Abstract
In the enhancement of Novel Sorghum bicolor × S. propinquum Hybrid utilization, optimal planting densities and silage methods remain elusive. This study assesses the effects of planting densities, cellulase (CE), Lactobacillus buchneri (LAB), and their combination (LC) on fermentation quality and bacterial diversity of the hybrid silage. The experiment was carried out in a completely random block design with four additives and five planting densities (M1, M2, M3, M4, M5) as follows (4 additives × 5 planting densities): a control group without additives (CK), a group treated with Lactobacillus buchneri (LAB), a group with cellulase (CE), and a group treated with a combination of LAB and CE (LC), maintaining triplicates per treatment. In this study, the additive treatment improved the fermentation quality of silage compared with the control. In the M2-LC group, the contents of crude protein (CP; 7.88%), ether extract (EE; 1.91%), and ash (7.76%) were the highest, while the pH (3.30) was the lowest. The water-soluble carbohydrate (WSC; 11.28%) content was the highest in the M3-CE group, the lactic acid (LA; 6.79%) content was the highest in the M4-CE group, and the acetic acid (AA; 7.71%) content was the highest in the M2-LAB group. Meanwhile, the neutral washing fiber (NDF; 53.17%) content was the lowest in the M5-CE group, the acid detergent fiber (ADF; 41.01%) content was the lowest in the M2-CE group, and the propionic acid (PA; 0.26%) content was the lowest in the M1-LAB group. Adding LC notably reduced bacterial diversity, boosted Lentilactobacillus, and curbed Proteobacteria. LAB and LC markedly improved amino acid metabolism over CE and CK. Conversely, beta-lactam resistance, flagellar assembly, and ascorbate/aldarate metabolism pathways were suppressed. In the future, we will explore a variety of additives and adjust the cutting height to improve its comprehensive quality, create an innovative path for silage production, promote the efficient use of agricultural resources, and provide high-quality feed for animal husbandry.
1. Introduction
The Novel Sorghum bicolor × S. propinquum Hybrid is a collection of Sorghum bicolor (L.) Moench and Sorghum propinquum (Kunth) Hitchc, a new breed of forage sorghum from distant cross-breeding. Compared to other forage sorghum varieties, it exhibits characteristics of perennial plants, higher productivity, and stronger resistance. However, the method of storing this forage has not been studied. Therefore, it is important to investigate the methods of processing and storing it. Studies have shown that sorghum has high water-soluble carbohydrates. However, it also has high fiber content, which reduces the utilization of nutrients by animals [1]. Silage is considered one of the most effective animal feeds, and additives play an important role in improving the quality and palatability of silage [2]. Through careful selection and the application of silage additives, it can not only effectively meet the specific challenges of sorghum as silage raw materials but also significantly improve the overall quality and nutritional value of silage and provide more high-quality and efficient feed solutions for animal production. In order to make efficient use of land resources, it is particularly important to select suitable planting density.
Lactobacillus buchneri, a widely utilized strain, is notable for its production of a substantial concentration of acetic acid. This activity not only reduces the pH of the feed but also enhances the storage longevity of the silage [3]. During the fermentation of Medicago sativa L. silage, the incorporation of Lactobacillus buchneri has been observed to decrease the abundance of Enterobacter, eliminate pathogenic organisms, restrain the degradation of crude protein, and augment the nutritional value of the silage [4]. Similarly, the synergistic addition of Pediococcus lactis and Lactobacillus buchneri to ryegrass (Lolium perenne L.) silage has significantly elevated the presence of Lactobacillus, increased the levels of acetic acid and propylene glycol, and diminished the incidence of yeasts and molds, thereby mitigating silage spoilage [5]. Furthermore, the application of cellulase enhances the availability of water-soluble carbohydrates (WSC) by breaking down cellulose, thereby offering substrates for fermentation [6]. This enzymatic intervention has also been shown to substantially boost the population of lactic acid bacteria while reducing the presence of aerobic bacteria within the silage [7] previously reported that cellulase addition to Pennisetum purpureum Schum. Silage not only increased the diversity indices, such as Simpson and Shannon, and the abundance of Firmicutes but also led to a decrease in Proteobacteria populations. Additional research has established that cellulase supplementation can enhance protein content and overall silage quality [8]. Despite these findings suggesting the broad applicability of additives across various forage types, the specific impact of cellulase and Lactobacillus buchneri on the fermentation quality of fresh Sorghum bicolor × S. propinquum remains to be elucidated.
