1. Introduction
The world possesses abundant marine and freshwater resources, with well-established shrimp and crab farming and fishing industries. The main shrimps and crabs include
Penaeus vannamei,
Macrobrachium rosenbergii, Jumbo Crab, and King Crab [
1]. According to the statistics of the Food and Agriculture Organization of the United Nations (FAO), the total output of shrimps and crabs caught in the world reached 2.185 million tons in 2021 and increased to 2.232 million tons in 2022, with a year-on-year increase of 2.2% [
2]. In terms of shrimp farming, the global production was about 5.6 million tons in 2022, slightly decreased (by 0.4%) to 5.56 million tons in 2023, and is expected to increase by 4.8% to 5.88 million tons in 2024 [
3]. Current shrimp and crab processing practices generate shell waste amounting to 30–40% of the raw material input. [
4]. According to the latest research estimates, more than 2.5 million tons of shrimp and crab shell waste are produced in the world every year [
5], of which only about 25% are used for feed production and other purposes, and most of them are directly discarded or landfilled, which not only causes environmental pollution but also wastes valuable biological resources. Therefore, the resource utilization of shrimp and crab shells has become a global research hotspot [
6].
Chitin, the main structural component in shrimp and crab shells, undergoes enzymatic modification through two principal pathways to generate bioactive derivatives [
7]. Chitin deacetylase (CDA) catalyzes the deacetylation of chitin to produce chitosan through selective removal of acetyl groups, while chitinases hydrolyze β-1,4-glycosidic bonds to yield chitooligosaccharides [
8]. The resulting chitosan exhibits unique cationic properties critical for antimicrobial applications and drug delivery systems, whereas chitooligosaccharides demonstrate immunomodulatory and prebiotic functions across medical and agricultural domains [
9]. This enzymatic specificity highlights CDA’s unique role in value-added chitin conversion.
Bacillus species have emerged as particularly prolific sources of chitinolytic enzymes, with several CDAs from this genus demonstrating remarkable industrial potential.
Bacillus spp. are superior for chitosanase (CDA) production due to their stress-resilient extracellular chitinolytic systems, driven by specialized chitinase gene clusters (chiA/B), enabling efficient, cofactor-free degradation of crustacean waste. Their GRAS status, compatibility with low-cost substrates, and industrial scalability further solidify their practicality for valorization applications. Recent studies have characterized novel CDAs from
Bacillus strains isolated from diverse environments. Liang et al. identified a CDA from
Bacillus aryabhattai TCI-16 isolated from mangrove soil, achieving 120.35 U/mL activity and demonstrating unique genomic organization of chitinolytic enzymes through complete genome sequencing [
10]. Subsequent heterologous expression of this enzyme revealed its ability to modify chitin structure through partial deacetylation (69.23% degree of deacetylation) while maintaining thermal stability up to 60 °C [
11]. Parallel work on
Bacillus cereus ZWT-08 demonstrated exceptional CDA performance (613.25 U/mL) through optimized fermentation, achieving > 90% deacetylation of colloidal chitin substrates. These
Bacillus-derived enzymes share common alkaline pH optima (pH 8.0–9.0) and moderate thermostability (50–60 °C) while exhibiting distinct substrate recognition patterns and genomic architectures that suggest evolutionary adaptation to specific ecological niches [
12]. The research of Sreekumar [
13] and Linhorst et al. [
14] shows that chitin deacetylase can realize the selective modification of chitin under specific conditions and obtain chitosan products with different physical and chemical properties. These findings provide a new enzymatic basis for the development of efficient chitin degradation processes. Compared to the
Bacillus species, Escherichia coli offers advantages in cost-effective large-scale production, avoids protease interference from native hosts, and provides a controllable platform for subsequent enzyme engineering (e.g., thermostability improvement), thereby balancing R&D efficiency with industrial adaptability requirements.
In the early stage, our group identified Bacillus strains with high chitinolytic potential from 151 isolates through functional genomics screening. In this study, we sequenced the genomes of 151 Bacillus strains and established a genome-directed screening approach that identified five high-yield CDA-producing strains. B. pumilus B866 showed exceptional potential, and its CDA was obtained through heterologous expression in E. coli. The enzyme exhibited optimal activity at 55 °C and pH 7.0–7.5, with a novel ferrous-ion-dependent catalytic mechanism. This research provides key enzymatic resources for developing efficient chitin biotransformation technologies.
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
As in the previous research [
15], all strains were isolated from coordinates 90°66′–91°80′ E, 41°99′–40°94′ N. In brief, 50 sampling points were selected within the coordinates 90°66′–91°80′ E, 41°99′–40°94′ N. Three samples (1 kg soil for each sample) were collected randomly within a 5 × 5 m area at each location, and each sample was collected at a depth of 10–15 cm from the surface. The soil sample (2 kg) was mixed well at each site after removing impurities obtained by the quartering method [
16] and stored at 4 °C within 24 h. Subsequently,
Bacillus were isolated and purified from the soil samples, and 151
Bacillus strains were isolated from samples collected in this region. Among these isolates,
Bacillus pumilus B866 was obtained through this isolation protocol and deposited in the China Center for Type Culture Collection (CCTCC) under accession number CCTCC M 2025385.
2.2. Genomic Library Preparation and Sequencing
Genomic DNA was isolated from
Bacillus cultures (2 × 10
6 cells) using a modified CTAB protocol [
17], with additional lysozyme treatment (10 mg/mL, 37 °C for 30 min) to enhance lysis of Gram-positive cell walls. Residual RNA was removed by DNase-free RNase A digestion, and DNA purity/quantity was assessed using a Qubit 3.0 fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) alongside agarose gel electrophoresis. Library construction was performed using the MGIEasy Micro DNA Library Preparation Kit (MGI, Shenzhen, China, cat. No. 1000011553). DNA was fragmented to 50–800 bp, which was performed using a Covaris E220 ultrasonicator (Brighton, UK), followed by size selection (200–500 bp) with AMPure XP beads (Beckman Coulter, Brea, CA, USA).
DNA ends were repaired using T4 DNA polymerase and polynucleotide kinase (Enzymatics, Beverly, MA, USA), and blunt-end adapters with T-overhangs were ligated. Libraries were amplified for 8 PCR cycles and circularized into single-stranded DNA templates. For sequencing, strain libraries were processed on a DNBSEQ-G400 platform (BGI, Shenzhen, China) using the DNBSEQ-G400RS High-throughput Rapid Sequencing Set (Item No.: 940-000231-00) to generate 100 bp paired-end reads.
2.3. Genome Assembly and Quality Control
Raw reads were filtered using SOAPnuke [
18] (v1.5.2; parameters “q 0.2 -l 0.2 -n 0.05 -d”) to eliminate low-quality bases (Phred score < 20), adapter sequences, and PCR duplicates. High-confidence reads were assembled de novo using SPAdes (v3.11.1) with iterative k-mer optimization (k = 43, 53, 63, 73, 83) and careful mismatch correction, specifically tuned for high-GC
Bacillus genomes. Contigs shorter than 500 bp were discarded to reduce fragmentation artifacts.
2.4. Gene Annotation and Biosynthetic Gene Cluster Prediction
Genome annotation for all 151 Bacillus strains was performed using Prokka (v1.14) (Prokka: Rapid prokaryotic genome annotation) with default parameters. For carbohydrate-active enzyme (CAZyme) annotation, the dbCAN2 meta server was employed to identify and classify enzymes into six major categories: glycoside hydrolases (GHs), glycosyltransferases (GTs), polysaccharide lyases (PLs), carbohydrate esterases (CEs), carbohydrate-binding modules (CBMs), and auxiliary activities (AAs). Special attention was given to CE4 family members (chitin deacetylases) and GH18/GH19 families (chitinases) due to their roles in chitin metabolism.
Secondary metabolite prediction was conducted through antiSMASH (antiSMASH 6.0: Improving cluster detection and comparison capabilities) using genomic data in FASTA format. Biosynthetic gene cluster (BGCs) information from each strain was individually stored in strain-specific folders for subsequent statistical analysis. Quantitative characteristics including secondary metabolite types, BGC numbers, gene counts, and cluster lengths were systematically recorded. BiG-SLiCE (A highly scalable tool maps the diversity of 1.2 million biosynthetic gene clusters) was subsequently employed for cluster classification using antiSMASH-generated GBK files.
2.5. Correlation Analysis and Difference Analysis
This study utilized GraphPad Prism 10 for correlation analysis and comparative analysis, with the analytical procedures structured as follows:
Correlation Analysis: Prior to analysis, data normality was assessed through either the Shapiro–Wilk test or Anderson–Darling test. For datasets conforming to normal distribution, Pearson correlation coefficients were calculated. For non-normally distributed data, Spearman correlation coefficients were employed. The selection of correlation methodology was strictly based on data distribution characteristics to ensure analytical appropriateness.
Comparative Analysis: The analysis comprised two primary scenarios: comparisons between two groups and comparisons involving three or more groups.
