Abstract
Hyaluronidase is a hydrolytic enzyme that cleaves β-1,4-glycosidic linkages in high-molecular-weight hyaluronic acid, generating low-molecular-weight oligosaccharides with enhanced biological functions. These products exhibit immunomodulatory, antioxidant, and tissue-regenerative properties, making them valuable in pharmaceutical, cosmetic, and functional food applications. However, most commercial hyaluronidases originate from pathogenic bacteria or recombinant hosts, raising concerns over their biosafety and regulatory acceptance, particularly in food-grade applications. In this study, we report the isolation and characterization of a novel non-pathogenic soil bacterium, Paenibacillus residui BSSK58, which produces an extracellular hyaluronidase. Whole-genome sequencing revealed the absence of known virulence factors and antibiotic resistance genes. Phenotypic safety evaluations confirmed that there was no hemolytic activity, biogenic amine production, or cytotoxicity against human intestinal epithelial cell lines (Caco-2 and HT-29). The purified BSSK58 hyaluronidase exhibited a molecular weight of approximately 170 kDa, with optimal activity at pH 8.0–9.0 and 50 °C. The enzyme showed broad substrate specificity toward hyaluronic acid, chondroitin sulfate, and alginate, and its depolymerizing activity was confirmed using gel permeation chromatography. Furthermore, a 13-week oral repeated-dose toxicity study under Good Laboratory Practice conditions demonstrated no adverse effects. These findings support the use of BSSK58 hyaluronidase as a safe, non-recombinant biocatalyst suitable for industrial applications under regulatory-compliant frameworks.
1. Introduction
Hyaluronic acid (HA) is a high-molecular-weight glycosaminoglycan that plays a vital structural and functional role in vertebrate connective tissues [1]. Due to its viscoelasticity, hydrophilicity, and excellent biocompatibility, HA is widely used in pharmaceutical, cosmeceutical, and nutraceutical formulations [1,2]. However, its large molecular size (1–3 MDa) significantly impedes paracellular absorption, thereby limiting its clinical and nutritional utility, particularly in oral and transdermal delivery systems [1,3]. In contrast, low-molecular-weight hyaluronic acid (LMW-HA), primarily obtained through enzymatic hydrolysis, demonstrates superior bioavailability and enhanced biological activities, such as antioxidant, immunomodulatory, and tissue-regenerative effects [2,3,4]. Compared to chemical depolymerization approaches, including acid hydrolysis, enzymatic processing is more selective, occurs under milder conditions, and reduces the number of undesirable side reactions, yielding structurally intact and functionally potent oligosaccharide products [5,6].
Hyaluronidase is the key enzyme responsible for cleaving β-1,4 glycosidic linkages within the HA backbone, thus facilitating the generation of low-molecular-weight hyaluronic acid [1]. Commercially available hyaluronidases are typically derived from mammalian tissues (e.g., bovine or ovine testes) or toxigenic bacteria such as Streptococcus pyogenes and Clostridium perfringens [7,8,9,10]. These sources have notable limitations in terms of production consistency, biosafety, and regulatory compliance, particularly for food-grade or cosmetic applications. While recombinant human hyaluronidase (rHuPH20), expressed in Chinese hamster ovary (CHO) cells, is used in injectable therapeutics, its deployment in food or cosmetic bioprocessing remains restricted due to its high production costs, allergenicity concerns, and regulatory barriers to using products from Genetically Modified Organisms (GMOs) [10,11].
Recent advances in microbial biotechnology have focused on developing non-recombinant, non-pathogenic microbial systems capable of producing industrially valuable enzymes under scalable fermentation conditions [12]. The genus Paenibacillus has attracted considerable interest due to its capacity to produce thermostable and pH-tolerant hydrolases such as cellulases and xylanases, positioning it as a viable candidate for use in enzyme-based bioprocessing platforms [12,13,14,15]. Furthermore, several Paenibacillus strains exhibit beneficial physiological attributes including probiotic activity, nitrogen fixation, and antimicrobial metabolite production, thereby enhancing their relevance to food and agricultural biotechnology [13,14,16]. Despite these advantages, hyaluronidase production in Paenibacillus species-particularly Paenibacillus residui-has not been reported, and its feasibility as a source of non-recombinant hyaluronidase for industrial use remains unexplored. Recent studies have reported microbial hyaluronidases with varying characteristics. A hyaluronidase from Paenibacillus aquistagni SH-7-A showed an optimum at pH 6 and 40 °C with a molecular weight of ~110 kDa, although its stability was limited to 30–40 °C [17]. Similarly, enzymes from Streptococcus mitis and Brevibacterium halotolerans were active mainly under acidic or neutral conditions and exhibited only moderate thermal stability [18,19]. To date, however, no hyaluronidase has been identified from P. residui, and none of the Paenibacillus-derived enzymes have been subjected to systematic in vivo safety evaluation.
In the present study, we hypothesized that a novel soil-derived strain of P. residui (designated as BSSK58) was capable of secreting extracellular hyaluronidase with enzymatic characteristics and a safety profile compatible with food-grade and industrial applications. To test this hypothesis, we isolated the strain P. residui BSSK58 and performed detailed biochemical characterization of the secreted hyaluronidase, including analysis of its catalytic efficiency, pH and thermal stability, and substrate specificity and the modulating effects of metal ions under industrially relevant conditions. Whole-genome sequencing confirmed the absence of known virulence factors and acquired antibiotic resistance genes. Additionally, we evaluated the biosafety of P. residui BSSK58 through hemolytic assays, cytotoxicity tests using human intestinal epithelial cell lines, biogenic amine detection, and a 13-week oral repeated-dose study of BSSK58 hyaluronidase’s toxicity in accordance with the Good Laboratory Practice (GLP) protocols.
2. Materials and Methods
2.1. Bacterial Isolation and Identification
Paenibacillus residui BSSK58 was isolated through enrichment culturing on minimal medium agar (MA) supplemented with 0.1% peptone, 0.5% NaCl, 0.02% K2HPO4, 0.02% KCl, 0.14% Na2HPO4·7H2O, 0.5% HA, and 2.0% agar [20]. The culture was streaked onto the MA and cultured at 35 °C for 2–3 days in an incubator. Colonies forming clear halos were visualized by applying cetylpyridinium chloride (CPC; Sigma-Aldrich, St. Louis, MO, USA) to the MA and subcultured to isolate strains exhibiting strong hyaluronidase activity. Microbial identification was performed via 16S rRNA gene sequencing (Solgent, Daejeon, Republic of Korea), and the strain was taxonomically classified based on NCBI/BLAST analysis and phylogenetic tree construction [21,22].
2.2. Genome Sequencing and Biosafety Profiling
To extract high-molecular-weight (HMW) genomic DNA, various commercially available HMW genomic DNA kits can be used including the Promega Wizard HMW DNA, Nanobind panDNA, and NEB Monarch HMW DNA extraction kits depending on the type of sample. Most extraction kits use a wide bore tip and inversion mixing to reduce the shearing force during the lysis, binding, and washing elution processes involved in extraction. The extracted HMW genomic DNA was quantified using a qubit HS reagent, and its size distribution was measured over 48 kb on an Agilent T4000 genomic tape. The genomic DNA was sheared to an average size suitable for long-read sequencing using a Covaris g-TUBE device and then purified using AMPure PB beads (Pacific Biosciences, Menlo Park, CA, USA). SMRTbell libraries were prepared using the SMRTbell Express Template Prep Kit 3.0 (Pacific Biosciences, Menlo Park, CA, USA), following the manufacturer’s protocol, and sequenced on the PacBio Revio system. The sequencing data were processed to generate raw long-read FASTQ files, which were analyzed for the presence of virulence genes using VirulenceFinder 2.0 with the default parameters.