During planting, density is paramount as a cultivation measure, standing as a pivotal factor that significantly impacts both yield and quality. In prior research, it was discovered that the corn’s contribution to crude protein (CP) in silage quality diminished as the corn planting density decreased in a mixed planting system with Lablab beans [9]. Upon thorough examination, the results indicated that the planting density did not exhibit a significant influence on the pH level of silage fermentation [10]. Therefore, the ideal planting density for enhancing the quality of silage produced from the Novel Sorghum bicolor × S. propinquum Hybrids remains elusive. The current study aims to determine this optimal density, ultimately benefiting both farmers and livestock producers through improvements in silage quality.
Therefore, this study aims to investigate the effects of planting density, Lactobacillus buchneri, and cellulase on the fermentation process of Sorghum bicolor × S. propinquum by examining fermentation parameters, chemical composition, and bacterial communities. The insights gained will offer valuable technical references for the industrial application and optimization of Sorghum bicolor × S. propinquum silage production.
2. Materials and Methods
2.1. Material Planting and Silage Preparation
The experimental material was planted in Changzhai Town, Changshun County, Guizhou Province, at five planting densities (M1: 10,000 plants/hm2; M2: 11,800 plants /hm2; M3: 14,300 plants/hm2; M4: 18,200 plants/hm2; M5: 25,000 plant/hm2). The samples were then harvested at maturity and cut into 2–3 cm pieces using a cutter. The sample pieces obtained from each planting density were then treated as follows: (1) no additive control (CK); (2) lactic acid bacteria (LAB: Lactobacillus buchneri (ATCC49374); dosage: 1 × 109 cfu/kg FM); (3) cellulase (1, 4-β-D-glucan glucoside hydrolase) (CE: dosage: 6 × 103 U/kg FM); (4) LAB + CE (LC; dosage: LAB: 1 × 109 cfu/kg FM; CE: 6 × 103 U/kg FM). The additives were mixed evenly with the harvested sample, divided into three parts of 1 kg each, packed in polyethylene bags (30 cm × 40 cm), and vacuum-sealed. A total of 60 bags (5 planting density × 4 treatments × 3 repetitions) were prepared and stored at ambient temperature (20–30 °C). After 45 days, the chemical composition, fermentation characteristics, and bacterial community were analyzed.
2.2. Fermentation Parameters and Chemical Composition Analysis
Take 10 g silage sample and mix it with 90 mL sterile water, shake well, and incubate overnight to obtain organic acids. Then, the pH was measured using an electrode pH meter (thunder magnetic). The organic acid extract was absorbed, filtered through a 0.22 µm nylon filter membrane, and injected into the liquid chromatograph for further analysis. The test solution obtained was used to assess the acetic acid (AA), lactic acid (LA), butyric acid (BA), and propionic acid (PA) contents.
Approximately 0.5 g of sample was weighed and mixed with the solution, digested at high temperature (420 °C), cooled, and transferred to a distillation flask. Further, the sodium hydroxide was added to the mixture, and the released ammonia was distilled and collected in boric acid. There were standard sulfuric acid titration, color change (blue-green to grey-purple) endpoint, and determination of crude protein content (CP). About 1.0 g of sample was ground, heated in a water bath for 25 min, and cooled to a constant volume. A total of 1.0 mL of the extract was mixed with water, anthrone reagent, and concentrated sulfuric acid and boiled in a water bath for 10 min. After cooling, the mixture’s absorbance at 620 nm was measured using a spectrophotometer, and the soluble sugar content was determined based on the standard curve (WSC). Weigh 5 g of the sample, soak it in petroleum ether, distill out the dissolved fat, and then dry and weigh the residue to obtain the ether extract content (EE). Further, to determine the acid detergent fiber (ADF) and neutral washing fibers (NDF) contents, 0.5 g of the sample was taken in a paper bag and mixed with an appropriate amount of neutral/acidic detergent. This mixture was placed in a fiber analyzer and digested at constant temperature for 1 h. After digestion, the remaining detergent was washed away, and the fibers were soaked in acetone to remove impurities. The sample was further dried in an oven to constant weight. About 5.0 g of the dried sample was weighed in a crucible and carbonized at a high temperature; further, the sample was placed in a Muffle furnace at 550 °C, burned to constant weight, and weighed to determine the ash content.