- (1)
Two-Group Comparison—Independent Samples: Normality testing was first conducted. For normally or approximately normal distributed data, an F-test for homogeneity of variances was performed. Non-paired t-test was applied when variance homogeneity was satisfied. Welch’s corrected unpaired t-test was utilized when variance heterogeneity was detected. For non-normally distributed data, the non-parametric Mann–Whitney U test was implemented.
- (2)
Two-Group Comparison—Paired Samples: Normality testing was performed on paired differences. For normally distributed differences, standard paired t-test was applied when differences demonstrated consistency. Ratio paired t-test was employed for inconsistent differences. Wilcoxon signed-rank test served as the non-parametric alternative for non-normal differences.
- (3)
Multi-Group Comparison (≥3 Groups): Joint assessment of normality (Shapiro–Wilk/Anderson–Darling) and variance homogeneity (Brown–Forsythe test) was conducted. For normally distributed data with homogeneous variances, one-way ANOVA was executed. For normal data with heterogeneous variances, Brown–Forsythe and Welch ANOVA tests were applied. For non-normal distributed data with variance heterogeneity, the Friedman test (non-parametric repeated measures ANOVA) was employed.
2.6. Pan-Genome Analysis
A pan-genome analysis of strains was conducted using BPGA (BPGA—an ultra-fast pan-genome analysis pipeline). The built-in USEARCH algorithm clustered homologous gene families from Prokka-annotated protein FASTA files with default sequence similarity threshold (50%). Core and pan-genome curves were generated through 500 combinatorial iterations. A neighbor-joining (NJ) tree was constructed using core/accessory genes. Functional annotation of core, accessory, and unique genes was performed through comparative analysis with the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Clusters of Orthologous Groups (COG) databases.
2.7. Average Nucleotide Identity Analysis
As a widely adopted method in microbial taxonomy, phylogenetics, and ecology, the average nucleotide identity analysis evaluates genomic similarity between microorganisms. The integrated prokaryotes genome and pan-genome analysis service (IPGA v1.09,
https://nmdc.cn) was utilized to perform ANI analysis on all 151
Bacillus genomes under default parameters.
2.8. Primary Screening and Secondary Screening of CDA Strain
The 120 strains to be screened were retrieved from cold storage and inoculated into LB medium for activation. Each strain was transferred to a 20 mL test tube containing 5 mL of LB liquid medium and incubated overnight at 30 °C with agitation at 180 rpm. Primary screening medium plates containing p-nitroacetanilide were prepared for detection of strains producing some deacetylases. The strains were streaked on the LB plate and cultured at 30 °C for 36 h. A single colony was selected and inoculated on the primary screening plate and cultured at 30 °C for 6 days to observe whether there was a yellow color circle around the colony, and the chitin deacetylase would deacetylate p-nitroacetanilide and change it from colorless to yellow. Bacteria with discolored culture medium were selected and transferred to LB liquid culture medium test tubes for culture and stored at 4 °C for rescreening.
The strains selected by primary screening were inoculated into the medium for secondary screening, and 50 mL of the medium was filled in a 250 mL triangular flask, and the medium was shaken at 30 °C and 180 rpm for 2 days to determine the enzyme activity of the inoculated strains. The activity of the CDA enzyme was determined by spectrophotometry at 400 nm using a MAPADA P4 spectrophotometer (Shanghai mapada instruments Co., Ltd., Shanghai, China) with a 1 cm pathlength quartz cuvette to select the appropriate strain.
For extracellular enzyme activity, cultures were centrifuged at 10,000 rpm for 15 min at 4 °C to separate bacterial cells from the fermentation broth. The cell-free supernatant was carefully collected and used directly for extracellular enzyme activity assays. For intracellular enzyme activity, the cell pellets obtained after centrifugation were washed twice with phosphate buffer (50 mM, pH 7.0) to remove residual extracellular enzymes. The washed cells were then resuspended in the same buffer and disrupted by sonication (6 cycles of 30 s on/30 s off at 40% amplitude) on ice. After sonication, the lysate was centrifuged at 12,000 rpm for 20 min at 4 °C to remove cell debris, and the resulting supernatant was used for intracellular enzyme activity measurements.
2.9. CDA Enzyme Activity Determination Method
Take a test tube and add 1 mL of 200 mg/L p-nitroacetanilide aqueous solution, 1 mL of enzyme solution with appropriate concentration, and 3 mL of 0.05 mol/L phosphate buffer pre-incubated at 50 °C to make the final volume of the reaction solution 5.0 mL. React in a water bath at 50 °C for 15 min, stop the enzymatic reaction in a boiling water bath, add water to a constant volume of 10 mL, mix well, centrifuge at 3000 rpm for 10 min, determine the absorbance value (A
400) of the supernatant, add 1 mL of the same concentration of inactivated enzyme solution (inactivated at high temperature) to the blank control system, and the rest are the same as above. The absorbance (A
0) of the supernatant was measured. Each sample corresponds to a blank. K-linear coefficient (0.0648) obtained from the standard calibration curve of p-nitroaniline, representing the slope of the linear regression between p-nitroaniline concentration and absorbance at 400 nm. This calibration curve is included as
Supplementary Figure S1. Definition of enzyme activity unit: under the above reaction conditions, the amount of enzyme required to produce 1 μg of p-nitroaniline per hour is defined as an enzyme activity unit. The calculation formula for enzyme activity is
where
A400—Absorbance value of enzymatic hydrolysate sample;
A0—absorbance value of blank;
T—enzymatic reaction time, h;
K—linear coefficient (0.0648).
2.10. Optimization of Fermentation Medium
The fermentation medium was systematically optimized based on the single factor rotation method. All experiments were repeated three times. First, in the basal medium (10 g/L peptone, 5 g/L NaCl, 10 g/L chitin), 10 g/L glucose, sucrose, lactose, starch, or fructose were used as carbon sources, respectively, and cultured at 37 °C and 160 rpm for 48 h to select the best carbon source. Then, the preferred carbon source was fixed, and the response of 10 g/L peptone, yeast extract, urea, (NH4)2 SO4, and NaNO3 to the effect of the nitrogen source was tested in turn. After the combination of carbon and nitrogen sources was determined, the effects of inorganic salts such as 5 g/L KH2PO4, MgSO4, NaCl, FeSO4, and CuSO4 were further evaluated, and the gradient addition of chitin from 2 to 12 g/L (30 °C, 180 rpm at this stage to enhance the induction effect) was also investigated. Finally, the optimal components (lactose, yeast extract, FeSO4, and 10 g/L chitin) were integrated. The optimized culture medium was prepared and fermented at 30 °C and 180 rpm for 48 h to verify the effect of enzyme production, and the significant difference was confirmed by t test (p < 0.05).
2.11. Heterologous Expression of BpCDA
Bacillus pumilus strain preserved in glycerol was activated on LB solid medium and cultured at 37 °C for 12 h. A single colony was picked and inoculated into 5 mL LB liquid medium and cultured overnight at 37 °C and 180 rpm. The genomic DNA of the strain was extracted according to the FastPure Bacteria DNA Isolation Mini Kit (Vazyme, Nanjing, China). The primers (F: CGGGATCCatgattaaattaattgtaaatgcag R: CCCAAGCTTttaaaactttacaagctctattcct) were designed according to the genetic sequence of CDA in NCBI database (accession numbers PV243451), and the target gene fragment underwent double restriction enzyme digestion with BamH I/Hind III, followed by verification through 1% agarose gel electrophoresis and target fragment recovery. The purified gene fragment was directionally ligated into pET-28a vector. The recombinant plasmid was transformed into competent E. coli BL21 (DE3) and spread on LB plates containing kanamycin for overnight culture at 37 °C. Transformants were randomly selected for PCR verification, and the recombinant strain BL21 (DE3)/pET-28a-CDA was stored at −80 °C after positive clones were confirmed.
2.12. BpCDA Activity Assay
The enzyme activity assay was mainly as previously described with some modifications [
19]. The reaction mixtures of 400 μL (GlcNAc)
2 (10 mg/mL) and 100 µL of
BpCDA (6 mg/mL) were added to 400 µL Tris-HCl buffer solution (10 mM, pH 7.0), and the reaction system was incubated at 40 °C for 6 h. The enzyme reaction was terminated by boiling at 100 °C for 10 min in a sealed tube prior. The supernatant was collected by centrifugation at 12,000 rpm for 5 min, and the amount of acetic acid in 10 µL supernatant was determined to calculate the enzyme activity through the A340 according to the K-ACET acetic acid determination kit (Megazyme, Bray, Ireland) [
20]. One unit of
BpCDA activity (U) was defined as the amount of enzyme required to produce 1 μmol of acetic acid per minute under the above conditions. Recombinant
E. coli BL21(DE3)/pET-28a-CDA was induced with 0.4 mM IPTG at 16 °C for 20 h, followed by cell lysis via sonication in binding buffer (20 mM Tris-HCl, 500 mM NaCl, 20 mM imidazole, pH 7.9) and purification using Ni-NTA.