Biosafety profiling was performed to assess the hemolytic activity on 5% horse blood agar (Sigma-Aldrich, St. Louis, MO, USA). The plates were incubated at 37 °C for 24 h, and the hemolytic zones were subsequently examined. Based on the zone characteristics, the isolates were classified as α-, β-, or γ-hemolytic. A greenish zone around Bacillus subtilis (B. subtilis) colonies indicated α-hemolysis, whereas Staphylococcus aureus (S. aureus) produced clear zones corresponding to β-hemolysis [23]. To evaluate biogenic amine production, the isolates were cultured on MA supplemented with 0.1% of one of three amino acids (L-tyrosine, L-histidine, or L-ornithine), followed by incubation at 37 °C for 48 h. P. residui BSSK58, Escherichia coli (E. coli), B. subtilis, and S. aureus were inoculated sequentially in a clockwise order, starting from the 9 o’clock position [24]. The biogenic amine production was assessed by monitoring the color changes around the colonies.
2.3. Antimicrobial Resistance and Cytotoxicity
The raw sequencing reads underwent quality control and trimming using fastp (v0.23.1) with the default parameters to remove low-quality bases and adapter sequences [25]. The trimmed reads were then used for genome assembly. Acquired antimicrobial-resistant (AMR) genes in the assembled genome were detected using RESFinder (v4.1.1) [26].
The cytotoxicity was assessed using a lactate dehydrogenase (LDH) assay kit (DG-LDH500, DLS2501; DoGenBio, Seoul, Republic of Korea). Caco-2 and HT-29 cells (Korean Cell Line Bank, Seoul, Republic of Korea) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; HyClone, Logan, UT, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Corning, NY, USA) under a humidified 5% CO2 atmosphere at 37 °C. The cells were seeded into 96-well plates at a density of 1 × 104 cells/well and incubated for 24 h [27]. After incubation, the cells were then treated with serial dilutions of a P. residui BSSK58 culture for 24 h. The cell viability was quantified by measuring the LDH release at 450 nm using a microplate reader (Spark; TECAN, Männedorf, Switzerland) [28,29].
2.4. Optimization of Enzyme Production in P. residui BSSK58 Strain
A medium containing 1.13% tryptone, 0.3% soytone, 0.57% peptone, 0.5% NaCl, 0.25% K2HPO4, and 0.5% hyaluronic acid was used to provide the optimal enzyme production conditions for the P. residui BSSK58 strain. The culture temperature was adjusted to various values: 30 °C, 40 °C, 45 °C, and 50 °C. To investigate the effect of the initial pH on hyaluronidase enzyme production, different pH values of 6.0, 6.5, 7.0, 7.5, 8.0, and 8.5 were used. All the flasks were incubated with shaking at 150 rpm for 24 to 96 h. Then, the culture medium was centrifuged at 8000 rpm for 30 min to obtain a cell-free supernatant, which was used for enzyme analysis.
2.5. Enzyme Production and Purification
The fermentation conditions utilized for maximum enzyme production were based on the flask conditions. To purify hyaluronidase extracted from the P. residui BSSK58 culture, the enzyme protein was first precipitated using a 70% saturated ammonium sulfate solution, as recommended by Reda and El-Shanawany [30]. The hyaluronidase precipitate was dissolved using a 10 mM sodium phosphate buffer (SPB, pH 7.2). The enzyme solution was dialyzed with the same buffer at 5 °C for 1 day and was then purified using an anion exchange resin (Q-Sepharose FF; GE Healthcare, Chicago, IL, USA). The purified active enzyme fraction was collected and dialyzed against a solution containing 30% (w/v) sodium chloride in 10 mM of SPB (pH 7.2). Finally, gel filtration (HiPrep 26/60 Sephacryl S-400 HR; GE Healthcare, Chicago, IL, USA) was performed to obtain a solution containing pure hyaluronidase. The purity of the final hyaluronidase preparation was assessed by Native-PAGE, and only fractions with >95% purity were used for subsequent biochemical characterization experiments [31].
2.6. Determining Protein Concentration and Molecular Weight
The protein concentration was determined using the bicinchoninic acid (BCA; Thermo Fisher Scientific, Waltham, MA, USA) method and bovine serum albumin (BSA; Thermo Fisher Scientific, Waltham, MA, USA) as a standard [32]. The purity and molecular weight of the purified hyaluronidase were measured using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) or native PAGE, following Laemmli’s method [33].
2.7. Biochemical Characterization of Hyaluronidase
The activity of the BSSK58 hyaluronidase was assessed using the modified Elson–Morgan colorimetric method [34,35]. At 35 °C and pH 8, a high-molecular-weight hyaluronic acid (HMW-HA, 1.2–2.2 MDa; Kewpie Corporation, Tokyo, Japan) substrate and dilute BSSK58 hyaluronidase were reacted for 1 h. Then, a 0.8 M potassium tetraborate tetrahydrate (Sigma-Aldrich, St. Louis, MO, USA) solution was added, and the mixture was heated and cooled to room temperature. It was then subjected to a color reaction with a 1% p-dimethylaminobenzaldehyde (p-DMAB, Sigma-Aldrich, St. Louis, MO, USA) solution in acetic acid. The absorbance of the products of this reaction was measured at 585 nm, and the solution’s N-acetylglucosamine (NAG) concentration was obtained by substituting the absorbance into an NAG standard curve for the same reaction. The NAG concentration was subsequently incorporated into the following formula to calculate the hyaluronidase activity unit (HAU).
N: calculated NAG concentration (μM); T: reaction time (min); D: dilution rate; E: volume of the reacted enzyme (mL); S: volume of the reacted substrate (mL).
In this method, 1 unit of hyaluronidase (HAU, unit/mL) was defined as the amount of enzyme required to produce 1 μM of NAG per minute under the aforementioned conditions.
The optimal pH and temperature promoting the enzymatic action of BSSK58 hyaluronidase were investigated. To obtain the optimal pH, 1% HMW-HA solutions were prepared from 100 mM buffers of sodium acetate (pH 3–6), sodium phosphate (pH 6–8), Tris-HCl (pH 7–9), and sodium carbonate (pH 9–11). Subsequently, the activity of BSSK58 hyaluronidase was assessed in HMW-HA substrate solutions with diverse pH ranges.
To determine the optimal reaction temperature, hyaluronidase reactions were conducted at 5 °C intervals in a range of 25–55 °C. The resulting hyaluronidase activity was measured and compared. Additionally, the thermal stability was evaluated by pre-incubating the enzyme at 50, 60, or 80 °C for predetermined periods.