2.3. DNA Extraction and PCR Sequencing
The silage samples were analyzed through a biotechnology platform (Paisenol). Bacterial DNA was extracted with DP302-02 DNA extraction kit (Tiangen, Beijing, China). The extracted DNA was evaluated by 0.8% agarose gel electrophoresis and quantified using an ultraviolet spectrophotometer. Further, the V5–V7 region of 16S rRNA was amplified from the total DNA using forward (5′-CCTACGGGNGGCWGCAG-3′) and reverse (5′-GACTACHVGGGTATCTAATCC-3′) primers, and the amplicon was purified with the BECKMAN AMPure XP (Beckman Coulter, Inc., Brea, CA, USA) beads. Our raw sequence data are available under the NCBI project PRJNA1119948 with the accession number SRP511592. The purified amplicon was further quantified using the Quant-iT PicoGreen dsDNA Assay Kit on a microplate reader (BioTek, FL× 800; Winooski, VT, USA). Then, the samples were mixed according to the data required for each sample. PCR products were analyzed on 2% agarose gel and recovered using the AMPure XT bead recovery kit (Beckman Coulter, Inc., Brea, CA). Finally, double-ended sequencing was carried out using the NovaSeq 6000 SP Reagent Kit (500 cycles) on an Illumina NovaSeq platform (Illumina, San Diego, CA, USA).
2.4. Bacterial Community Sequencing Analysis
The reads were primed, quality filtered, denoised, spliced, and mosaiced using the DADA2 method. The obtained information on the ASV abundance was flattened with the “qiime feature-table rarefy” function of QIIME2 (version 2019.4), setting the flattening depth to 95% of the minimum sample sequence size. Then, the “qiime diversity alpha-rarefaction” command was used for the unflattened ASV to calculate the alpha diversity (α-diversity) index. Simultaneously, using the flattened ASV table, the “qiime diversity core-metrics-phylogenetic” or “qiime diversity core-metrics” command was called based on whether the tree file is phylogenetic, the Bray–Curtis distance matrix was calculated, and PCoA was performed. The obtained Bray–Curtis distance matrix file was used to perform the “permanova” inter-group difference analysis with Python’s scikit-bio package, setting the permutations to 999. Here, utilizing R software and QIIME2, a comprehensive Beta diversity analysis was conducted, employing Bray–Curtis and Jaccard distance metrics, to delve into the variations in microbial community structure among diverse samples. The findings of this analysis were then graphically represented through Principal Coordinate Analysis (PCoA) and Non-metric Multidimensional Scaling (NMDS), providing a visual perspective on the complexities and patterns within the microbial communities. Further, the clustering of each sample and each taxon species was assessed using the R language and pheatmap package. Finally, LEfSe analysis was performed to determine the differences using the Python LEfSe package and the R ggtree package (LDA > 3.5). PICRUSt2 was used for the Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KO) classification based on the 16S rRNA-labeled gene sequences.
2.5. Statistical Analysis
Data were analyzed using the SPSS program (version 26.0, SPSS Inc., Chicago, IL, USA). One-way analysis of variance (ANOVA) was performed to assess the differences in the fermentation products, chemical composition, microbial population, and KEGG pathways. The model is as follows:
represents the observed value, ( = LAB, CE, LC or M1, M2, M3, M4, M5) represents the average value, and is the error. Duncan’s multipass method was adopted to determine the statistical significance, and differences at p < 0.05 were considered significant. Spearman correlation coefficient was calculated to assess the correlation. The closer the Spearman correlation coefficient is to 1 or −1, the stronger the correlation between the two variables. All graphs were produced using the derived Novon platform and Origin.
3. Results
3.1. Dry Matter Content and Chemical Composition
An initial analysis focused on the nutrient composition of freshly harvested Sorghum bicolor × S. propinquum, cultivated at varied densities (Table 1). Notably, the dry matter (DM) content from plants at the M4 density (35.24% of fresh mass [FM]) was significantly higher than that observed at other densities (p < 0.05).

Table 1.
Chemical composition of Sorghum bicolor × S. propinquum.