2.13. Enzymatic Properties of BpCDA
The optimal temperature for BpCDA was determined by measuring enzyme activity at 5 °C increments from 35 °C to 65 °C under standard conditions. Reactions in Tris-HCl buffer (10 mM, pH 7.0) were incubated for 2 h at test temperatures, with relative activities normalized to maximum observed activity (100%). For the thermal stability assay, aliquots of the enzyme were placed at 50, 55, and 60 °C after being incubated at 4, 8, 12, 16, 20, and 24 h to determine the temperature stability of BpCDA. Three replicate experiments were set for each group.
The effects of pH on BpCDA activity were assessed using citrate (pH 3.0–5.0), phosphate (pH 5.0–7.0), Tris-HCl (pH 7.0–9.0), and glycine-NaOH (pH 9.0–10.0) buffers (all 10 mM). Enzyme activity was measured after 2 h incubation at 55 °C, with results expressed as percentage relative to maximum activity. For the pH stability assessment, enzyme-substrate mixtures in test buffers were incubated at 55 °C for 10 h in a water bath before residual activity measurement. All experiments included three biological replicates.
To investigate metal ion effects on enzymatic activity, purified BpCDA was incubated with Fe3+, Cu2+, Ca2+, Mn2+, Fe2+, Mg2+, Zn2+, Pb2+, K+, Na+ (1 mM or 10 mM final concentrations) alongside EDTA controls in Tris-HCl buffer (10 mM, pH 8.0) at 55 °C for 6 h. Pre-experimental validation confirmed that Fe2+ stability was rigorously maintained throughout the 6-h incubation at 55 °C; reaction systems were supplemented with 1 mM sodium ascorbate (a reducing agent) and rigorously purged with nitrogen prior to sealing, effectively preventing significant oxidation (<5% conversion to Fe3+, verified by ferrozine assay in parallel controls). Whereas control groups received equivalent volumes of deionized water, residual activities were quantified relative to metal-free controls (defined as 100% activity). Finally, triplicate biological replicates were performed for all experimental conditions.
3. Results
3.1. Genomic Overview and Diversity
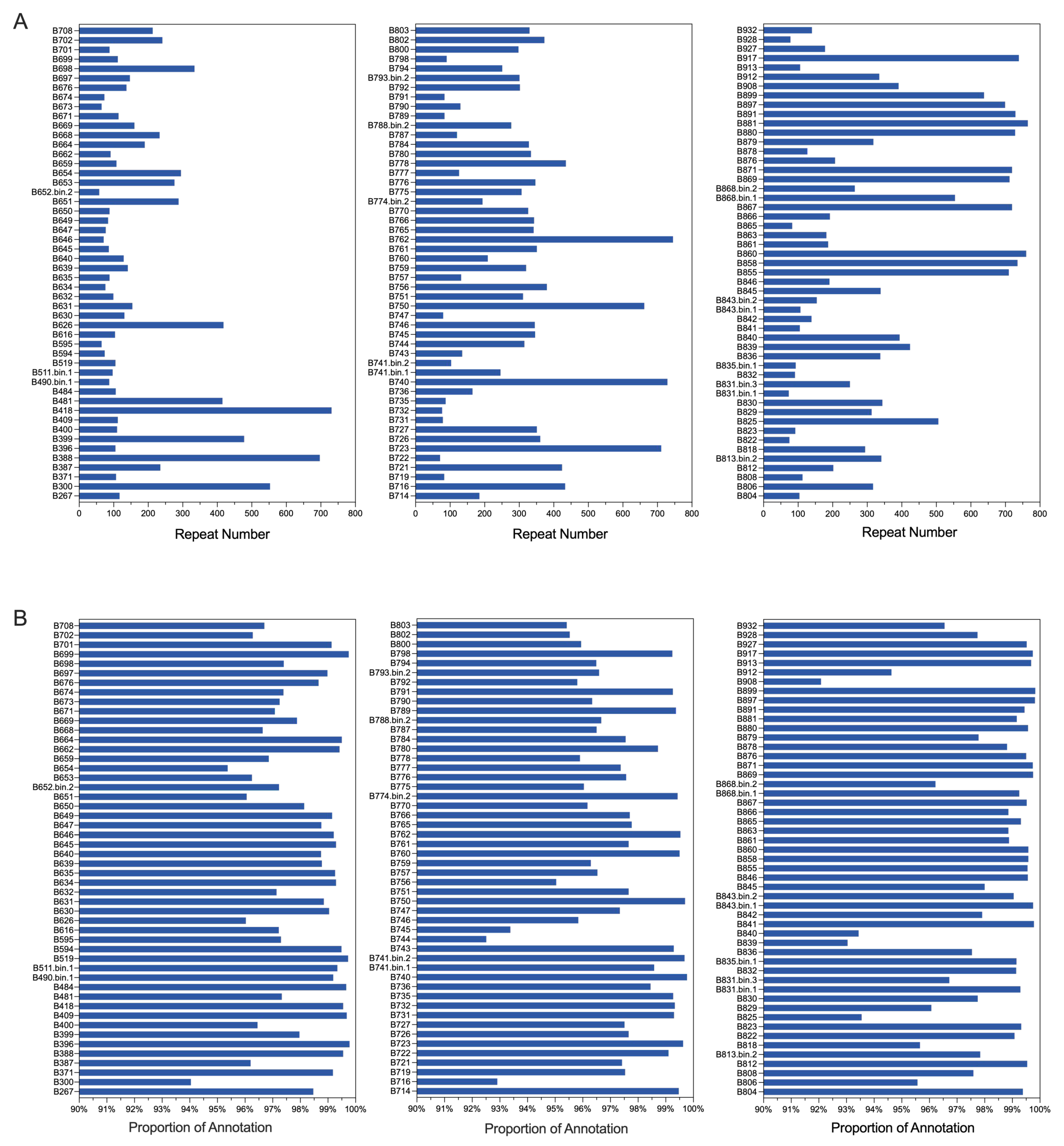
Among the 151 strains of
Bacillus that have been sequenced, we performed genome analysis on all of them, with results shown in
Table S1, including genome completeness and contamination levels. The size of the
Bacillus genomes ranged between 2.49 million base pairs (Mb) (B399) and 9.38 Mb (B778), with an average size of 4.75 ± 0.08 Mb. The G + C content varied from 32.50% to 73.06% (average 40.50 ± 0.38%), reflecting the genetic diversity within the genus. The number of coding sequences (CDS) ranged from 2265 (B399) to 9047 (B778), with an average of 4769.18 ± 83, and contig numbers varied between 10 (B843) and 2952 (B778), with an average of 90.21 ± 23.25 (
Supplementary Table S1). Gene annotation ratios were consistently high (>90%) across all strains, indicating high-quality genome sequencing and annotation (
Figure 1B). Notable differences in repetitive sequences were observed, with strains B881 (765), B860 (760), and B762 (745) containing the highest numbers (
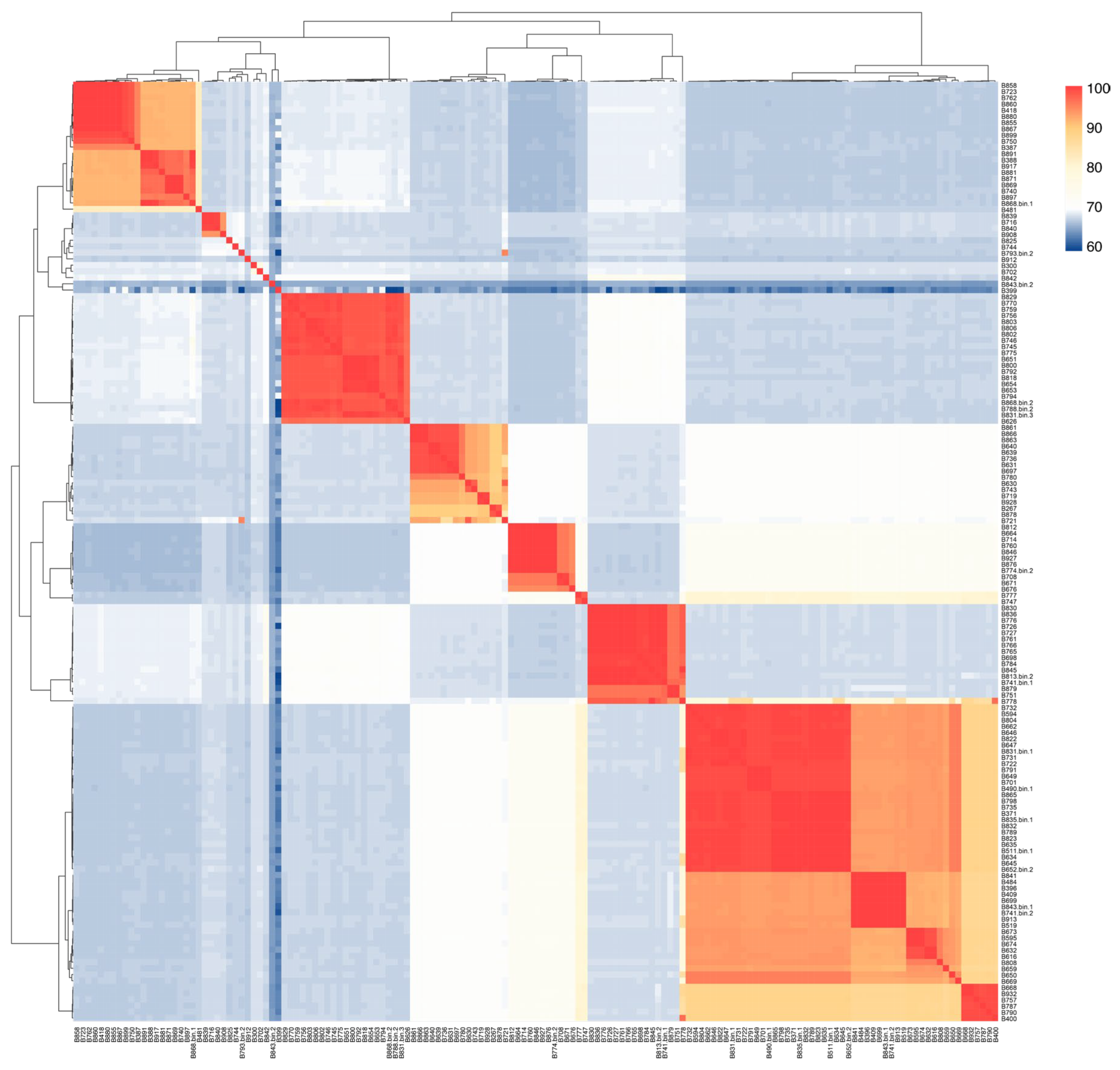
Figure 1A). Average nucleotide identity (ANI) analysis revealed both similarities and differences among the 151 strains, with a mean ANI value of 72.63% ± 10.82%. While most ANI values distributed between 60% and 70%, some strains showed remarkable similarity, such as B371 and B647 (99.17%), B594 and B822 (99.18%), indicating close evolutionary relationships. These findings highlight the genomic diversity within the
Bacillus species and provide important insights into their genetic relationships and evolutionary history (
Figure 2).
