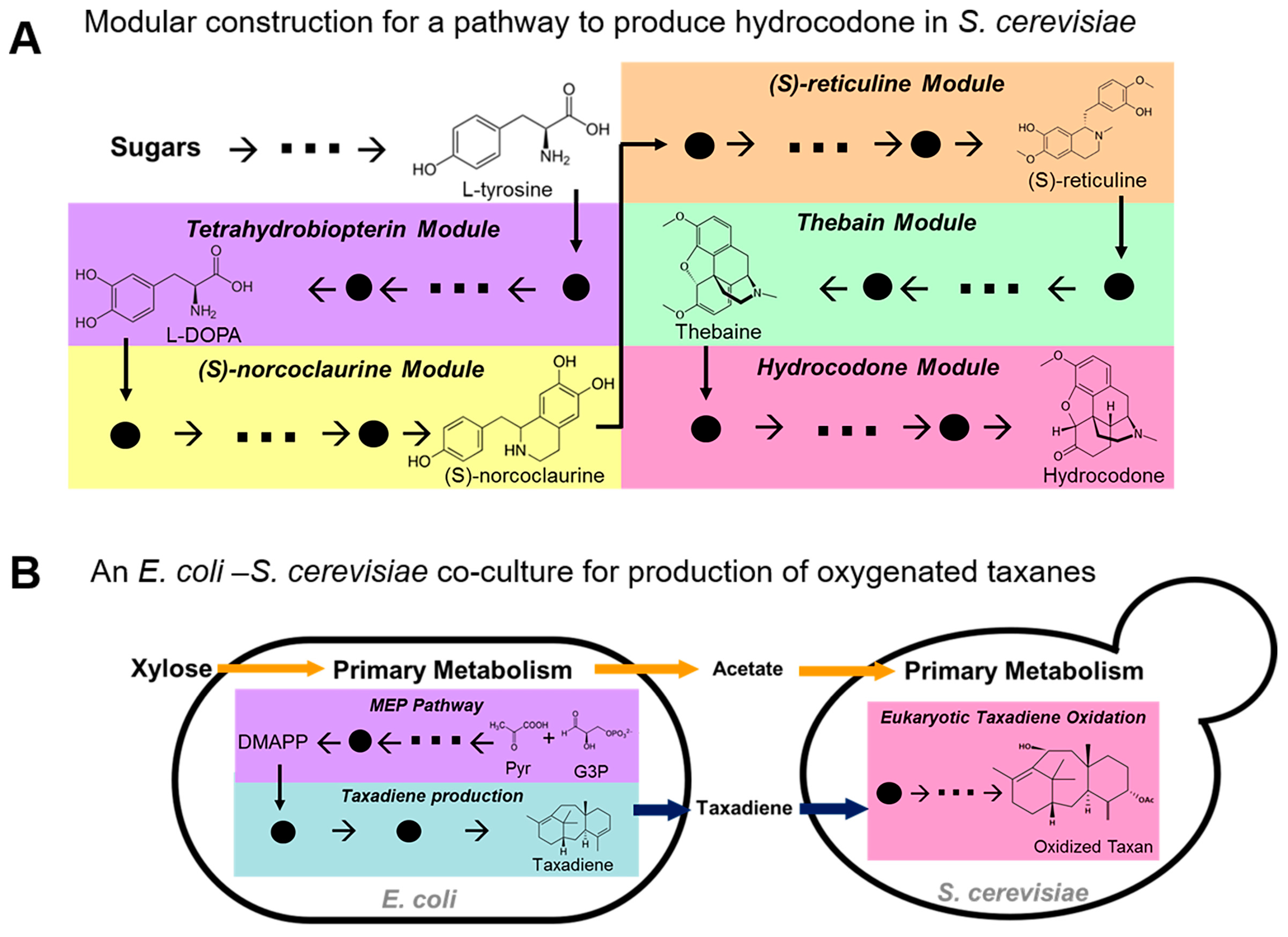
3.1. Long Biosynthesis Pathway
3.2. Poor Enzyme Expression
4. Pathway Optimizations
4.1. Pathway Bottlenecks
4.2. Transport Limitation of Intermediates
5. Summary and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schreiber, S.L.; Halpern, J. Organic synthesis toward small-molecule probes and drugs. Proc. Natl. Acad. Sci. USA 2011, 108, 6699–6702. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.M.; Behnken, S.; Hertweck, C. Genomics-inspired discovery of natural products. Curr. Opin. Chem. Biol. 2011, 15, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J. Natural products as leads to potential drugs: An old process or the new hope for drug discovery? J. Med. Chem. 2008, 51, 2589–2599. [Google Scholar] [CrossRef] [PubMed]
- Galm, U.; Shen, B. Expression of biosynthetic gene clusters in heterologous hosts for natural product production and combinatorial biosynthesis. Expert Opin. Drug Discov. 2006, 1, 409–437. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Li, B.-Z.; Liu, D.; Zhang, L.; Chen, Y.; Jia, B.; Zeng, B.-X.; Zhao, H.; Yuan, Y.-J. Engineered biosynthesis of natural products in heterologous hosts. Chem. Soc. Rev. 2015, 44, 5265–5290. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.M.; Tang, Y. Synthetic biological approaches to natural product biosynthesis. Curr. Opin. Biotechnol. 2012, 23, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Zadran, S.; Levine, R.D. Perspectives in metabolic engineering: Understanding cellular regulation towards the control of metabolic routes. Appl. Biochem. Biotechnol. 2013, 169, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.A.; Keasling, J.D.; Kirby, J.; Shiba, Y.; Paradise, E.M.; Fisher, K.J.; Ro, D.-K.; Ndungu, J.M.; Chang, M.C.Y.; Newman, K.L.; et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 2006, 440, 940–943. [Google Scholar]
- Thodey, K.; Galanie, S.; Smolke, C.D. A microbial biomanufacturing platform for natural and semisynthetic opioids. Nat. Chem. Biol. 2014, 10, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Ajikumar, P.K.; Xiao, W.-H.; Keith, E.J.T.; Wang, Y.; Simeon, F.; Leonard, E.; Mucha, O.; Phon, T.H.; Pfeifer, B.; Stephanopoulos, G. Isoprenoid Pathway Optimization for Taxol Precursor Overproduction in Escherichia coli. Science 2010, 330, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Cobb, R.E.; Zhao, H.M. Direct cloning of large genomic sequences. Nat. Biotechnol. 2012, 30, 405–406. [Google Scholar] [CrossRef] [PubMed]
- Guerra-Bubb, J.; Croteau, R.; Williams, R.M. The early stages of taxol biosynthesis: An interim report on the synthesis and identification of early pathway metabolites. Nat. Prod. Rep. 2012, 29, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Piškur, J.; Schnackerz, K.D.; Andersen, G.; Björnberg, O. Comparative genomics reveals novel biochemical pathways. Trends Genet. 2007, 23, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Dellas, N.; Thomas, S.T.; Manning, G.; Noel, J.P. Discovery of a metabolic alternative to the classical mevalonate pathway. ELIFE 2013, 2, e00672. [Google Scholar] [CrossRef] [PubMed]
- Alexander, R.W. Teasing apart the Taxol pathway. Trends Biochem. Sci. 2001, 26, 152–153. [Google Scholar] [CrossRef]
- Sato, M.; Yamada, H.; Hotta, K.; Watanabe, K. Elucidation of the shanorellin biosynthetic pathway and functional analysis of associated enzymes. Medchemcomm 2015, 6, 425–430. [Google Scholar] [CrossRef]