Further analysis revealed that higher planting densities negatively impacted the contents of crude protein (CP), ether extract (EE), and ash. This outcome supports the hypothesis that intensified competition at higher densities not only limits nutrient availability but also hampers the synthesis of proteins and fats. Additionally, the observed reduction in ash content can be attributed to a prioritization of resources towards growth and development over the accumulation of inorganic substances. Despite these variations, the neutral detergent fiber (NDF) and acid detergent fiber (ADF) contents remained relatively stable across the studied densities (60.02–61.00% and 44.33–46.80%, respectively). These findings suggest that M3 and M4 balance between nutrient competition and the production of desirable chemical compositions for cultivating Sorghum bicolor × S. propinquum.
3.2. Fermentation Characteristics and Chemical Composition of the Silage
The chemical composition of Sorghum bicolor × S. propinquum silage was assessed after a fermentation period of 45 days (Figure 1). This analysis indicated significant variances in dry matter (DM), water-soluble carbohydrates (WSC), crude protein (CP), ether extract (EE), ash, neutral detergent fiber (NDF), and acid detergent fiber (ADF) contents, contingent upon the use of additives. To be specific, a reduction in DM content (ranging from 28.95% to 32.28% of fresh mass [FM]) was observed post fermentation, and the retention of dry matter in CK was the lowest at the same density (28.21–30.66%). Notably, silage treated with cellulase (CE) and the combination of cellulase and Lactobacillus buchneri (LC) demonstrated significantly elevated levels of WSC and CP in comparison to the control group (CK) (p < 0.05). The content of EE in the LC group was notably higher in comparison to the CK group (p < 0.05). Among the tested groups, the WSC of M3 stood at the highest level of 11.28%, while the CP of M2 reached 7.88%, and the EE of M2 peaked at 1.91%. Concurrently, the introduction of Lactobacillus buchneri (LAB) enhances the production of organic acids, facilitating the growth of beneficial micro-organisms. A significant reduction in the contents of NDF and ADF was observed in the CE and LC groups compared to CK (p < 0.05), and the contents were the lowest in M2 (41.01%) and M5 (53.17%). Conversely, an increase in ash content post fermentation was noted.
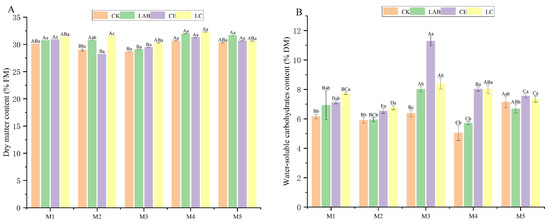
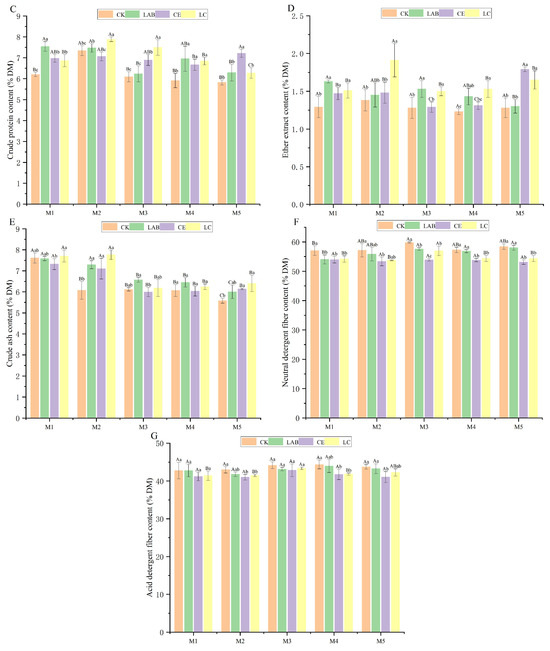
Figure 1.
Effects of additives and planting density on the chemical composition of the silage. Different capital letters indicate significant differences among different planting densities under the same treatment (p < 0.05); different lowercase letters indicate significant differences among different treatments under the same planting density (p < 0.05); the same letter indicates not significant (p > 0.05). CK, control; LAB, Lactobacillus buchneri; CE, cellulase; LC, Lactobacillus buchneri + cellulase. M1, M2, M3, M4, and M5 represent five planting densities adopted in this study. (A) dry matter content; (B) water-soluble carbohydrates content; (C) crude protein content; (D) ether extract content; (E) crude ash content; (F) neutral detergent fiber content; (G) acid detergent fiber content.