3.2. Metabolic Analysis
According to
Figure 3, we can further study the metabolic mechanism of
Bacillus through analysis of their carbohydrate-active enzymes (CAZymes). CAZymes include glycoside hydrolases (GHs), glycoside transferases (GTs), polysaccharide lyases (PLs), carbohydrate esterases (CEs), carbohydrate-binding modules (CBMs), and auxiliary activities (AAs). This CAZy enzyme annotation analysis of radiation-resistant
Bacillus sp. strains revealed their remarkable diversity and environmental adaptability potential in carbohydrate metabolism (
Figure 3).
Among the six major CAZy categories, GHs were the most abundant, with GH family counts ranging from 19 to 164 across strains, highlighting their central role in carbohydrate degradation. Notably, strains B721 (164), B387 (146), and B778 (130) contained the most glycoside hydrolases, suggesting exceptional potential in carbohydrate degradation. The GH18 and GH19 families, encoding chitinases responsible for chitin hydrolysis, were widely distributed among multiple strains, likely correlating with the ecological niche of Bacillus sp. in chitin-rich environments (e.g., soil or insect residues), where chitin serves as a critical carbon and nitrogen source.
The annotation of CEs revealed a significant prevalence of CE4 family members (chitin deacetylases) across strains (3–40 per strain). Chitin deacetylases catalyze the deacetylation of chitin to chitosan, a process that modifies substrate chemistry and enhances biodegradability. For instance, strains B778 and B651 contained 40 and 23 CEs, respectively, indicating their pronounced capability in chitin modification. The synergistic interaction between GHs and CEs likely further enhances chitin degradation efficiency in these strains.
Regarding other CAZy categories, the observed GTs (12–67 per strain) suggest potential involvement in extracellular polysaccharide synthesis, possibly linked to stress tolerance in extreme environments. The abundance of CBMs (10–72 per strain) reflects robust substrate-binding capacity, which may facilitate adaptation to complex carbohydrate environments, with strain B387 (72 CBMs) exhibiting exceptional proficiency in substrate recognition and binding.
Interestingly, almost all Bacillus strains had relatively few auxiliary activities (AAs), with B778 (6) and B378 (5) containing the most. AAs usually include enzymes that participate in redox reactions during biodegradation processes, such as ligninolytic and cellulolytic enzymes. This relative scarcity may reflect a simplification of oxidoreductase-associated metabolic pathways in these Bacillus species.
Taken together, this in-depth study of Bacillus CAZymes has revealed their diverse metabolic mechanisms, especially their potential in carbohydrate degradation and modification. The distribution and abundance patterns of various CAZy families underscore their environmental adaptability and provide an important reference for future applications of these microorganisms in agriculture, pharmaceuticals, waste degradation, and biotechnological processes like chitin valorization.
3.3. Analysis and Comparison of Biosynthetic Gene Cluster
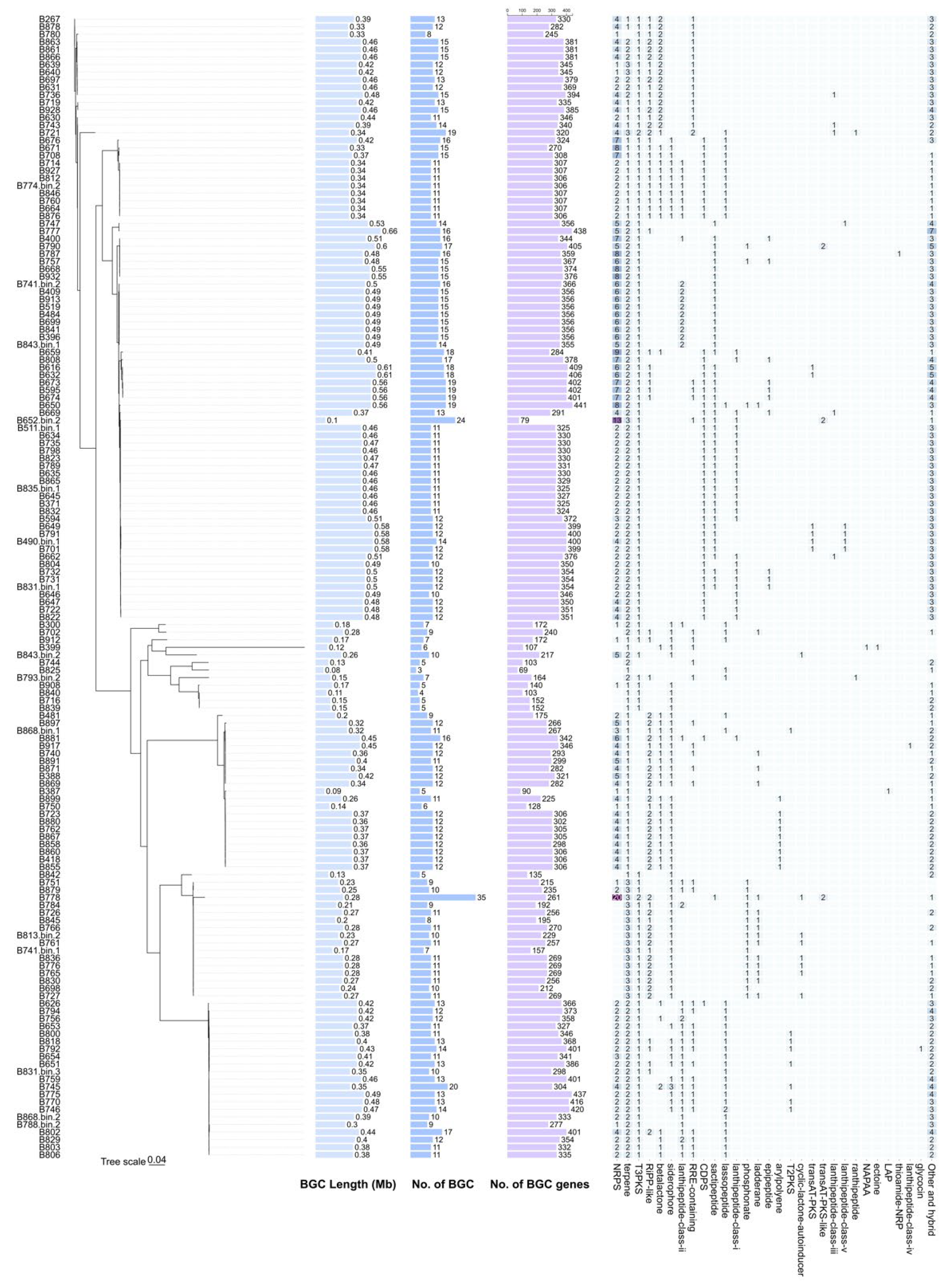
As shown in
Figure 4 and
Supplementary Figure S2, the genome size of 61 of the 151
Bacillus strains exceeded 5 Mb, but not all of these strains with large genomes showed a corresponding increase in gene clusters. Interestingly, several strains with relatively compact genomes harbor extensive gene clusters. Notably,
Bacillus strains B721 and B778 possess 8.6 Mb and 9 Mb, respectively, which is the longest among all strains. However, even these strains with expanded genomes do not possess the largest gene clusters, suggesting that factors beyond genome size influence gene cluster development. The secondary metabolic gene clusters in most
Bacillus strains range from 0.1 Mb~0.6 Mb. Among them, B777 strain had the largest gene cluster, reaching 0.66 Mb. Additionally, 23 strains feature gene clusters exceeding 0.5 Mb, which indicated that these strains had higher secondary metabolic potential. These findings highlight the importance of these strains as primary materials for microbial secondary metabolite studies and bacterial natural product mining. Statistical analysis reveals a weak correlation between total cluster length and genome size (R
2 = 0.01,
p < 0.0001), indicating that cluster development in
Bacillus strains operates independently from genome size constraints. The secondary metabolic potential of
Bacillus strains may be more affected by other factors, which provides useful clues for further study of microbial secondary metabolic mechanisms.