To test the substrate specificity of BSSK58 hyaluronidase for diverse polysaccharides—including bovine- and shark cartilage-derived chondroitin sulfate, sodium heparin, alginate, xanthan, and agarose—in addition to hyaluronic acid, the following experiments were conducted on the aforementioned substrates. Each substrate was prepared using 250 mM of an SPB at a concentration of 10 mg/mL. A total of 100 μL of each substrate solution was mixed with 600 μL of distilled water, 200 μL of the buffer, and 100 μL of the enzyme solution, which had an activity of 350 HAU/mL. The mixed solutions were reacted at 35 °C and 150 rpm for 12 h, then heated at 100 °C for 10 min to stop the reaction.
In addition to performing the preceding experiment, we also calculated the reaction rate constants (Km and Vmax) of BSSK58 hyaluronidase with substrates of HMW-HA and bovine- and shark cartilage-derived chondroitin sulfate using the Lineweaver–Burk equation [36].
2.8. Analysis of BSSK58 Hyaluronidase’s Amino Acid Sequence
The amino acid sequence of hyaluronidase was confirmed through the comparative analysis of the N-terminal 10-amino-acid sequences (ESPEPDNLLQ) obtained from purified protein using the Edman degradation method [37]. The amino acid sequence homology analysis was performed using NCBI/BLAST software.
2.9. Cloning and Expression of Recombinant Hyaluronidase
The genomic DNA of P. residui BSSK58 was extracted using TRIzol (Sigma-Aldrich, St. Louis, MO, USA), which was then used as a template for polymerase chain reaction (PCR) amplification. The primers were designed based on the whole-genome sequencing (Macrogen, Seoul, Republic of Korea) of P. residui BSSK58, using the forward primer F (5′-TTTGCTAGCATGATTCCAAGGAAATGGTTTTCCG-3′) and reverse primer R (5′-TTTTCTCGAGATCACCGTTCTTCGGACCG-3′) (underlined sections indicate the addition of Nhe I and Xho I sites). The PCR program was as follows: initial denaturation at 95 °C for 10 min, followed by 30 cycles of 95 °C for 1 min, 58 °C for 1 min, and 72 °C for 4 min, with a final extension step at 72 °C for 10 min. The PCR product was cloned into TA-cloning vector (Intron Bio, Seongnam-si, Republic of Korea), and then the BSSK58 hyaluronidase gene was isolated and purified by treatment with Nhe I and Xho I restriction enzymes. The isolated BSSK58 hyaluronidase gene was ligated into the pET-24a (Merck, Darmstadt, Germany) vector using DNA ligase enzymes, and the recombinant vector was transformed into E. coli BL21 (DE3) and cultured in a Luria–Bertani (LB) medium containing kanamycin until the absorbance at 600 nm reached approximately 0.6. Then, 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) was added, and the cells were cultured overnight at 30 °C before being centrifuged to form a pellet. The cells were suspended in a phosphate buffer (20 mM sodium phosphate, 500 mM NaCl, 20 mM imidazole, pH 7.2) and sonicated, the extract was centrifuged (13,000 rpm), and the supernatant was collected and filtered through a 0.45 μm syringe filter (Sartorius, Göttingen, Germany). The extracted proteins were purified using affinity chromatography and a HiScreen Ni FF column (GE Healthcare, Chicago, IL, USA).
2.10. Functional Applications and Stability
LMW-HA and chondroitin sulfate were produced by incubating 1% and 10% (w/v) substrate solutions, respectively, with BSSK58 hyaluronidase (35 HAU/mL) at 35 °C and 200 rpm. The degradation products were analyzed through gel permeation chromatography (GPC) using an OHpak SB-804 HQ column (Shodex, Yokohama, Japan). To evaluate their storage stability, the enzyme aliquots were stored at low (4 °C), room (25 °C), and extreme temperatures (35 °C) for up to 24 weeks, and their enzymatic activity was assessed biweekly.
2.11. 13-Week Repeated Oral Dose Toxicity Study
A 13-week repeated oral dose toxicity study of BSSK58 hyaluronidase was conducted in accordance with the Good Laboratory Practice (GLP) guidelines (MFDS Notification No. 2022-93) and OECD Test Guideline 408. The study was performed at Biotoxtech (Chungbuk, Republic of Korea), an AAALAC-accredited facility, and approved by the Institutional Animal Care and Use Committee (IACUC approval no. 241200000036). The test article (BSSK58 hyaluronidase, 170 kDa, 350 HAU, pH 7.2) was supplied as a liquid formulation and administered without dilution. The preparation was obtained through an industrially relevant purification process and ensured a purity level greater than 50%. Specific pathogen-free Sprague—Dawley rats (Crl:CD(SD), 5 weeks old) were obtained from Orient Bio (Seongnam-si, Republic of Korea) and acclimated for 7 days. The animals were housed in polycarbonate cages under controlled environmental conditions (temperature: 19–25 °C; humidity: 30–70%; 12 h light/dark cycle) and provided with an irradiated standard rodent diet and sterilized water ad libitum. Based on a prior 4-week dose range finding study, doses of 0, 1250, 2500, and 5000 mg/kg/day were selected. Each group consisted of 10 males and 10 females, and the test substance was administered once daily by oral gavage for 91 consecutive days. The control group received an equivalent volume of sterile distilled water. The animals’ clinical signs, mortality, body weight, and food consumption were monitored weekly. Ophthalmological examinations and functional observational battery tests were conducted in week 13. At the study’s termination, urinalysis, hematology, serum biochemistry, and coagulation tests were performed. The animals were euthanized, and gross necropsy, organ weight measurements, and histopathological examinations were conducted for the major tissues. Histopathology was performed on all the control- and high-dose-group animals and extended to those in lower-dose groups if treatment-related findings were observed.
2.12. Statistical Analysis
Statistical comparisons between the treatment groups and controls were performed using a one-way analysis of variance (ANOVA), followed by Dunnett’s multiple comparisons test. The analyses were conducted using GraphPad Prism 5 version 5.01 (GraphPad Software, San Diego, CA, USA). Differences were considered statistically significant at p < 0.05.
3. Results
3.1. Isolation and Phylogenetic Identification of Paenibacillus residui BSSK58 Strain
Soil samples collected from Gyeonggi-do, Republic of Korea, were subjected to enrichment culturing on MA (0.1% peptone, 0.5% NaCl, 0.02% K2HPO4, 0.02% KCl, 0.14% Na2HPO4·7H2O, 0.5% HA, and 2.0% agar). The colonies were screened for hyaluronidase activity using 2% cetylpyridinium chloride (CPC), which precipitated undegraded HA, thereby visualizing halo zones around the active colonies. Among the screened isolates, the strain showing the most prominent and consistent halo formation was designated as P. residui BSSK58 (Figure 1A,B). This isolate demonstrated strong extracellular hyaluronic acid-degrading activity, indicating robust hyaluronidase production.