Further examination was conducted on the fermentation characteristics of Sorghum bicolor × S. propinquum silage, as documented in Figure 2. This segment of the study revealed that under the planting densities of M2 and M3, the pH levels of the LAB (3.56, 3.68) and LC (3.3, 3.69) groups were significantly lower than those of the CK group (3.83, 3.94), with statistical significance at p < 0.05. Notably, the concentration of lactic acid (LA) in the CK group was significantly lower than that in the treatment groups (p < 0.05). The LA levels in the M2 and M3 groups were relatively elevated, with the LC group exhibiting significantly higher LA concentrations in the M2 (6.06%), M3 (5.94%), and M4 (6.02%) groups compared to the M1 (4.82%) and M5 (4.85%) groups. Furthermore, the acetic acid (AA) levels in the LAB group peaked at M2, reaching a notable concentration of 7.71% DM. The butyric acid (BA) content exhibited variability across the different treatment groups. The concentration of propionic acid (PA) in the LAB group M1 (0.26%) and the LC group M2 (0.37%) was significantly lower than that of the CK group (0.38%, 0.54%), further demonstrating a decrement compared to other planting densities.
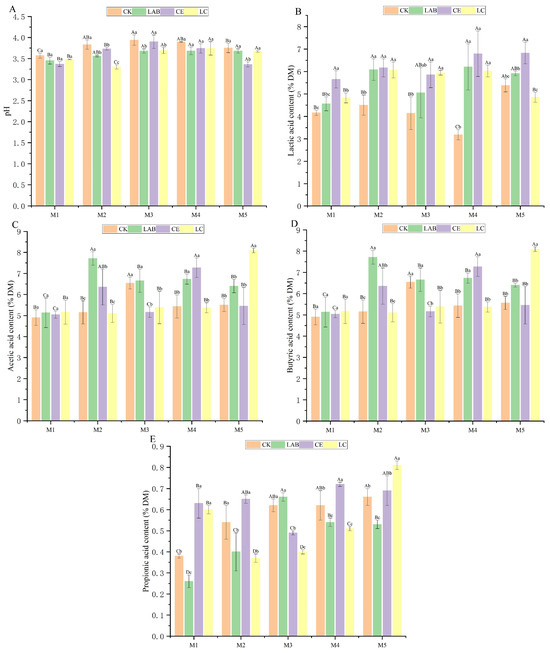
Figure 2.
Effects of additives and planting density on fermentation characteristics of the silage. Different capital letters indicate significant differences among different planting densities under the same treatment (p < 0.05); different lowercase letters indicate significant differences among different treatments under the same planting density (p < 0.05); the same letter indicates not significant (p > 0.05). CK, control; LAB, Lactobacillus buchneri; CE, cellulase; LC, Lactobacillus buchneri + cellulase. M1, M2, M3, M4, and M5 represent five planting densities adopted in this study. (A) pH value; (B) lactic acid content; (C) acetic acid content; (D) butyric acid content; (E) propionic acid content.
3.3. Bacterial Diversity and Community Composition of the Silage
The alpha diversity of dynamic Sorghum bicolor × S. propinquumsilage silage bacterial communities is shown in Figure 3. The coverage index, ranging from 0.997 to 0.999, confirmed the reliability of the microbial community data, underscoring the accuracy of the bacterial diversity measurements. Planting density exerted a significant influence on the indices Chao1, Shannon, Simpson, and Observed_species, revealing that M2 or M3 treatments resulted in significantly lower values compared to other density variations (p < 0.05). Notably, the relative abundance of these indices in the LAB and LC treatment groups was significantly diminished compared to the CK group (p < 0.05). Specifically, under the M3 planting density, the LC treatment group exhibited the lowest relative abundance for the Chao1 index (41.81) and Observed_species index (41.13). Meanwhile, under the M1 planting density, the LC treatment group showed the lowest relative abundance for the Shannon index (1.13) and Simpson index (0.48).
Figure 3.