- Tang, Y.J.; Martin, H.G.; Myers, S.; Rodriguez, S.; Baidoo, E.E.; Keasling, J.D. Advances in analysis of microbial metabolic fluxes via (13)C isotopic labeling. Mass Spectrom. Rev. 2009, 28, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Sulikowski, G.A.; Pongdee, R. Elucidation of the Biosynthetic Pathway Leading to the Complex Nonadride Phomoidride B. Synlett 2006, 2006, 354–363. [Google Scholar] [CrossRef]
- Nonaka, K.; Ahlert, J.; Thorson, J.S.; Bachmann, B.O.; Zazopoulos, E.; Shen, B.; Huang, K.; Staffa, A.; Liu, W.; Farnet, C.M. A genomics-guided approach for discovering and expressing cryptic metabolic pathways. Nat. Biotechnol. 2003, 21, 187–190. [Google Scholar]
- Mattheus, W.; Gao, L.-J.; Herdewijn, P.; Landuyt, B.; Verhaegen, J.; Masschelein, J.; Volckaert, G.; Lavigne, R. Isolation and Purification of a New Kalimantacin/Batumin-Related Polyketide Antibiotic and Elucidation of Its Biosynthesis Gene Cluster. Chem. Biol. 2010, 17, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Gross, H.; Stockwell, V.O.; Henkels, M.D.; Nowak-Thompson, B.; Loper, J.E.; Gerwick, W.H. The Genomisotopic Approach: A Systematic Method to Isolate Products of Orphan Biosynthetic Gene Clusters. Chem. Biol. 2007, 14, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.F.; Zhao, H.N.; Barrero, R.A.; Zhang, B.H.; Sun, G.L.; Wilson, I.W.; Xie, F.L.; Walker, K.D.; Parks, J.W.; Bruce, R.; et al. Genome sequencing and analysis of the paclitaxel-producing endophytic fungus Penicillium aurantiogriseum NRRL 62431. BMC Genom. 2014, 15, 69. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhong, Y.; Yuan, H.; Wang, J.; Zheng, H.; Wang, Y.; Cen, X.; Xu, F.; Bai, J.; Han, X.; et al. Complete genome sequence of the rifamycin SV-producing Amycolatopsis mediterranei U32 revealed its genetic characteristics in phylogeny and metabolism. Cell Res. 2010, 20, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Hagel, J.M.; Facchini, P.J. Dioxygenases catalyze the O-demethylation steps of morphine biosynthesis in opium poppy. Nat. Chem. Biol. 2010, 6, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Hao, D.C.; Ge, G.B.; Xiao, P.G.; Zhang, Y.Y.; Yang, L. The First Insight into the Tissue Specific Taxus Transcriptome via Illumina Second Generation Sequencing. PLoS ONE 2011, 6, e21220. [Google Scholar] [CrossRef] [PubMed]
- Ziemert, N.; Ishida, K.; Liaimer, A.; Hertweck, C.; Dittmann, E. Ribosomal synthesis of tricyclic depsipeptides in bloom-forming cyanobacteria. Angew. Chem. 2008, 47, 7756–7759. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Reynolds, K.A.; Kersten, R.D.; Ryan, K.S.; Gonzalez, D.J.; Nizet, V.; Dorrestein, P.C.; Moore, B.S. Direct cloning and refactoring of a silent lipopeptide biosynthetic gene cluster yields the antibiotic taromycin A. Proc. Natl. Acad. Sci. 2014, 111, 1957–1962. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, R.; Yuan, L. Directed evolution of metabolic pathways. Trends Biotechnol. 2006, 24, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Brakhage, A.A.; Schuemann, J.; Bergmann, S.; Scherlach, K.; Schroeckh, V.; Hertweck, C. Activation of fungal silent gene clusters: A new avenue to drug discovery. Prog. Res. Fortschr. Arzneim. Prog. Rech. Pharm. 2008, 66, 1–12. [Google Scholar]
- Yaegashi, J.; Oakley, B.R.; Wang, C.C.C. Recent advances in genome mining of secondary metabolite biosynthetic gene clusters and the development of heterologous expression systems in Aspergillus nidulans. J. Ind. Microbiol. Biotechnol. 2014, 41, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.M.; Szewczyk, E.; Davidson, A.D.; Keller, N.; Oakley, B.R.; Wang, C.C.C. A Gene Cluster Containing Two Fungal Polyketide Synthases Encodes the Biosynthetic Pathway for a Polyketide, Asperfuranone, in Aspergillus nidulans. J. Am. Chem. Soc. 2009, 131, 2965–2970. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.-M.; Lo, H.-C.; Strauss, J.; Wang, C.C.C.; Davidson, A.D.; Oakley, B.R.; Reyes-Dominguez, Y.; Watanabe, K.; Szewczyk, E.; Sanchez, J.F.; et al. Chromatin-level regulation of biosynthetic gene clusters. Nat. Chem. Biol. 2009, 5, 462–464. [Google Scholar]