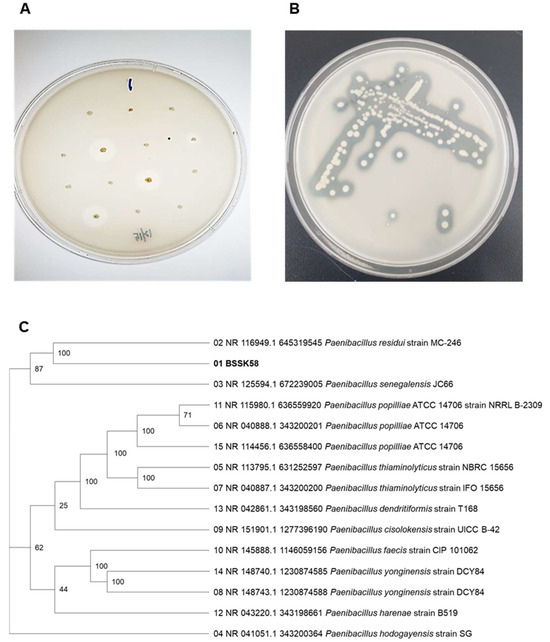
Figure 1.
Isolation and identification of P. residui BSSK58. (A) Clear zone surrounding selected P. residui BSSK58 colony on MA. (B) Growth performance of P. residui BSSK58 colonies treated with CPC on MA. (C) Phylogenetic trees based on 16S rRNA sequences of strain P. residui BSSK58 and other bacillus species. Numbers at nodes represent bootstrap support values from 1000 replicates (only values >50% are shown).
16S rRNA gene sequencing, followed by BLAST analysis, revealed that the isolate had a 99% identity with the type strain P. residui. Phylogenetic tree construction using the neighbor-joining method further supported its classification within the genus Paenibacillus, being closely related to P. residui (100%) and more distantly to P. senegalensis and P. hodogayensis (both 87%) (Figure 1C). This confirmed the isolate’s taxonomic identity to be P. residui BSSK58.
3.2. Whole-Genome Sequencing and Safety Characterization
Virulence gene screening using VirulenceFinder 2.0 identified no virulence-associated genes in the assembled genomic data. This suggests that the sequenced sample did not contain known virulence factors detectable using the VirulenceFinder 2.0 database and algorithm (Table S1).
The hemolytic activity was assessed on 5% horse blood agar. P. residui BSSK58 exhibited γ-hemolysis, characterized by the absence of visible lysis around the colonies (Figure 2B), whereas positive controls (S. aureus and B. subtilis) showed clear β- and greenish α-hemolytic zones, respectively (Figure 2A).
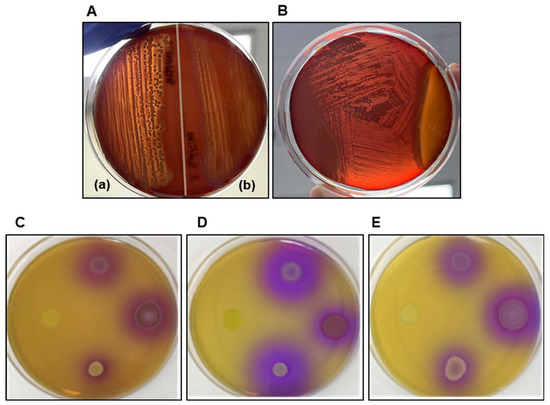
Figure 2.
Assessment of hemolytic activity and biogenic amine production of P. residui BSSK58 and control strains (S. aureus, B. subtilis, and E. coli). (A) Hemolytic activity on 5% horse blood agar of S. aureus (a), exhibiting β-hemolysis, and B. subtilis (b), showing α-hemolysis. (B) Evidence of non-hemolytic phenotype of P. residui BSSK58, categorized as γ-hemolytic. (C–E) Biogenic amine production test on amino acid-supplemented agar: P. residui BSSK58 induced no color changes (yellow) in media containing (C) L-tyrosine, (D) L-histidine, and (E) L-ornithine, indicating absence of biogenic amine production.
P. residui BSSK58′s biogenic amine production was evaluated on modified MA plates supplemented with 0.1% L-tyrosine, L-histidine, or L-ornithine. Each plate was simultaneously inoculated in a clockwise manner with P. residui BSSK58, E. coli, B. subtilis, and S. aureus, starting from the 9 o’clock position. After incubation at 37 °C for 48 h, the positive control strains induced a color change in the surrounding medium from yellow to purple, indicating the decarboxylation of amino acids and subsequent biogenic amine production. In contrast, the region surrounding P. residui BSSK58 remained yellow in all three amino acid media, confirming this strain’s inability to produce biogenic amines (Figure 2C–E). Therefore, P. residui BSSK58 does not produce hemolysin or biogenic amines, ensuring food safety and proving to be a strain that can be utilized industrially.
3.3. Antimicrobial Resistance and Cytotoxicity Profiling
After the whole-genome sequencing (WGS) of the P. residui isolate, an analysis using ResFinder detected no AMR genes (Table S2). The cytotoxicity of viable P. residui BSSK58 cells was evaluated in human intestinal epithelial cell lines (Caco-2 and HT-29) using a lactate dehydrogenase (LDH) release assay (Figure 3). The cells were exposed to P. residui BSSK58 at concentrations ranging from 4.1 × 105 to 4.1 × 108 CFU/mL for 24 h. As shown in Figure 3A,B, no notable increase in the LDH release was observed at concentrations of up to 4.1 × 107 CFU/mL, with the cell viability remaining above 85% in both the cell lines. However, exposure to 4.1 × 108 CFU/mL resulted in an increase in the LDH release exceeding 15%, indicating potential cytotoxicity at this elevated dose. These findings indicate that P. residui BSSK58 does not exert cytotoxic effects at concentrations of up to 4.1 × 107 CFU/mL. Therefore, it can be understood that P. residui BSSK58 does not cause harm to the human body at concentrations below 4.1 × 107 CFU/mL and shows compatibility with intestinal cells, supporting its biosafety for potential applications.
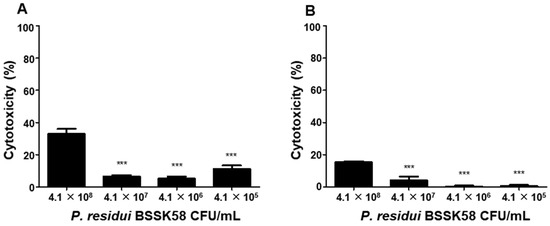
Figure 3.
Assessment of P. residui BSSK58′s cytotoxicity in human intestinal epithelial cells using LDH release assay. (A) Caco-2 cells; (B) HT-29 cells. Cells were exposed to increasing concentrations of viable P. residui BSSK58 (4.1 × 105 to 4.1 × 108 CFU/mL) for 24 h. The data are expressed as the percentage normalized to the untreated control cells (0%). Values are the means ± SD (n = 3), as determined by one-way ANOVA Dunnett’s multiple comparison test. *** Means with different letters are significantly different (p < 0.0001).
3.4. Optimization of Enzyme Production and Purification
The optimal conditions for hyaluronidase production were determined through factorial testing. The maximum enzyme activity was observed at 30–45 °C and pH 7.5–8.0, with the peak yield achieved after 48–72 h incubation (Figure 4A,B). Based on these results, the optimal conditions for the production of BSSK58 hyaluronidase were identified. The enzyme production was closely linked to the cell growth kinetics, with the stationary phase correlating with maximal extracellular activity. The fermentation medium containing 0.5% HA provided the best conditions. Native PAGE analysis of the purified enzyme revealed a major band at 170 kDa, consistent with the presence of a single isoform (Figure 4D). A multi-step purification scheme involving ammonium sulfate precipitation, Q-Sepharose ion exchange, and gel filtration chromatography significantly enhanced the enzyme’s purity and specific activity (Figure 4C,D). The amino acid sequence was analyzed using pure hyaluronidase obtained by removing impurities using the above purification method.