Using antiSMASH to mine secondary metabolic gene clusters in 151 Bacillus genomes, we found 1853 secondary metabolic gene clusters. Among them, 344 secondary metabolic gene clusters were classified as “other-hybrid” type, mainly because the types of secondary metabolites synthesized by these gene clusters remained uncharacterized. Our analysis revealed that non-ribosomal peptide synthetases (NRPS), terpenes, and T3PKS (type III PKS) are almost universally distributed in the genomes of all Bacillus groups. Specifically, NRPS is usually involved in the synthesis of peptide products, terpene is involved in the synthesis of terpenoid compounds, and T3PKS is mainly involved in the synthesis of type III polyketide products. The wide distribution of these secondary metabolic gene clusters indicates that they play an important role in the biochemical synthesis of Bacillus. In addition to these common secondary metabolic gene clusters, 26 secondary metabolic gene clusters were found to exist almost exclusively in some specific Bacillus genomes. This may reflect differences in secondary metabolite synthesis potential and adaptability between different Bacillus strains. In addition, by analyzing the correlation between the number of genes in the genome and the number of genes in the secondary metabolic gene cluster, it was found that the correlation between them was poor (R2 = 0.08, p = 0.0006). This means that the number of genes does not directly predict the number of secondary metabolic gene clusters, suggesting that secondary metabolism may be regulated by other multiple factors in Bacillus. This provides an indirect clue for the further study of the secondary metabolic mechanism of Bacillus. Overall, the correlation between total cluster length and genome size was poor (R2 = 0.01, p < 0.0001), indicating that the cluster size of Bacillus strains is not directly limited by their genome size.
3.4. Screening of CDA Strains
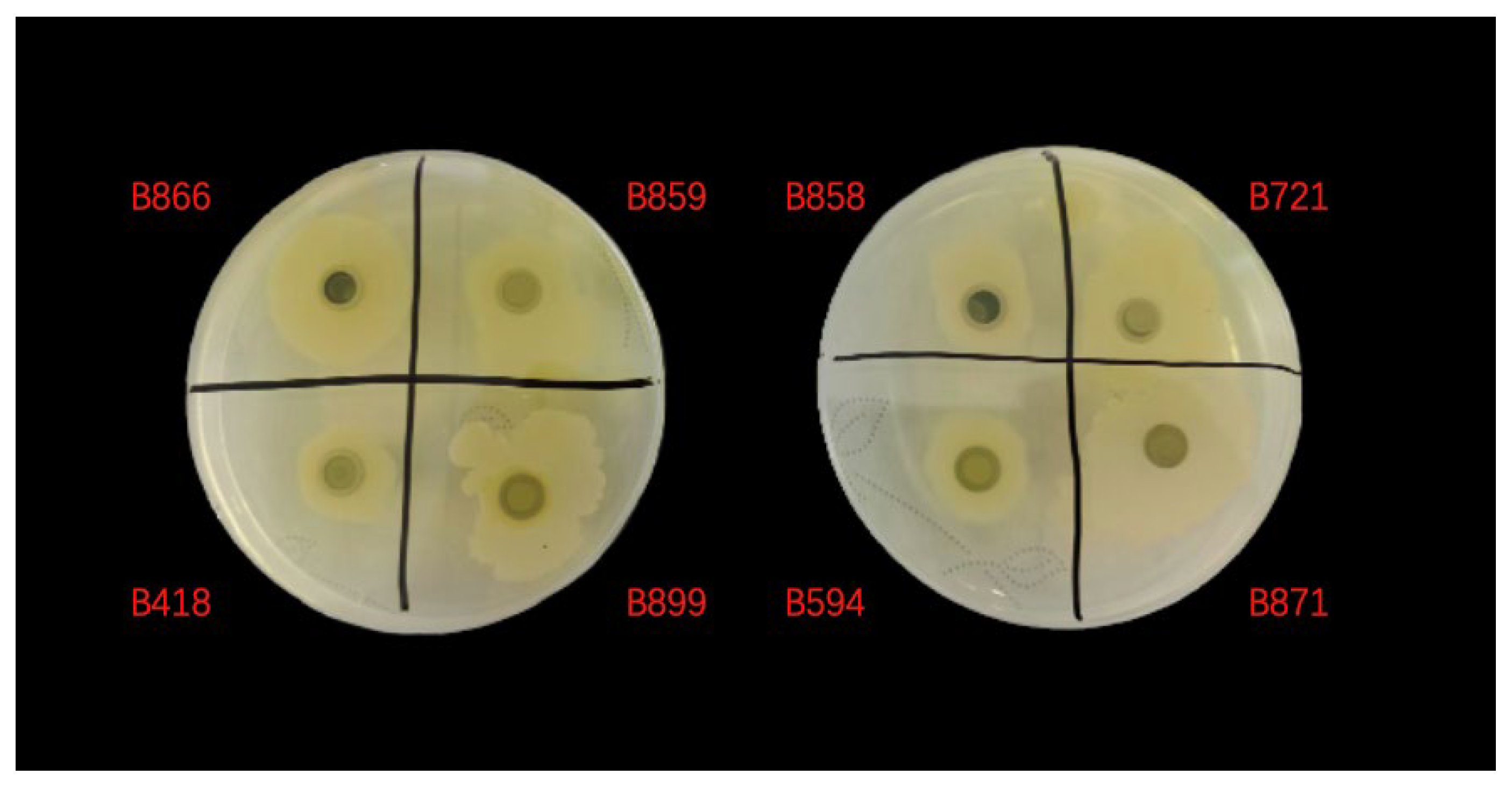
Based on the comprehensive genome analysis, we identified 120 strains containing CDA-encoding genes from the 151
Bacillus strains. We then designed a screening method based on the chemical reaction where CDA catalyzes the deacetylation of p-nitroacetanilide to produce p-nitroaniline, resulting in a distinctive color change from colorless to yellow. Using this approach, we conducted an initial screening of all 120 strains, which revealed that 95 exhibited measurable enzymatic activity following cultivation. Further secondary screening identified five strains with exceptional CDA activity: B721, B858, B866, B871, and B899 (
Figure 5). For confirmatory analysis, we inoculated these five high-performing strains into fresh selection medium and incubated them at 30 °C with agitation at 180 rpm for 48 h. Following incubation, we measured sample absorbance using an ultraviolet spectrophotometer and calculated the corresponding CDA activity. The quantitative results are presented in
Table 1.
Through this approach, we identified Bacillus pumilus B866 as the optimal strain. Enzyme activity assays revealed predominantly intracellular CDA activity across tested strains, suggesting CDA functions primarily as an intracellular enzyme. Consequently, all subsequent enzyme activity calculations focused on intracellular measurements. After identifying this promising CDA-producing strain, we conducted medium optimization experiments to enhance enzyme production and determine optimal growth conditions.