- Moody, S.C. Microbial co-culture: Harnessing intermicrobial signaling for the production of novel antimicrobials. Fut. Microbiol. 2014, 9, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Brasch, J.; Horter, F.; Fritsch, D.; Beck-Jendroschek, V.; Tröger, A.; Francke, W. Acyclic sesquiterpenes released by Candida albicans inhibit growth of dermatophytes. Med. Mycol. 2014, 52, 46–55. [Google Scholar] [PubMed]
- Dashti, Y.; Grkovic, T.; Abdelmohsen, U.R.; Hentschel, U.; Quinn, R.J. Production of Induced Secondary Metabolites by a Co-Culture of Sponge-Associated Actinomycetes, Actinokineospora sp. EG49 and Nocardiopsis sp. RV163. Mar. Drugs 2014, 12, 3046–3059. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Huang, H.; Liang, J.; Wang, M.; Lu, L.; Shao, Z.; Cobb, R.E.; Zhao, H. Activation and characterization of a cryptic polycyclic tetramate macrolactam biosynthetic gene cluster. Nat. Commun. 2013, 4, 2894. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.-R. Modification of natural products for drug discovery. Acta Pharm. Sin. 2012, 47, 144–157. [Google Scholar]
- Chen, J.C.; Li, W.L.; Yao, H.Q.; Xu, J.Y. Insights into drug discovery from natural products through structural modification. Fitoterapia 2015, 103, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Nobili, S.; Landini, I.; Mazzei, T.; Mini, E. Overcoming tumor multidrug resistance using drugs able to evade P-glycoprotein or to exploit its expression. Med. Res. Rev. 2012, 32, 1220–1262. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yang, J.; Ran, Q.; Wang, L.; Wang, Z.; Liu, J.; Wu, X.; Hua, W.; Yuan, S.; Zhang, L.; et al. Synthesis and biological evaluation of novel 1- O- and 14- O-derivatives of oridonin as potential anticancer drug candidates. Bioorg. Med. Chem. Lett. 2008, 18, 4741–4744. [Google Scholar] [CrossRef] [PubMed]
- Boehm, M.; Fuenfschilling, P.C.; Krieger, M.; Kuesters, E.; Struber, F. An improved manufacturing process for the antimalaria drug coartem. Part I. Org. Process Res. Dev. 2007, 11, 336–340. [Google Scholar] [CrossRef]
- Westfall, P.J.; Pitera, D.J.; Lenihan, J.R.; Eng, D.; Woolard, F.X.; Regentin, R.; Horning, T.; Tsuruta, H.; Melis, D.J.; Owens, A.; et al. Production of amorphadiene in yeast, and its conversion to dihydroartemisinic acid, precursor to the antimalarial agent artemisinin. Proc. Natl. Acad. Sci. USA 2012, 109, E111–E118. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Liang, D.; Ning, M.; Wang, Q.; Meng, X.; Li, Z. Semi-synthesis of neomangiferin from mangiferin. Tetrahedron Lett. 2014, 55, 3083–3086. [Google Scholar] [CrossRef]
- Kong, J.; Yang, Y.; Wang, W.; Cheng, K.; Zhu, P. Artemisinic acid: A promising molecule potentially suitable for the semi-synthesis of artemisinin. RSC Adv. 2013, 3, 7622–7641. [Google Scholar] [CrossRef]
- Moura, M.; Broadbelt, L.; Tyo, K. Computational tools for guided discovery and engineering of metabolic pathways. Methods Mol. Biol. 2013, 985, 123–147. [Google Scholar] [PubMed]