Effects of additives and planting densities on bacterial alpha diversity of Sorghum bicolor × S. propinquum silage. Different capital letters indicate significant differences among different planting densities under the same treatment (p < 0.05); different lowercase letters indicate significant differences among different treatments under the same planting density (p < 0.05); the same letter indicates not significant (p > 0.05). CK, control; LAB, Lactobacillus buchneri; CE, cellulase; LC, Lactobacillus buchneri + cellulase. M1, M2, M3, M4, and M5 represent five planting densities adopted in this study. (A) Chao1 index; (B) Goods_coverage index; (C) Observed_species index; (D) Shannon index; (E) Simpson index.
The changes in the microbial community of the silage were further verified by beta diversity analysis using Principal Coordinates Analysis (PCoA) and Non-metric Multidimensional Scaling (NMDS) (Figure 4). Further analytical techniques, such as Principal Coordinates Analysis (PCoA) and Non-metric Multidimensional Scaling (NMDS), were employed to assess the bacterial communities, utilizing the Bray–Curtis distance matrix based on AVS/OTU. These analyses demonstrated a distinct separation between the bacterial communities associated with CK and CE treatments and those treated with LAB and LC (Figure 4A,B).
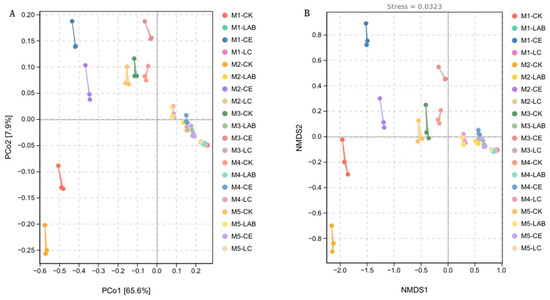
Figure 4.
Analysis of bacterial diversity in Sorghum bicolor × S. propinquum silage under different fermentation additives and planting densities. (A) Differences in bacterial community structure; (B) differences in bacterial community structure. CK, control; LAB, Lactobacillus buchneri; CE, cellulase; LC, Lactobacillus buchneri + cellulase. M1, M2, M3, M4, and M5 represent the five planting densities adopted in this study.
Subsequent analysis focused on the taxonomic composition of the bacterial communities present in silage subjected to different treatments, specifically examining the phylum and genus levels. The predominant phyla identified were Firmicutes and Proteobacteria. Notably, Firmicutes accounted for over 96% of the bacterial composition (Figure 5A). Furthermore, the analysis revealed that the CK group possessed a greater abundance of Proteobacteria compared to other treatment groups, with M1-CK exhibiting the highest percentage, standing at 5.07%. At the genus level, the abundance of Lentilactobacillus in the LAB and LC treatments surpassed all others under varying planting densities, whereas the control group (CK) displayed a comparatively lower abundance (Figure 5B). Interestingly, the abundance of Lactiplantibacillus was the highest in CK, and its abundance decreased with an increase in planting density. Levilactobacillus showed the highest abundance in the cellulase (CE)-treated group, and it was the lowest under M4 (2.35%) and M5 (1.85%) densities. Conversely, Paenibacillus was most abundant in CK and absent in the Lactobacillus buchneri (LAB) treatment. Lactococcus and Leuconostoc, identified only in CK and CE, are noted for their poor acid resistance.
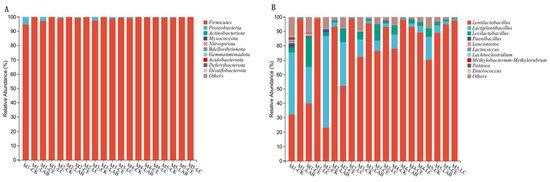
Figure 5.
Bacterial communities in the silage samples. (A) Bacterial community at the phylum level; (B) Bacterial communities at the genus level. CK, control; LAB, Lactobacillus buchneri; CE, cellulase; LC, Lactobacillus buchneri + cellulase. M1, M2, M3, M4, and M5 represent the five planting densities adopted in this study.
To further elucidate the microbial community differences attributable to the use of additives across varying planting densities, LEfSe analysis (Linear discriminant analysis Effect Size) was conducted, with a focus on genus-level bacteria exhibiting an LDA score greater than 3.5 (Figure 6). This analysis confirmed that Lactiplantibacillus was predominantly enriched in CK across all densities. Conversely, Lentilactobacillus showed significant enrichment in treatments involving LC and LAB, while Levilactobacillus was notably enriched in the CE treatment group. Paenibacillus, Pantoea, Lachnoclostrdium, Methylobacteria-methylorubrum, and Enterococcus were found in CK and CE but not M3 and M5. M2 and M4 had only partial traces, and M1 had both. These results underscore the distinct microbial community structures induced by different silage additives.