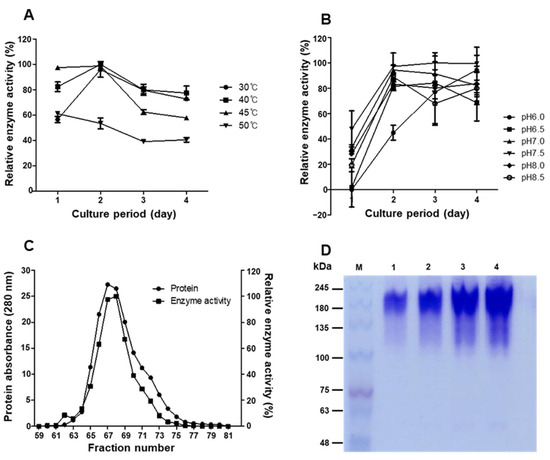
Figure 4.
Optimization of production and purification of enzyme from P. residui BSSK58. (A) Optimal temperature. (B) Optimal pH. (C) Elution profile of BSSK58 hyaluronidase obtained using gel filtration chromatography. (D) Native PAGE of BSSK58 hyaluronidase. Protein marker is shown in lane M, and lanes 1–4 (5, 10, 20, and 30 µg) show purified BSSK58 hyaluronidase with relative molecular weight of 170 kDa.
3.5. The Cloning and Sequence Analysis of the P. residui BSSK58 Hyaluronidase Gene
The amino acid sequence of hyaluronidase was confirmed by comparing the N-terminal 10-amino-acid sequences, ESPEPDNLLQ, obtained through an Edman sequencing analysis of the purified protein. The analysis of hyaluronidase’s full-length amino acid sequence revealed a similarity to proteins containing Ig-like domains (Figure S1). Two primers were designed based on the N-terminus and whole-genome sequencing. The genes were amplified through a PCR, with the primers designed using extracted chromosomal P. residui BSSK58 DNA as a template. As a result, the amplified hyaluronidase gene was found to show a band corresponding to more than 3000 bp (Figure 5A). After the TA cloning of the amplified hyaluronidase gene, it was isolated through treatment with Nhe I and Xho I restriction enzymes. The isolated hyaluronidase was cloned into a pET-24a expression vector (Figure 5B). E. coli BL21 (DE3) was transformed using the recombinant vector and subjected to gene induction using IPTG in the culture medium. The recombinant hyaluronidase protein expressed in the transformed E. coli was purified using affinity chromatography. The purified protein was found to be approximately 170 kDa using SDS-PAGE (Figure 5C).
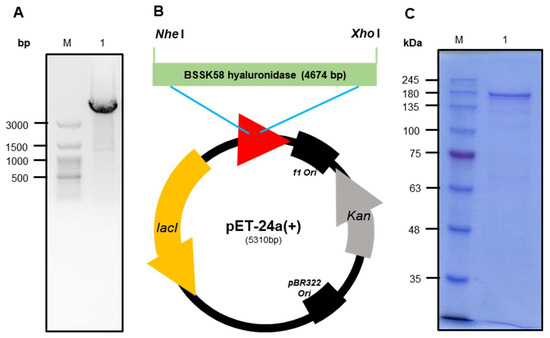
Figure 5.
Cloning of hyaluronidase gene from P. residui BSSK58 and SDS–PAGE showing affinity purification fractions of BSSK58 hyaluronidase. (A) Lane M, DNA ladder marker; lane 1, BSSK58 hyaluronidase gene. (B) pET-24a/BSSK58 hyaluronidase recombinant vector diagram. (C) Lane M shows protein marker; lane 1 shows purified hyaluronidase with relative molecular weight of 170 kDa.
3.6. Enzyme Activity and Thermal Stability
The catalytic activity of BSSK58 hyaluronidase was evaluated over a wide pH range using appropriate buffers for each pH (pH 3.0–11.0). The enzyme showed a distinct optimum pH between 8.0 and 9.0, retaining over 50% of its activity at pH 7.0 and 11.0 (Figure 6A). The optimum temperature was confirmed to be 50 °C (Figure 6B). The thermal stability assays demonstrated the time and temperature dependent inactivation of the hyaluronidase; after 1 h of incubation at 50 °C, 60% of the activity remained; at 60 °C, the activity decreased to 15% within an hour and 0 within 2 h (Figure 6C). Rapid inactivation was observed at 80 °C, with a complete loss of activity within 10 min. These results indicate that BSSK58 hyaluronidase is moderately thermostable under optimal reaction conditions, but prolonged exposure to elevated temperatures compromises its activity.
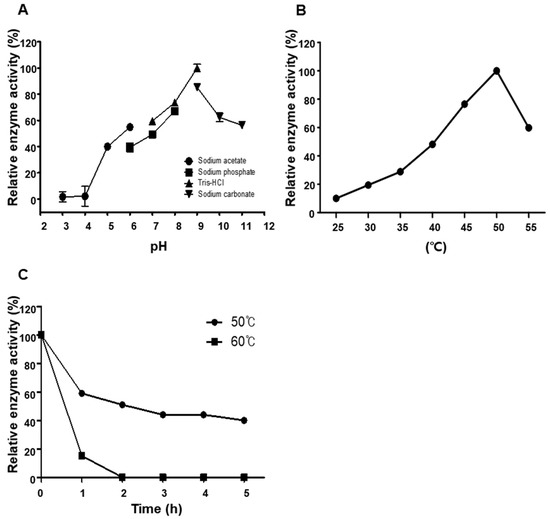
Figure 6.
Optimal pH and temperature and thermal stability of BSSK58 hyaluronidase. (A) Enzyme activity across pH range of 3.0–11.0. (B) Activity at different temperatures (25–55 °C). (C) Thermal stability after 1 h of incubation at 50 °C and 60 °C.
3.7. Effects of Metal Ions and Chelating Agents
Enzyme activity assays conducted in the presence of various metal ions and ethylenediaminetetraacetic acid (EDTA) revealed that Ca2+ most significantly enhanced the activity (173% of the control), followed by Ba2+ (128%), Ni2+ (122%), and Mg2+ (128%) (Table 1). EDTA also increased the activity to 116%, suggesting potential removal of inhibitory metal contaminants or conformational stabilization. In contrast, the activity was markedly reduced by Ag2+ (6%), Cu2+ (27%), and Fe2+ (46%). Other ions, such as Mn2+, Na+, Zn2+, and K+, had variable effects, ranging from moderate to negligible (Table 1). These findings suggest that divalent cations may differentially modulate the enzyme’s conformation or catalytic residues.

Table 1.
Effects of metal ions and chelating agents on the activity of hyaluronidase.