- Brunk, E.; Neri, M.; Tavernelli, I.; Hatzimanikatis, V.; Rothlisberger, U. Integrating computational methods to retrofit enzymes to synthetic pathways. Biotechnol. Bioeng. 2012, 109, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Pleiss, J. Protein design in metabolic engineering and synthetic biology. Curr. Opin. Biotechnol. 2011, 22, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Kushnir, S.; Sundermann, U.; Yahiaoui, S.; Brockmeyer, A.; Janning, P.; Schulz, F. Minimally Invasive Mutagenesis Gives Rise to a Biosynthetic Polyketide Library. Angew. Chem. Int. Ed. 2012, 51, 10664–10669. [Google Scholar] [CrossRef] [PubMed]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.S.; Chen, Y.; Metcalf, W.W.; Zhao, H.; Kelleher, N.L. Directed Evolution of the Nonribosomal Peptide Synthetase AdmK Generates New Andrimid Derivatives In Vivo. Chem. Biol. 2011, 18, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Liu, Z.; Zhao, H.; El, A. Recent advances in combinatorial biosynthesis for drug discovery. Drug Des. Dev Ther. 2015, 2015, 823–833. [Google Scholar]
- Liu, T.; Chiang, Y.M.; Somoza, A.D.; Oakley, B.R.; Wang, C.C.C. Engineering of an “Unnatural” Natural Product by Swapping Polyketide Synthase Domains in Aspergillus nidulans. J. Am. Chem. Soc. 2011, 133, 13314–13316. [Google Scholar] [CrossRef] [PubMed]
- Galanie, S.; Thodey, K.; Trenchard, I.J.; Interrante, M.F.; Smolke, C.D. Complete biosynthesis of opioids in yeast. Science 2015, 349, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Qiao, K.; Edgar, S.; Stephanopoulos, G. Distributing a metabolic pathway among a microbial consortium enhances production of natural products. Nat. Biotechnol. 2015, 33, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.C.; Gust, B.; Kulik, A.; Heide, L.; Buttner, M.J.; Bibb, M.J. Phage P1-Derived Artificial Chromosomes Facilitate Heterologous Expression of the FK506 Gene Cluster. PLoS ONE 2013, 8, e69319. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Shah, S.; Chung, L.; Carney, J.; Katz, L.; Khosla, C.; Julien, B. Cloning and Heterologous Expression of the Epothilone Gene Cluster. Science 2000, 287, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Engler, C.; Gruetzner, R.; Kandzia, R.; Marillonnet, S. Golden Gate Shuffling: A One-Pot DNA Shuffling Method Based on Type IIs Restriction Enzymes. PLoS ONE 2009, 4, e5553. [Google Scholar] [CrossRef] [PubMed]
- Bruckner, K.; Tissier, A. High-level diterpene production by transient expression in Nicotiana benthamiana. Plant Methods 2013, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Zhao, H. DNA assembler, an in vivo genetic method for rapid construction of biochemical pathways. Nucleic Acids Res. 2009, 37, e16. [Google Scholar] [CrossRef] [PubMed]
- Fossati, E.; Narcross, L.; Ekins, A.; Falgueyret, J.P.; Martin, V.J.J. Synthesis of Morphinan Alkaloids in Saccharomyces cerevisiae. PLoS ONE 2015, 10, e0124459. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.C.; Gross, F.; Zhang, Y.; Fu, J.; Stewart, A.F.; Müller, R. Heterologous Expression of a Myxobacterial Natural Products Assembly Line in Pseudomonads via Red/ET Recombineering. Chem. Biol. 2005, 12, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Cobb, R.E.; Ning, J.C.; Zhao, H. DNA assembly techniques for next-generation combinatorial biosynthesis of natural products. J. Ind. Microbiol. Biotechnol. 2014, 41, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Tokmakov, A.A.; Kurotani, A.; Takagi, T.; Toyama, M.; Shirouzu, M.; Fukami, Y.; Yokoyama, S. Multiple Post-translational Modifications Affect Heterologous Protein Synthesis. J. Biol. Chem. 2012, 287, 27106–27116. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, C.; Govindarajan, S.; Minshull, J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004, 22, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Bernaudat, F.; Frelet-Barrand, A.; Pochon, N.; Dementin, S.; Hivin, P.; Boutigny, S.; Rioux, J.B.; Salvi, D.; Seigneurin-Berny, D.; Richaud, P.; et al. Heterologous Expression of Membrane Proteins: Choosing the Appropriate Host. PLoS ONE 2011, 6, e29191. [Google Scholar] [CrossRef] [PubMed]