3.5. Results of Medium Composition on Chitin Deacetylase Production
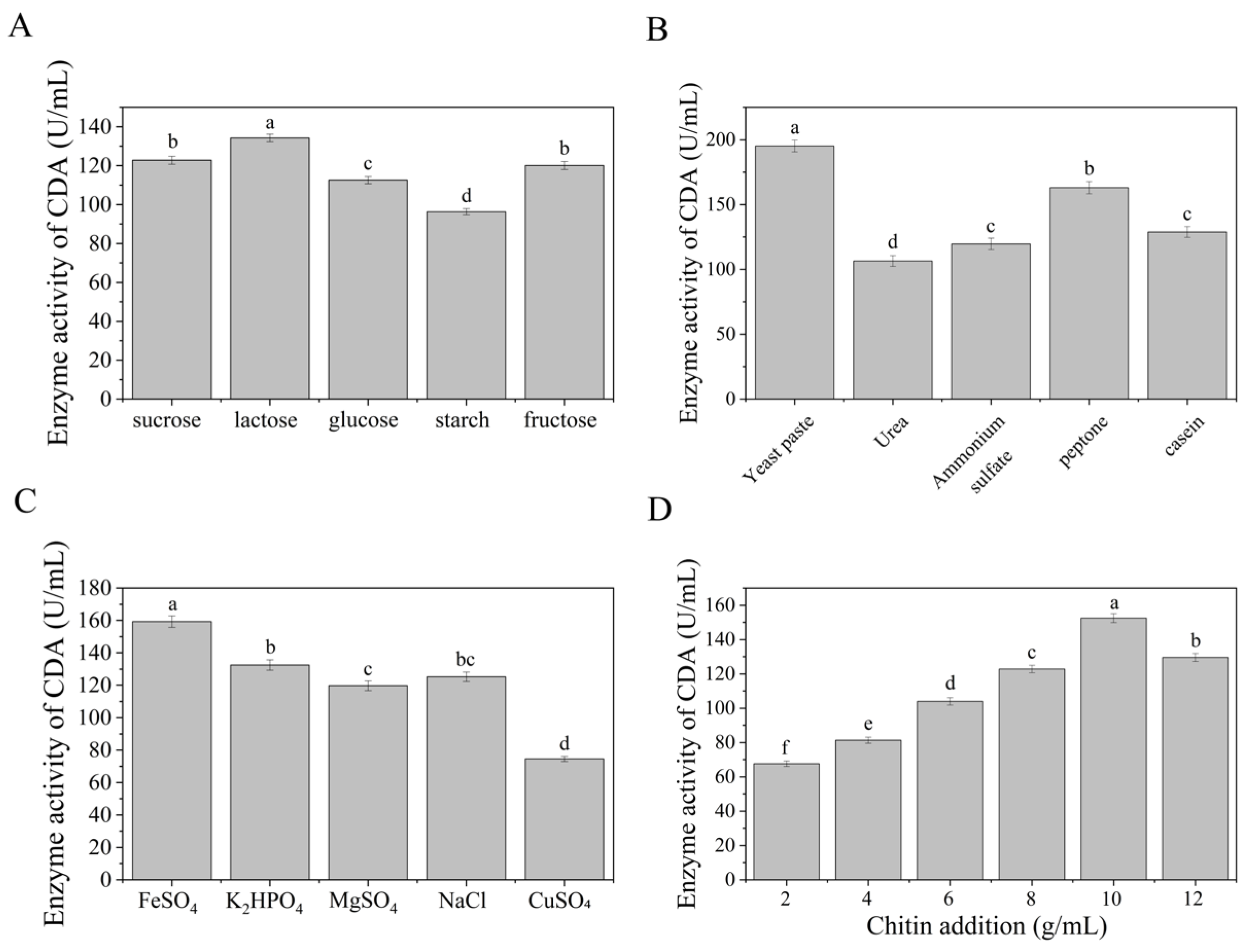
To enhance CDA production in
Bacillus pumilus B866, we systematically evaluated the effects of various carbon sources, nitrogen sources, inorganic salts, and chitin concentrations on enzyme activity. Carbon source optimization showed that lactose could significantly promote the synthesis of CDA. By detecting the enzyme activity in the supernatant of fermentation broth, it was found that the enzyme activity reached 134.27 U/mL when lactose was used as the carbon source (
Figure 6A). Compared with other carbon sources (such as glucose, sucrose, etc.), lactose was selected as the optimal carbon source. Nitrogen source screening experiments showed that yeast extract had the most prominent effect on the synthesis of CDA. In the medium containing yeast extract, the enzyme activity reached 187.38 U/mL (
Figure 6B), which was higher than that of other nitrogen sources (such as peptone, ammonium sulfate, etc.). About 20–40%, indicating that yeast extract can effectively support cell metabolism and enzyme expression by providing abundant organic nitrogen components. It was found that ferrous sulfate played a key role in maintaining the enzyme activity. After the addition of ferrous sulfate, the activity of CDA increased to 158.32 U/mL (
Figure 6C), suggesting that it may participate in the conformational stabilization of the enzyme by regulating osmotic pressure or as a cofactor. In further investigation of chitin concentration on enzyme production, the results showed that when the addition of chitin was 10 g/L, the enzyme activity reached the peak (147.91 U/mL,
Figure 6D). The substrate limitation effect was significant at low concentration (<10 g/L), while high concentration (>10 g/L) may lead to substrate inhibition or insufficient dissolved oxygen, resulting in the decrease in enzyme activity. Based on the above results, the optimal medium formula was determined as follows: lactose (carbon source), yeast extract (nitrogen source), ferrous sulfate (inorganic salt), and 10 g/L chitin. The verification experiment demonstrated that the CDA enzyme activity of the optimized medium was 191.32 U/mL (
Figure 7), which was 2.39 times higher than that of the original medium and was significantly higher than the highest value (187.38 U/mL) when each single variable was optimized, indicating that the multi-component collaborative optimization had a superimposed gain effect on the enzyme activity.
3.6. Results of Heterologous Expression of BpCDA and Its Enzymatic Properties
Comparative analysis with chitin deacetylase homologs from NCBI reveals significant structural divergence in the novel enzyme
BpCDA. While all sequences share the conserved catalytic core,
BpCDA exhibits critical substitutions at two functionally essential positions within this motif, suggesting altered substrate interaction or metal coordination. Notably,
BpCDA’s shorter open reading frame and acidic isoelectric point contrasts with the typically basic pIs of aligned CDAs. Further differentiation includes the absence of a signal peptide and reduced N-glycosylation sites. These collective variations—particularly the catalytic site substitutions and altered charge profile—position
BpCDA as a functionally divergent member of the family (
Figure 8).
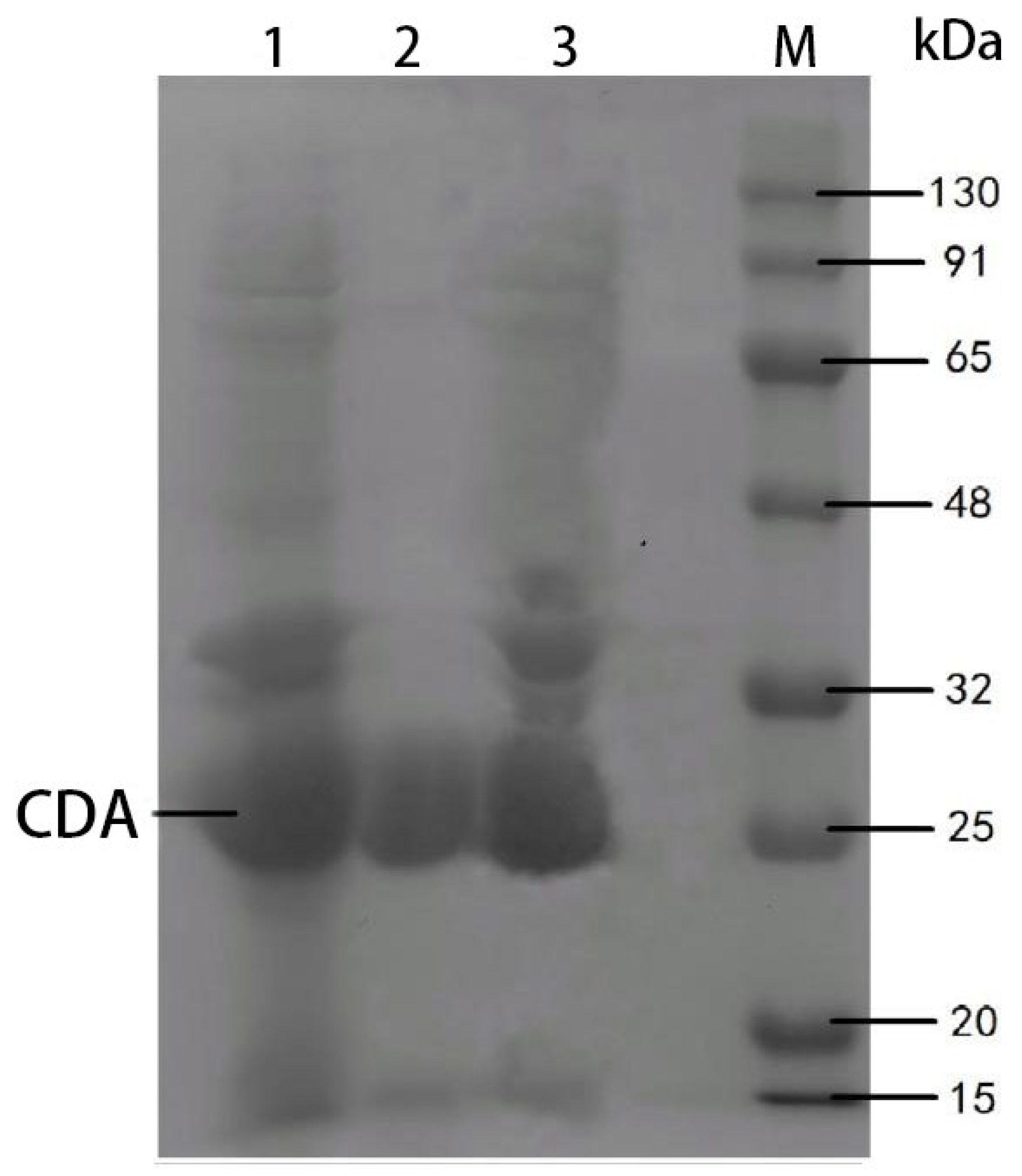
Based on the gene resources of the optimized strain, the recombinant pET-28a-CDA vector was constructed by
BamH I/
Hind III double digestion, and the correct insertion of the gene was confirmed by PCR and double digestion verification. SDS-PAGE showed that the molecular weight of the recombinant protein was about 25 kDa (
Figure 9), and the enzyme activity reached 123.27 U/mL; the CDA gene was successfully expressed in