3.8. Substrate Specificity and Enzyme Kinetics
The substrate scope analysis revealed that BSSK58 hyaluronidase hydrolyzed HA (1% w/v) and chondroitin sulfate (10% w/v, both bovine- and shark-derived) with high efficiency. Partial activity was observed for alginate, heparin, and xanthan, while no activity was detected for agarose (Table 2). The high activity toward glycosaminoglycans suggests potential applications in biomedical degradation processes. The kinetic parameters determined using Lineweaver–Burk plots indicated the following rate constants: for HA, Km = 5.78 mM/mL and Vmax = 28.38 mM/mL/min; for bovine CS, Km = 4.93 mM/mL and Vmax = 1.12 mM/mL/min; and for shark CS, Km = 8.06 mM/mL and Vmax = 6.78 mM/mL/min. These values reflect moderate substrate affinity and differential catalytic efficiency depending on the glycosidic structure.
Table 2.
Catalytic activity of BSSK58 hyaluronidase for various polysaccharide substrates.
3.9. Substrate Depolymerization Efficiency and Long-Term Stability
The depolymerization of HMW-HA and chondroitin sulfate was confirmed using GPC following treatment with 35 HAU BSSK58 hyaluronidase at 35 °C. The chromatographic profiles (Figure 7) showed a clear shift toward LMW fractions over time, with distinct peaks observed within 1–2 h of the enzymatic reaction commencing. This progressive breakdown supports the potential use of BSSK58 hyaluronidase in the controlled production of oligosaccharide derivatives.
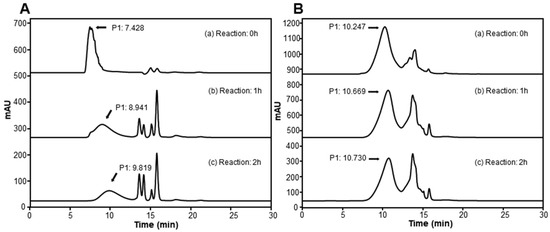
Figure 7.
GPC analysis of BSSK58 hyaluronidase’s enzyme activity for HMW-HA and chondroitin sulfate. (A) In the enzymatic reaction with HMW-HA, the major retention time (RT) peak (P1) was found to shift from 7.4 min to 9.8 min. (B) In the reaction with chondroitin sulfate, the major RT peak (P1) was found to shift from 10.2 min to 10.7 min as the reaction progressed.
In the storage stability experiments, the enzyme aliquots retained >90% of their initial activity after 24 weeks of storage at 4 °C and 25 °C, while a significant decline was observed after 12 weeks at 35 °C (Figure S2). These data underscore the importance of temperature management for preserving enzyme function during long-term storage.
3.10. 13-Weeks Repeated Oral Dose Toxicity Study
To evaluate the systemic safety of BSSK58 hyaluronidase, a 13-week repeated oral dose toxicity study was conducted in Sprague—Dawley rats under GLP-compliant conditions and following OECD Test Guideline 408. The test article was administered once daily by oral gavage at dose levels of 0 (vehicle control), 1250, 2500, and 5000 mg/kg/day, and each group in the study comprised 10 males and 10 females. No test article-related mortality or clinical signs were observed in any dose group throughout the study period. Body weight, food consumption, and ophthalmological assessments revealed no significant differences between the rats administered the test article and the controls (Table S3). Neurological examinations, including a functional observational battery and assessment of spontaneous locomotor activity, showed no treatment-related abnormalities (Table S3). Moreover, the urinalysis, hematological, and serum biochemical parameters demonstrated no toxicologically meaningful alterations attributable to the test article. Finally, the necropsy findings, including the gross pathology, organ weight measurements, and results from a comprehensive histopathological evaluation, revealed no treatment-related lesions in either sex at any dose level (Table S3). Based on these findings, the no-observed-adverse-effect level (NOAEL) for BSSK58 hyaluronidase was determined to be 5000 mg/kg/day in both male and female rats (Table S3).
4. Discussion
In this study, we isolated a novel hyaluronidase-producing strain, Paenibacillus residui BSSK58, from soil collected in Gyeonggi-do, Republic of Korea, using a selective screening method employing hyaluronic acid and cetylpyridinium chloride. The strain exhibited strong extracellular hyaluronidase activity and was taxonomically identified through 16S rRNA sequencing and phylogenetic analysis (Figure 1). Importantly, whole-genome screening against the VFDB and CARD databases revealed no virulence or antibiotic resistance genes (Tables S1 and S2). Additional safety assays confirmed the absence of hemolytic activity, biogenic amine production, and cytotoxicity in human intestinal epithelial cell lines (Caco-2 and HT-29) (Figure 2 and Figure 3).
P. residui BSSK58 also demonstrated susceptibility to EFSA-recommended antibiotics, suggesting its potential safety for use in food and pharmaceutical settings. Several Paenibacillus species are known to produce thermostable hydrolytic enzymes such as cellulases, xylanases, and proteases under cost-effective and scalable fermentation conditions [12,13,14,15]. These bacteria have attracted attention due to their environmental adaptability and low pathogenic risk. However, to date, no reports have described the production of hyaluronidase by P. residui. The discovery of BSSK58 hyaluronidase therefore not only expands the known enzymatic spectrum of this genus but also provides a safer and non-recombinant alternative to commercial hyaluronidases derived from mammalian organs or toxigenic strains such as Clostridium perfringens and Streptococcus pyogenes [5,6,7,8,9,10]. The biochemical analysis of the purified BSSK58 hyaluronidase revealed optimal catalytic activity at pH 8.0–9.0 and 50 °C (Figure 6), with considerable stability under a broad range of physicochemical conditions. The enzyme retained over 90% of its activity after 24 weeks of storage at 4 °C and 25 °C and tolerated an acidic pH, indicating its feasibility for use with oral delivery systems. At 50 °C, the half-life (t1/2) of the purified hyaluronidase was determined to be 2 h, whereas at 60 °C, the t1/2 was reduced to 0.5 h. These results indicate that the enzyme retains substantial thermal stability at moderate temperatures, which may be advantageous for industrial applications requiring prolonged activity [38].
The metal ion modulation studies showed that Ca2+, Ba2+ and Mg2+ enhanced the enzymatic activity, while Cu2+ and Ag2+ strongly inhibited it (Table 1), consistent with the presence of redox-sensitive catalytic residues.
These findings suggest that the enzyme’s performance can be tuned to diverse process conditions. Functionally, BSSK58 hyaluronidase efficiently depolymerized HMW-HA and chondroitin sulfate, as confirmed using gel permeation chromatography (Figure 7). This depolymerization resulted in the production of low-molecular-weight (LMW) derivatives, which are known to exhibit superior biological properties compared to their native macromolecular counterparts. Previous studies have shown that LMW-HA possess enhanced antioxidant, anti-inflammatory, and tissue-penetrative effects [2,3,4]. Compared with animal derived hyaluronidases that present safety and stability issues, and recombinant human hyaluronidase (rHuPH20), which requires costly mammalian expression systems and faces regulatory limitations, BSSK58 offers a safer, non-recombinant, and stable alternative. Microbial enzymes such as the hyaluronidase from Paenibacillus aquistagni SH-7-A act under acidic and less stable conditions, whereas BSSK58 performs optimally under alkaline environments and retains long-term activity, highlighting its broader industrial applicability. From a manufacturing perspective, BSSK58 hyaluronidase is secreted extracellularly and can be purified through a simple centrifugation–microfiltration–ultrafiltration process that scales efficiently from laboratory to pilot production. Its alkaline optimum and thermal stability (50 °C, >90% activity retained for 24 weeks at 4 °C and 25 °C) ensure robustness during concentration and storage. Compared with animal- or mammalian-cell-derived enzymes, the non-recombinant microbial process, use of inexpensive media, and reduced cold-chain requirements indicate clear cost advantages, supporting the feasibility of industrial application. Therefore, the use of BSSK58 hyaluronidase represents not only a catalytic transformation but also a strategic approach to generating bioactive ingredients for high-value applications such as oral nutraceuticals, injectable dermal fillers, and wound-healing biomaterials.