- Thanapipatsiri, A.; Claesen, J.; Gomez-Escribano, J.P.; Bibb, M.; Thamchaipenet, A. A Streptomyces coelicolor host for the heterologous expression of Type III polyketide synthase genes. Microb. Cell Factories 2015, 14, 145. [Google Scholar] [CrossRef] [PubMed]
- Condon, A.; Thachuk, C. Efficient codon optimization with motif engineering. J. Discret. Algorithms 2012, 16, 104. [Google Scholar] [CrossRef]
- Gvritishvili, A.G.; Leung, K.W.; Tombran-Tink, J. Codon Preference Optimization Increases Heterologous PEDF Expression. PLoS ONE 2010, 5, e15056. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Lee, S.-H.; Baek, K.; Kim, B.-G. Heterologous expression of tyrosinase (MelC2) from Streptomyces avermitilis MA4680 in E. coli and its application for ortho-hydroxylation of resveratrol to produce piceatannol. Appl. Microbiol. Biotechnol. 2015, 99, 7915–7924. [Google Scholar] [CrossRef] [PubMed]
- Okesli, A.; Cooper, L.E.; Fogle, E.J.; van der Donk, W.A. Nine Post-translational Modifications during the Biosynthesis of Cinnamycin. J. Am. Chem. Soc. 2011, 133, 13753–13760. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, B.A.; Admiraal, S.J.; Gramajo, H.; Cane, D.E.; Khosla, C. Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science 2001, 291, 1790–1792. [Google Scholar] [CrossRef] [PubMed]
- DeLoache, W.C.; Russ, Z.N.; Narcross, L.; Gonzales, A.M.; Martin, V.J.; Dueber, J.E. An enzyme-coupled biosensor enables (S)-reticuline production in yeast from glucose. Nat. Chem. Biol. 2015, 11, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Li, J. Precursor engineering and cell physiological regulation for high level rapamycin production by Streptomyces hygroscopicus. Ann. Microbiol. 2013, 63, 1371–1378. [Google Scholar] [CrossRef]
- Ryu, Y.G.; Butler, M.J.; Chater, K.F.; Lee, K.J. Engineering of primary carbohydrate metabolism for increased production of actinorhodin in Streptomyces coelicolor. Appl. Environ. Microbiol. 2006, 72, 7132–7139. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Xia, M.; Li, S.; Wen, J.; Jia, X. Enhancement of FK506 production by engineering secondary pathways of Streptomyces tsukubaensis and exogenous feeding strategies. J. Ind. Microbiol. Biotechnol. 2013, 40, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Wasylenko, T.M.; Stephanopoulos, G. Metabolomic and 13C-metabolic flux analysis of a xylose-consuming Saccharomyces cerevisiae strain expressing xylose isomerase. Biotechnol. Bioeng. 2015, 112, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Varman, A.M.; He, L.; You, L.; Hollinshead, W.; Tang, Y.J. Elucidation of intrinsic biosynthesis yields using 13C-based metabolism analysis. Microb. Cell Factor. 2014, 13, 42. [Google Scholar] [CrossRef] [PubMed]
- Zamboni, N.; Fendt, S.M.; Rühl, M.; Sauer, U. 13C-based metabolic flux analysis. Nat. Protoc. 2009, 4, 878–892. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Page, L.; Rubens, J.; Chircus, L.; Colletti, P.; Pakrasi, H.B.; Tang, Y.J. Bridging the gap between fluxomics and industrial biotechnology. J. Biomed. Biotechnol. 2010, 2010, 460717. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Jia, X.; Wen, J.; Wang, G.; Yu, G.; Caiyin, Q.; Chen, Y. Metabolic Flux Analysis and Principal Nodes Identification for Daptomycin Production Improvement by Streptomyces roseosporus. Appl. Biochem. Biotechnol. 2011, 165, 1725–1739. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-J.; Blount, J.W.; Steele, C.L.; Dixon, R.A. Bottlenecks for Metabolic Engineering of Isoflavone Glycoconjugates in Arabidopsis. Proc. Natl. Acad. Sci. USA 2002, 99, 14578–14583. [Google Scholar] [CrossRef] [PubMed]