E. coli, providing a standardized enzyme preparation for the study of enzymatic properties.
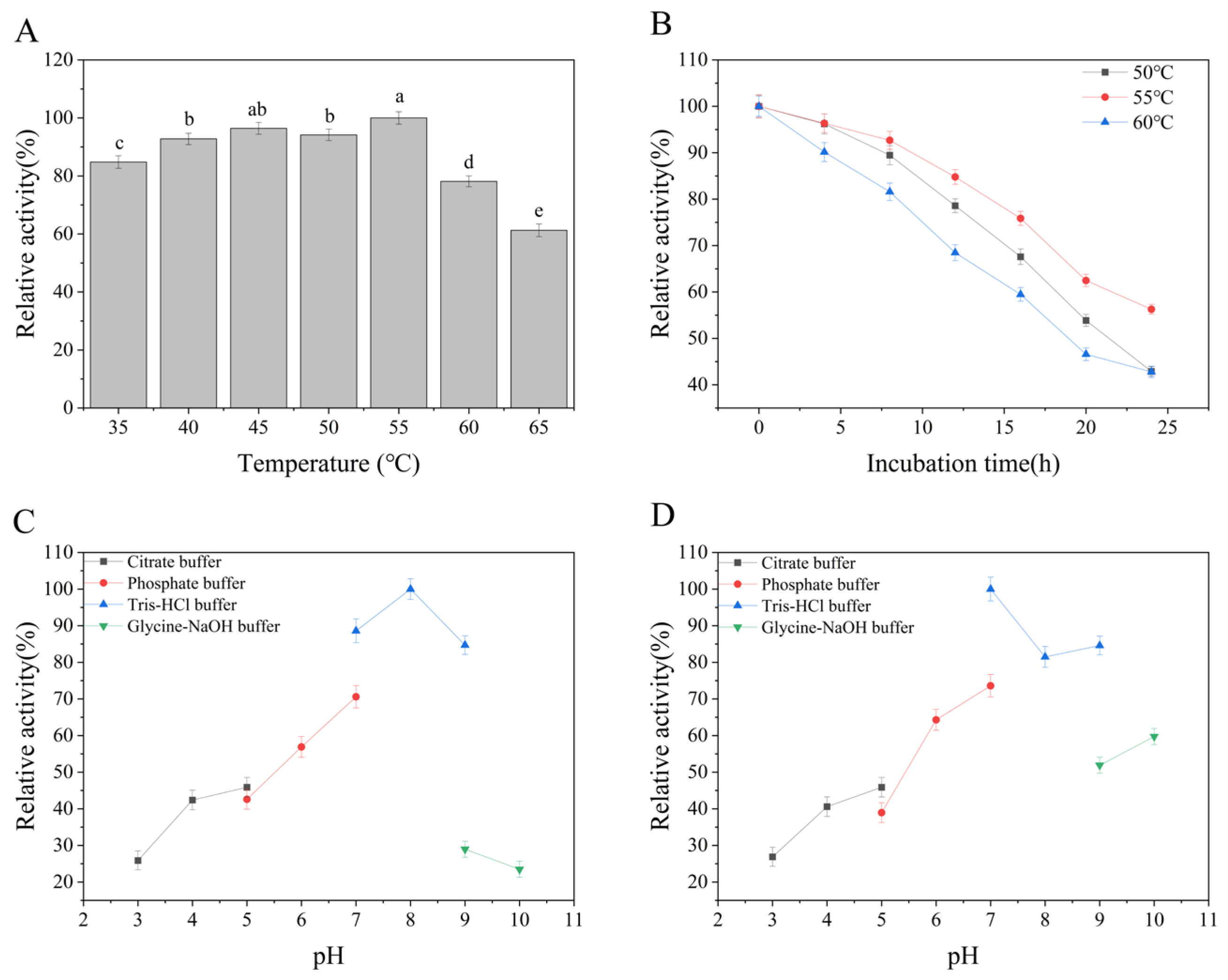
The enzymatic characterization revealed distinct thermal and pH adaptation patterns. Temperature profiling demonstrated maximal activity at 55 °C (100% relative activity,
Figure 10A), with activity retention exceeding 94% between 45 and 55 °C. However, a sharp decline occurred beyond 60 °C, reaching only 61.2% residual activity at 65 °C, suggesting thermophilic adaptation rather than mesophilic properties as initially hypothesized. Thermal stability analysis (
Figure 10B) showed differential patterns: At 55 °C, the enzyme retained moderate thermal stability, preserving >42.9% residual activity over a 24-h incubation period despite progressive deactivation. However, 60 °C treatment showed progressive inactivation, with activity dropping below 69% within 12 h and continuing to decline linearly, ultimately retaining only 42.8% activity after 24 h.
pH optimization studies (
Figure 10C) revealed maximal enzymatic activity at pH 7.0 in Tris-HCl buffer, with >84.7% activity retained across the pH 7.0–9.0 range (encompassing Tris-HCl buffer systems). Severe activity reduction (<29%) occurred under extreme pH conditions (pH 3 in citrate buffer or pH > 9 in glycine-NaOH). pH stability profiling (
Figure 10D) highlighted buffer-dependent variations, with Tris-HCl demonstrating optimal stability at pH 7.0–9.0 (>81.4% residual activity after 10 h incubation; peak stability: 100% at pH 7.0). Notably, the pH stability range (7.0–9.0) exhibited minimal overlap with the enzyme’s activity-optimal pH (8.0), prompting critical evaluation of its functional resilience under industrial pH fluctuations.
3.7. Effects of Metal Ions on BpCDA Enzymatic Activity
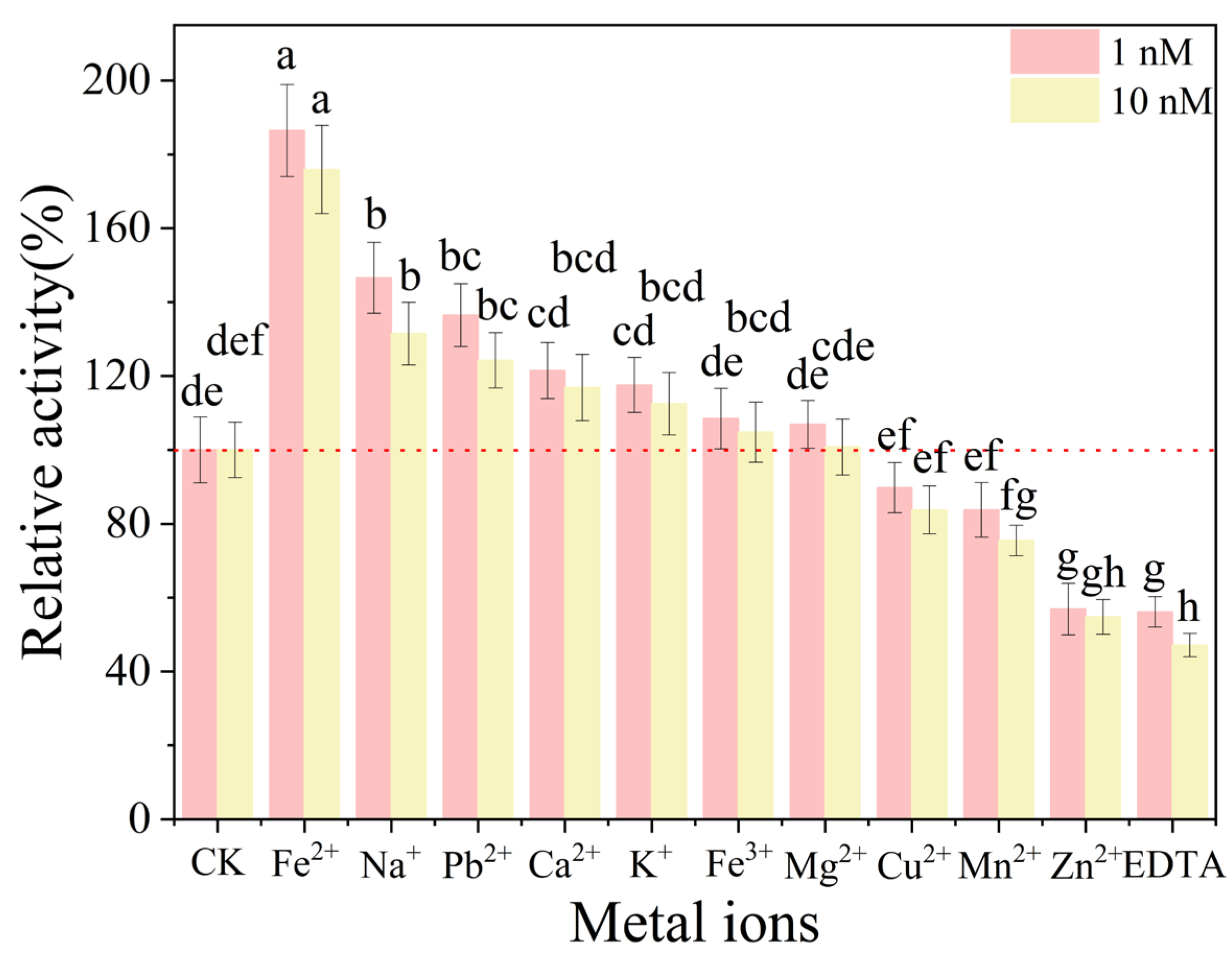
To assess
BpCDA’s metal ion stability, enzyme activity was measured with Fe
3+, Cu
2+, Ca
2+, Mn
2+, Fe
2+, Mg
2+, Zn
2+, Pb
2+, K
+, Na
+, and EDTA(1 nM and 10 nM) (
Figure 11). Fe
2+ demonstrated exceptional activation potential, elevating relative activity to 186.4% at 1 nM compared to control. Divalent cations exhibited differential responses: Na
+ showed substantial enhancement (146.6% at 1 nM), while Zn
2+ caused significant suppression (54.7% at 10 nM), contrasting with Ca
2+’s neutral response (103.2–107.5%).