To further validate the functional identity of the hyaluronidase encoded by P. residui BSSK58, N-terminal sequencing and full-length gene cloning were conducted. The sequence analysis revealed that the gene shares partial sequence similarity with known hyaluronidases, particularly within conserved domains involved in HA degradation, while also exhibiting distinct regions suggestive of novel structural features. The heterologous expression of the cloned gene in E. coli successfully yielded a recombinant enzyme exhibiting catalytic activity comparable to that of the native form (Figure 5).
Our results provide strong evidence that the identified gene encodes a functionally active hyaluronidase and supports its suitability for application in non-pathogenic and scalable enzyme production using a microbial system. A 13-week repeated-dose oral toxicity study performed under GLP conditions verified that the administration of BSSK58 hyaluronidase did not cause systemic toxicity in vivo. All hematological, biochemical, and histopathological parameters remained within normal ranges, supporting the enzyme’s regulatory suitability for food, cosmetic, and biomedical applications.
In conclusion, we present the first characterization of a functionally active hyaluronidase from P. residui BSSK58. The strain exhibited a favorable safety profile and produced an extracellular enzyme capable of efficiently depolymerizing HMW-HA and chondroitin sulfate to produce low-molecular-weight derivatives. These products are known to exhibit enhanced biological activity and bioavailability, suggesting the enzyme may have utility in generating high-value functional glycosaminoglycan ingredients. This work highlights the potential of using P. residui BSSK58 as a non-pathogenic microbial platform for safe, scalable, and targeted biocatalysis. Future research will focus on structural elucidation and protein engineering to further enhance its catalytic properties for customized industrial applications.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/fermentation11090519/s1, Table S1: This table shows the presence or absence of virulence genes conferring virulence to five organism in the P. residui isolate’s genotype.; Table S2: This table shows the presence or absence of genes conferring resistance to ten antibiotics in the P. residui isolate’s genotype.; Table S3: Summary of findings from 13-week repeated oral dose toxicity study of BSSK58 hyaluronidase.; Figure S1: Analysis of amino acid sequence homology and gene sequence homology of P. residui BSSK58 strain based on NCBI/BLAST software.; Figure S2: Change of BSSK58 hyaluronidase activity through long-term stability assay.
Author Contributions
Conceptualization, J.-W.P. and J.R.; methodology, J.-S.P., H.C. and J.S.K.; investigation, J.-S.P., H.C., J.S.K. and S.L.; data curation, J.-S.P., H.C., J.S.K., S.L., J.-W.P. and J.R.; validation, J.-W.P. and J.R.; writing—original draft, J.-S.P., H.C., J.S.K. and S.L.; resources, J.-W.P. and J.R.; writing—review and editing, H.H.L., J.-W.P. and J.R.; funding acquisition, H.H.L. and J.-W.P.; supervision, J.-W.P. and J.R. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Republic of Korea Institute of Planning and Evaluation for Technology in Food, Agriculture and Forestry (IPET) through High Value-added Food Technology Development Program, funded by Ministry of Agriculture, Food and Rural Affairs (MAFRA) (RS-2024-00393708).
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Animal Care and Use Committee (Approval Code: 241200000036, Approval Date: 17 December 2024).
Data Availability Statement
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding authors.
Conflicts of Interest
Authors are employed by the company “Biostream Co., Ltd.”. But for purposes of this investigation, there was no financing relationship with the company; therefore, there are no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript.
| HA | Hyaluronic acid |
| HMW | High-molecular-weight |
| LMW | Low-molecular-weight |
| CS | Chondroitin sulfate |
| CHO | Chinese hamster ovary |
| GLP | Good Laboratory Practice |
| MA | Minimal medium agar |
| CPC | Cetylpyridinium chloride |
| WGC | Whole-genome sequencing |
| LDH | Lactate dehydrogenase |
| p-DMAB | p-dimethylaminobenzaldehyde |
| NAG | N-acetylglucosamine |
| HAU | Hyaluronidase activity unit |
| IPTG | Isopropyl β-D-1-thiogalactopyranoside |
| EDTA | Ethylenediaminetetraacetic acid |
References
- Fraser, J.R.; Laurent, T.C.; Laurent, U.B. Hyaluronan: Its nature, distribution, functions and turnover. J. Intern. Med. 1997, 242, 27–33. [Google Scholar] [CrossRef]
- Fallacara, A.; Baldini, E.; Manfredini, S.; Vertuani, S. Hyaluronic Acid in the Third Millennium. Polymers 2018, 10, 701. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Abatangelo, G. Functions of hyaluronan in wound repair. Wound Repair Regen. 1999, 7, 79–89. [Google Scholar] [CrossRef]
- Neuman, M.G.; Nanau, R.M.; Oruña-Sanchez, L.; Coto, G. Hyaluronic acid and wound healing. J. Pharm. Pharm. Sci. 2015, 18, 53–60. [Google Scholar] [CrossRef]
- Krishna Perumal, P.; Huang, C.Y.; Chen, C.W.; Anisha, G.S.; Singhania, R.R.; Dong, C.D.; Patel, A.K. Advances in oligosaccharides production from brown seaweeds: Extraction, characterization, antimetabolic syndrome, and other potential applications. Bioengineered 2023, 14, 2252659. [Google Scholar] [CrossRef]
- Stern, R.; Kogan, G.; Jedrzejas, M.J.; Soltés, L. The many ways to cleave hyaluronan. Biotechnol. Adv. 2007, 25, 537–557. [Google Scholar] [CrossRef] [PubMed]
- Knowles, S.P.; Printz, M.A.; Kang, D.W.; LaBarre, M.J.; Tannenbaum, R.P. Safety of recombinant human hyaluronidase PH20 for subcutaneous drug delivery. Expert Opin. Drug Deliv. 2021, 18, 1673–1685. [Google Scholar] [CrossRef]
- Emmart, E.W.; Cole, L.J. Studies on streptococcal hyaluronidase and antihyaluronidase. J. Bacteriol. 1955, 70, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Benchetrit, L.C.; Avelino, C.C.; Barrucand, L.; St. Amand, S.F.; de Oliveira, C.M. Hyaluronidase production by groups A, B, C, and G streptococci: A statistical analysis. Zentralblatt Für Bakteriol. Mikrobiol. Und Hygiene. 1. Abt. Originale. A Med. Mikrobiol. Infekt. Und Parasitol. 1984, 257, 27–37. [Google Scholar] [CrossRef]