- Cluis, C.P.; Ekins, A.; Narcross, L.; Jiang, H.; Gold, N.D.; Burja, A.M.; Martin, V.J.J. Identification of bottlenecks in Escherichia coli engineered for the production of CoQ(10). Metabol. Eng. 2011, 13, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.L.; Becker, J.; de Souza Lima, A.O.; Porto, L.M.; Wittmann, C. Systems metabolic engineering of Escherichia coli for gram scale production of the antitumor drug deoxyviolacein from glycerol. Biotechnol. Bioeng. 2014, 111, 2280–2289. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhao, J.; Li, L.; Chen, Z.; Wen, Y.; Li, J. The pathway-specific regulator AveR from Streptomyces avermitilis positively regulates avermectin production while it negatively affects oligomycin biosynthesis. Mol. Genet. Genom. 2010, 283, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Venayak, N.; Anesiadis, N.; Cluett, W.R.; Mahadevan, R. Engineering metabolism through dynamic control. Curr. Opin. Biotechnol. 2015, 34, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Alper, H.; Fischer, C.; Nevoigt, E.; Stephanopoulos, G.; Langer, R. Tuning Genetic Control through Promoter Engineering. Proc. Natl. Acad. Sci. USA 2005, 102, 12678–12683. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.Z.; Carothers, J.M.; Keasling, J.D. Design of a dynamic sensor-regulator system for production of chemicals and fuels derived from fatty acids. Nat. Biotechnol. 2012, 30, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.C.; Averesch, N.J.H.; Winter, G.; Plan, M.R.; Vickers, C.E.; Nielsen, L.K.; Krömer, J.O. Quorum-sensing linked RNA interference for dynamic metabolic pathway control in Saccharomyces cerevisiae. Metab. Eng. 2015, 29, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Dahl, R.H.; Zhang, F.; Alonso-Gutierrez, J.; Baidoo, E.; Batth, T.S.; Redding-Johanson, A.M.; Petzold, C.J.; Mukhopadhyay, A.; Lee, T.S.; Adams, P.D.; et al. Engineering dynamic pathway regulation using stress-response promoters. Nat. Biotechnol. 2013, 31, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Eckdahl, T.T.; Campbell, A.M.; Heyer, L.J.; Poet, J.L.; Blauch, D.N.; Snyder, N.L.; Atchley, D.T.; Baker, E.J.; Brown, M.; Brunner, E.C.; et al. Programmed Evolution for Optimization of Orthogonal Metabolic Output in Bacteria. PLoS ONE 2015, 10, e0118322. [Google Scholar] [CrossRef] [PubMed]
- Skretas, G.; Kolisis, F.N. Combinatorial approaches for inverse metabolic engineering applications. Comput. Struct. Biotechnol. J. 2012, 3, e201210021. [Google Scholar] [CrossRef] [PubMed]
- Dudley, Q.M.; Karim, A.S.; Jewett, M.C. Cell-free metabolic engineering: Biomanufacturing beyond the cell. Biotechnol. J. 2015, 10, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.-M.; Jones, D.P. Redox compartmentalization in eukaryotic cells. BBA Gen. Subj. 2008, 1780, 1273–1290. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.H.; Silver, P.A. Designing biological compartmentalization. Trends Cell Biol. 2012, 22, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Saxena, B.; Subramaniyan, M.; Malhotra, K.; Bhavesh, N.S.; Potlakayala, S.D.; Kumar, S. Metabolic engineering of chloroplasts for artemisinic acid biosynthesis and impact on plant growth. J. Biosci. 