Monovalent ions revealed divergent effects—Pb2+ enhanced activity to 136.4% at 1 nM, whereas K+ maintained neutral status (112.4–117.6%). Notably, EDTA exhibited strong inhibition (47.1% residual activity), while Fe3+ (108.5% at 1 nM; 104.8% at 10 nM) and Mg2+ (100.7–106.9%) showed concentration-independent neutral responses. Cu2+ displayed moderate suppression (83.7–89.7%). These results establish critical benchmarks for metalloenzyme optimization in bioprocessing applications.
4. Discussion
As a unique extreme ecosystem, a high saline and alkaline environment has been proven to be rich in microbial resources in recent years [
21]. Studies both domestically and internationally have shown that there are a large number of saline–alkali tolerant microorganisms in salt lakes [
22], salt pans [
23], and saline–alkali deserts [
24], and the functional enzymes produced by them often have special ion tolerance. Daoud et al. [
25] isolated
Bacillus US193 from Chott Eldjerid, Kerkennah Islands, which grew best at pH 9.7 and 50 °C. Zhang et al. [
26] purified and partially characterized the thermostable xylanase from the 90,462 of halophilic thermophilic
Bacillus YIM. The purified xylanase was purified at 80 °C and pH 6.0 has the best activity. A notable achievement in Sieiro’s study [
27] was the successful heterologous expression of the HschiA1 gene, derived from the archaeon
Halobacterium salinarum CECT 395 in
E. coli host cells. This demonstration of cross-domain genetic transfer between archaeal and bacterial systems represents a significant technical advancement in recombinant protein expression. The Ebinur Lake Basin in Xinjiang is a typical saline–alkali desert area with a soil salinity gradient of 0.8–12.3%, the annual rainfall is 89.9–169.7 mm [
28], and the extreme environment shapes a unique microbial community. Our group collected soil samples from a typical saline–alkali desert area (90°66′–91°80′ E, 41°99′–40°94′ N) in Xinjiang, characterized by extreme aridity (<20 mm annual precipitation), high soil salinity (1.5–15.8%), and significant temperature fluctuations. From this harsh environment, we isolated 151
Bacillus strains. Genome sequencing revealed these strains maintain a stable core genome while exhibiting considerable diversity in accessory and unique genes, reflecting environmental adaptation. Notably, 120 strains (79.5%) contained chitin deacetylase genes with significant sequence variations compared to non-extreme environment strains. This genetic diversity from Northwest China’s saline–alkaline soils provides a valuable resource of novel genes and enzymes with properties adapted to extreme conditions, supporting that harsh environments serve as reservoirs for biotechnologically relevant genetic resources.
In recent years, advances in sequencing technology and the development of powerful gene-mining tools have enabled researchers to reveal the
Bacillus multifunctional lifestyle [
29]. In this study, genome annotation and functional gene analysis of 151
Bacillus strains were carried out, which provided in-depth information for understanding the diversity, adaptability and synthesis of bioactive metabolites of
Bacillus [
21] When performing genome annotation and mining in
Bacillus, we may face some challenges and shortcomings, which may affect the overall understanding of genes and functions. First, genome annotation may contain errors or missing information. Some genes may be incorrectly annotated as pseudogenes, while other real genes may be ignored or missed. This can lead to misunderstandings about gene function and biological processes [
30]. Still other genes may be annotated as having an unknown function, making it difficult to understand their exact role in biological processes [
31]. This increases the need for experimental validation and functional studies. Part of the secondary metabolic gene cluster is classified as “other” type, indicating that the nature and function of these products are not yet clear. This may require further chemical and biological experiments to identify and understand these products [
32].
Bacillus is a very diverse genus, and there may be significant genomic differences between strains [
33]. The results of annotation and mining may be different between different strains, which requires more comparative analysis between strains to obtain comprehensive information. Our mining of secondary metabolic gene clusters using tools such as antiSMASH may be limited by tool limitations. Some gene clusters may be omitted due to special structures or regulatory mechanisms. The use of a combination of tools may help improve the sensitivity and accuracy of excavation [
34,
35]. In our study, we found that the correlation between the number of genes and the number of secondary metabolic gene clusters was low, which may indicate that other factors play an important role in the regulation of secondary metabolism [
35]. This requires more in-depth research to understand these regulatory factors. To solve these problems, we need to adopt comprehensive methods, including comparative analysis of different strains, experimental verification, and the development of more accurate bioinformatics tools. This will contribute to a more comprehensive understanding of the annotation of the
Bacillus genome and the mining of secondary metabolic gene clusters [
36].
Chitin deacetylase (CDA) catalyzes the conversion of chitin into chitosan by deacetylating N-acetyl-D-glucosamine residues, representing a sustainable approach for high-value utilization of crustacean waste. According to the chemical phenomenon that CDA can deacetylate p-nitro-N-acetanilide to produce p-nitroaniline (changing from colorless to yellow), we designed a colorimetric screening method for CDA-producing microorganisms and identified
B. pumilus B866 as the optimal candidate. While initial culture optimization experiments were conducted, we recognized that direct CDA production from protoplasts presents significant challenges including difficult purification and unstable yield [
37]. Therefore, to obtain CDA efficiently and in large quantities, we pursued heterologous expression in
E. coli. The CDA-encoding gene from
B. pumilus B866 was PCR-amplified using designed primers, cloned into an expression vector [
38,
39], and transformed into
E. coli for protein production. The recombinant enzyme (
BpCDA) was purified and confirmed by SDS-PAGE [
40], exhibiting a specific activity of 123.27 U/mL with optimal catalytic conditions at 55 °C and pH 8.0. Notably, ferrous ions significantly enhanced enzyme performance, suggesting a novel catalytic activation mechanism distinct from previously characterized
Bacillus CDAs.
Compared to previously reported enzymes, our findings illustrate several distinct advantages. For instance, the recently characterized CDA from
Bacillus aryabhattai TCI-16 showed a lower specific activity of 120.35 U/mL [
10], while
BpCDA from our study provides comparatively higher catalytic activity, highlighting its potential industrial relevance. Furthermore, our enzyme exhibited favorable catalytic characteristics, including a relatively superior temperature adaptability around 55 °C that is crucial for industrial processes. Meanwhile,
B. aryabhattai TCI-16 CDA typically required conditions at approximately 40 °C [
11]. Interestingly, unlike the Mg
2+-dependency reported for
B. aryabhattai TCI-16 CDA,
BpCDA identified in our current study uniquely demonstrated Fe
2+ dependency for activity enhancement. This novel metal-ion-dependent activation mechanism expands our understanding of
Bacillus CDA catalytic diversities and potential evolutionary adaptations.
Overall, this research advances our knowledge of CDA enzymatic activities and expands resources for the bioconversion of chitin-rich wastes into valuable biopolymers. The comprehensive investigation into enzyme properties—including metal ion effects, temperature, and pH stability—positions BpCDA as a promising candidate for the efficient, sustainable industrial production of chitosan.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}