- Frost, G.I. Recombinant human hyaluronidase (rHuPH20): An enabling platform for subcutaneous drug and fluid administration. Expert Opin. Drug Deliv. 2007, 4, 427–440. [Google Scholar] [CrossRef]
- Wunderlich, S.; Gatto, K.A. Consumer perception of genetically modified organisms and sources of information. Adv. Nutr. 2015, 6, 842–851. [Google Scholar] [CrossRef]
- Bampidis, V.; Azimonti, G.; de Lourdes Bastos, M.; Christensen, H.; Durjava, M.; Dusemund, B.; Kouba, M.; Lopez-Alonso, M.; Puente, S.L.; Marcon, F.; et al. Safety and efficacy of a feed additive consisting of endo-1,4-β-d-mannanase produced by Paenibacillus lentus DSM 33618 (Hemicell® HT/HT-L) for chickens and turkeys for fattening, chickens reared for laying, turkeys reared for breeding, minor poultry species to point of lay, pigs for fattening, weaned piglets and minor porcine species (Elanco GmbH). EFSA J. 2023, 21, e07878. [Google Scholar] [CrossRef] [PubMed]
- Grady, E.N.; MacDonald, J.; Liu, L.; Richman, A.; Yuan, Z.C. Current knowledge and perspectives of Paenibacillus: A review. Microb. Cell Fact. 2016, 15, 203. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, S.; Li, Y.; Hao, T.; Chen, S. Nitrogen fixation by Paenibacillus polymyxa WLY78 is responsible for cucumber growth promotion. Plant Soil 2022, 473, 507–516. [Google Scholar] [CrossRef]
- Siddiqui, M.A.H. Optimization, isolation and characterization of cellulase-free thermostable xylanase from Paenibacillus sp. Am. J. Life Sci. 2016, 4, 11. [Google Scholar] [CrossRef]
- Moon, S.G.; Kothari, D.; Lee, W.D.; Kim, J.I.; Kim, K.I.; Kim, Y.G.; Ga, G.W.; Kim, E.J.; Kim, S.K. Potential probiotic acceptability of a novel strain of Paenibacillus konkukensis SK 3146 and its dietary effects on growth performance, intestinal microbiota, and meat quality in broilers. Animals 2022, 12, 1471. [Google Scholar] [CrossRef]
- Li, H.; Zhang, X.; Zou, W.; Wang, T.; He, W.; Wang, L. Hyaluronidase from Paenibacillus aquistagni SH-7-A: Identification and characterization. Int. Biodeter Biodegrad. 2024, 187, 105798. [Google Scholar] [CrossRef]
- Sahoo, S.; Panda, P.K.; Mishra, S.R.; Nayak, A.; Dash, S.K.; Ellaiah, P. Optimization of Physical and Nutritional Parameters for Hyaluronidase Production by Streptococcus mitis. Indian. J. Pharm. Sci. 2008, 70, 661–664. [Google Scholar] [CrossRef]
- Patil, S.P.; Shirsath, L.P.; Chaudhari, B.L. A halotolerant hyaluronidase from newly isolated Brevibacterium halotolerans DC1: Purification and characterization. Int. J. Biol. Macromol. 2021, 166, 839–850. [Google Scholar] [CrossRef]
- Lu, Z.; Guo, W.; Liu, C. Isolation, identification and characterization of novel Bacillus subtilis. J. Vet. Med. Sci. 2018, 80, 427–433. [Google Scholar] [CrossRef]
- Thapa, A.; Budhathoki, A.; Sapkota, A.; Sainju, M.; Shrestha, P. Isolation, identification and screening of Bacillus species with antimicrobial activity from different soil samples of Kathmandu Valley. Nepal J. Biotechnol. 2021, 9, 1–6. Available online: https://nepjol.info/index.php/NJB/article/view/41892 (accessed on 28 July 2025). [CrossRef]
- Xu, B.H.; Lu, Y.Q.; Ye, Z.W.; Zheng, Q.W.; Wei, T.; Lin, J.F.; Guo, L.Q. Genomics-guided discovery and structure identification of cyclic lipopeptides from the Bacillus siamensis JFL15. PLoS ONE 2018, 13, e0202893. [Google Scholar] [CrossRef]
- Amoah, K.; Dong, X.H.; Tan, B.P.; Zhang, S.; Kilbourne, F.K.A.; Chi, S.Y.; Yang, Q.H.; Liu, H.Y.; Zhang, H.T.; Yang, Y.Z. In vitro assessment of the safety and potential probiotic characteristics of three Bacillus strains isolated from the intestine of hybrid grouper (Epinephelus fuscoguttatus × Epinephelus lanceolatus). Front. Vet. Sci. 2021, 8, 675962. [Google Scholar] [CrossRef]
- Kostandinovska, S.; Kungulovski, D.; Atanasova-Pancevska, N. Potential Probiotic Bacillus Strains Isolated from Contaminated Soil in North Macedonia: Salmonella Growth Inhibition. Biol. Life Sci. Forum. 2024, 31, 17. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesøe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Ezhilarasi, A.A.; Vijaya, J.J.; Kaviyarasu, K.; Maaza, M.; Ayeshamariam, A.; Kennedy, L.J. Green synthesis of NiO nanoparticles using Moringa oleifera extract and their biomedical applications: Cytotoxicity effect of nanoparticles against HT-29 cancer cells. J. Photochem. Photobiol. B Biol. 2016, 164, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Raje Urs, V.S.; Ramlal, S.; Batra, H.V.; Naika, M.; Jeyabalaji, J.K. An in-vitro screening for biogenic amines producing microorganisms from fermented foods and its degradation by bacteria from canine saliva. J. Pure Appl. Microbiol. 2019, 13, 271–280. [Google Scholar] [CrossRef]
- Van den Bossche, S.; Vandeplassche, E.; Ostyn, L.; Coenye, T.; Crabbe, A. Bacterial interference with lactate dehydrogenase assay leads to an underestimation of cytotoxicity. Front. Cell Infect. Microbiol. 2020, 10, 494. [Google Scholar] [CrossRef]
- Reda, F.; El-Shanawany, S. Characterization and immobilization of a novel hyaluronidase produced by Streptomyces roseofulvus. Egypt. J. Bot. 2020, 60, 213–224. [Google Scholar] [CrossRef]
- Ebraheem, M.A.; El-Fakharany, E.M.; Husseiny, S.M.; Mohammed, F.A. Purification and characterization of the produced hyaluronidase by Brucella Intermedia MEFS for antioxidant and anticancer applications. Microb. Cell Fact. 2024, 23, 200. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, C.R.; Bower, L.M.; Warren, C. Measurement of hyaluronidase activity in normal human serum. J. Pharm. Biomed. Anal. 1996, 14, 707–712. [Google Scholar] [CrossRef]
- Takahashi, T.; Ikegami-Kawai, M.; Okuda, R.; Suzuki, K. A fluorimetric Morgan–Elson assay method for hyaluronidase activity. Anal. Biochem. 2003, 322, 257–263. [Google Scholar] [CrossRef]
- Lineweaver, H.; Burk, D. The determination of enzyme dissociation constants. J. Am. Chem. Soc. 1934, 56, 658–666. [Google Scholar] [CrossRef]
- Edman, P.; Begg, G. A protein sequenator. Eur. J. Biochem. 1967, 1, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Kang, Z.; Zhang, N.; Du, G.; Chen, J. High-yield novel leech hyaluronidase to expedite the preparation of specific hyaluronan oligomers. Sci. Rep. 2014, 4, 4471. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).