2014, 39, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Herr, A.; Fischer, R. Improvement of Aspergillus nidulans penicillin production by targeting AcvA to peroxisomes. Metab. Eng. 2014, 25, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Farhi, M.; Marhevka, E.; Masci, T.; Marcos, E.; Eyal, Y.; Ovadis, M.; Abeliovich, H.; Vainstein, A. Harnessing yeast subcellular compartments for the production of plant terpenoids. Metab. Eng. 2011, 13, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Quin, M.B.; Sanders, M.A.; Johnson, E.T.; Schmidt-Dannert, C. Engineered protein nano-compartments for targeted enzyme localization. PLoS ONE 2012, 7, e33342. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.C.; Prather, K.L.J.; Malmirchegini, G.R.; Petzold, C.J.; Ullal, A.V.; Dueber, J.E.; Moon, T.S.; Keasling, J.D. Synthetic protein scaffolds provide modular control over metabolic flux. Nat. Biotechnol. 2009, 27, 753–759. [Google Scholar]
- Zhou, Y.J.; Gao, W.; Rong, Q.X.; Jin, G.J.; Chu, H.Y.; Liu, W.J.; Yang, W.; Zhu, Z.W.; Li, G.H.; Zhu, G.F.; et al. Modular Pathway Engineering of Diterpenoid Synthases and the Mevalonic Acid Pathway for Miltiradiene Production. J. Am. Chem. Soc. 2012, 134, 3234–3241. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-F.; Xiong, Z.-Q.; Li, S.-Y.; Wang, Y. Enhancing isoprenoid production through systematically assembling and modulating efflux pumps in Escherichia coli. Appl. Microbiol. Biotechnol. 2013, 97, 8057–8067. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.; Kuster, B.; Culhane, A.C.; Gholami, A.M. A multivariate approach to the integration of multi-omics datasets. BMC Bioinform. 2014, 15, 162. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.G.; de Mey, M.; Lim, C.G.; Ajikumar, P.K.; Stephanopoulos, G. The future of metabolic engineering and synthetic biology: Towards a systematic practice. Metab. Eng. 2012, 14, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Zalatan, J.G.; Lee, M.E.; Almeida, R.; Gilbert, L.A.; Whitehead, E.H.; La Russa, M.; Tsai, J.C.; Weissman, J.S.; Dueber, J.E.; Qi, L.S.; et al. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell 2015, 160, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Li, L.; Zhang, F.; Stephanopoulos, G.; Koffas, M. Improving fatty acids production by engineering dynamic pathway regulation and metabolic control. Proc. Natl. Acad. Sci. USA 2014, 111, 11299–11304. [Google Scholar] [CrossRef] [PubMed]
- Sparkman-Yager, D.; Correa-Rojas, R.A.; Carothers, J.M. Kinetic folding design of aptazyme-regulated expression devices as riboswitches for metabolic engineering. Methods Enzymol. 2015, 550, 321. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Step | Challenge | Solution | Host | Drug(s) | Achievement | Study |
|---|---|---|---|---|---|---|
| Pathway Discovery | Unknown Route | 13C-assisted pathway analysis | Unidentified Pleosporales | Phomoidrides | identified pathway intermediates | [18] |
| Unknown Route | Comparative genomics | Amycolatopsis mediterranei | Rifamycin | gene cluster identified | [23] | |
| Unknown Route | Sequence database search | Multiple | Isopredoids | ubiquitous alternate pathway discovered | [14] | |
| Silent Pathway | New promoters for transcription factors | Aspergillus nidulans | Asperfuranone | activation of silent pathway/discovery of new drug | [31] | |
| Silent Pathway | Deletion of heterochromatin factors | A. nidulans | Monodictyphenone | activation of silent pathway/discovery of new drug | [32] | |
| Silent Pathway | Co-culture with competitors | Candida albicans | Dihydrofarnesol | activation of silent pathway/discovery of new drug | [34] | |
| Silent Pathway | Introducing new promoters | Streptomyces griseus | Polycyclic tetramate macrolactams (PTMs) | activation of silent pathway/discovery of new drug | [36] | |
| Unknown Enzyme | Directed evolution of enzymes | Pantoea agglomerans | Andrimid Deriviatives | new derivative and enzyme produced | [50] | |
| Unknown Enzyme | Enzyme domain swapping | A. nidulans | 1,4-Naphthaquinone Derivative | new derivative produced | [52] | |
| Unknown Enzyme | Rational site-specific mutagenesis | Streptomyces cinnamonensis | Polyketide Derivatives | library of derivatives generated | [48] | |
| Pathway Assembly | Long Pathway | Integrative and replicative plasmids | Streptomyces coelicolor | Epothilone | 56 kb cluster transferred | [56] |
| Long Pathway | Red/ET recombination | Pseudomonas putida | Myxochromide S | 43 kb cosmid assembled | [61] | |
| Long Pathway | Homologous recombination | S. cerevisiae | Morphinan | one step multicloning in Eukaryotes | [60] | |
| Long Pathway | Golden gate assembly | Nicotiana benthamiana viaAgrobacterium tumefaciens | Diterpenes | one step multicloning in Prokaryotes | [58] | |
| Long Pathway | Artificial chromosome | S. coelicolor | Tacrolimus | 55 µg/mL (500% increase) | [55] | |
| Enzyme Expression | Codon optimization and enzyme engineering | E. coli | Piceatannol | 18.9 µg/mL (170% increase over natural producer) | [69] | |
| Enzyme Expression | Introduction of PTM genes | E. coli | Cinnamycin | qualitative assay confirmation | [70] | |
| Enzyme Expression | Codon optimization | E. coli | Pigment Epithilium Derived Factor (PEDF) | ~186.3 µg/mL * | [68] | |
| Enzyme Expression | Introduction of PTM genes | E. coli | 6-Deoxyerythronolide B | 23.2 µg/mL | [71] | |
| Enzyme Expression | PCR mutagenesis | S. ceverisiae | benzylisoquinoline alkaloids (BIAs) | 80.6~104.6 µg/mL | [72] | |
| Pathway Optimization | Bottlenecks | DSRS | E. coli | Fatty Acids | 4000 µg/mL | [106] |
| Bottlenecks | Dynamic quorum sensing | S. ceverisiae | Para-hydroxybenzoic acid (PHBA) | 151.9 µg/mL | [88] | |
| Bottlenecks | Combinatorial overexpression | Streptomyes tsukubaensis | FK506 | 457.5 µg/mL | [75] | |
| Bottlenecks | “Programmed” evolution | E. coli | Theophylline | 80 µg/mL | [90] | |
| Bottlenecks | Stress response regulation | E. coli | Amorphadiene | 1600 µg/mL (~100% increase, ~50% of theoretical yield **) | [89] | |
| Bottlenecks | Modular pathway engineering | E. coli | Taxadiene | 1000 µg/mL (~25% of theoretical yield **) | [10] | |
| Transport Limitations | Efflux pumps | E. coli | Kaurene | 250 µg/mL (37% increase) | [101] | |
| Transport Limitations | Protein scaffold | E. coli | Glucaric Acid | 500 µg/mL (7700% increase) | [99] | |
| Transport Limitations | Fusion proteins | S. ceverisiae | Miltiradien | 365 µg/mL | [100] | |
| Transport Limitations | Localization to mitochondria | S. ceverisiae | Valencene | 1.4 µg/mL | [97] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huttanus, H.M.; Sheng, J.; Feng, X. Metabolic Engineering for Production of Small Molecule Drugs: Challenges and Solutions. Fermentation 2016, 2, 4. https://doi.org/10.3390/fermentation2010004
Huttanus HM, Sheng J, Feng X. Metabolic Engineering for Production of Small Molecule Drugs: Challenges and Solutions. Fermentation. 2016; 2(1):4. https://doi.org/10.3390/fermentation2010004
Chicago/Turabian StyleHuttanus, Herbert M., Jiayuan Sheng, and Xueyang Feng. 2016. "Metabolic Engineering for Production of Small Molecule Drugs: Challenges and Solutions" Fermentation 2, no. 1: 4. https://doi.org/10.3390/fermentation2010004
APA StyleHuttanus, H. M., Sheng, J., & Feng, X. (2016). Metabolic Engineering for Production of Small Molecule Drugs: Challenges and Solutions. Fermentation, 2(1), 4. https://doi.org/10.3390/fermentation2010004