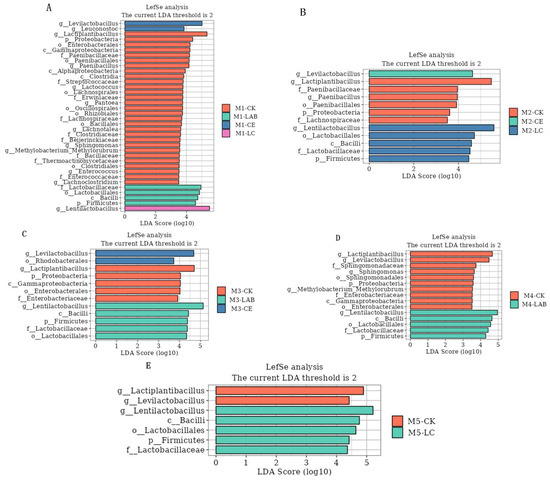
Figure 6.
The microbial community differences attributable to the use of additives across varying planting densities (genus-level). (A) Silage at M1 density; (B) silage at M2 density; (C) silage at M3 density; (D) silage at M4 density; (E) silage at M5 density. CK, control; LAB, Lactobacillus buchneri; CE, cellulase; LC, Lactobacillus buchneri+ cellulase. M1, M2, M3, M4, and M5 represent the five planting densities adopted in this study.
3.4. Correlation between Fermentation Parameters and Bacterial Communities
The exploration into the relationship between microbial communities at the genus level and fermentation characteristics yielded insightful correlations, as depicted in a heatmap based on Spearman correlation coefficients (Figure 7). Notably, Lentilactobacillus exhibited a positive correlation with lactic acid (LA) levels, and Lactiplantibacillus and Levilactobacillus are negatively correlated with LA. Additionally, both Lentilactobacillus and Levilactobacillus showed negative correlations with pH, whereas Lactiplantibacillus was positively correlated with pH. These correlations align with the observation of a higher pH in the control group (CK) compared to those in the LAB-, LC-, and cellulase (CE)-treated groups (Figure 2). Levilactobacillus was positively correlated with WSC but negatively with CP and NDF. On the other hand, Lentilactobacillus was negatively correlated with WSC but positively with CP. Moreover, Paenibacillus, Pantoea, Lachnoclostridium, Methylobacteria-methylotrophus, and Enterococcus were positively correlated with pH.
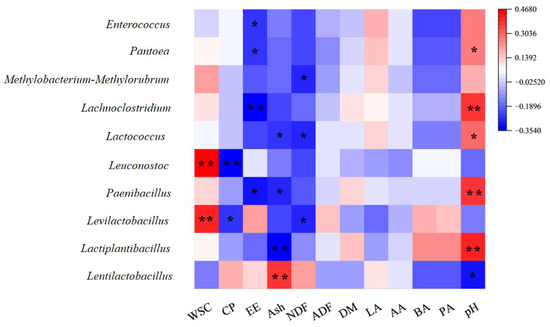
Figure 7.
Correlation between the bacterial genera and fermentation parameters of different silage groups. Heat map based on Spearman correlation coefficients. Blue indicates a negative correlation, and red indicates a positive correlation. WSC, water-soluble carbohydrate; CP, crude protein; EE, crude fat; Ash, coarse ash; NDF, neutral detergent fiber; ADF, acid detergent fiber. (*) indicates p < 0.05; (**) indicates p < 0.01.
3.5. Metabolic Pathways Based on Genes
Investigating the flora structure’s impact on silage quality at a higher and whole-genome level provides valuable insights into the metabolic pathways that influence silage fermentation. This study employed PICRUSt2 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) technology utilizing the Kyoto Encyclopedia of Genes and Genomes (KEGG) database to analyze the effects of different additive treatments on the metabolic pathways within Sorghum bicolor × S. propinquum silage.
The analysis identified distinct modifications in metabolic pathways at the third level, including the biosynthesis of ansamycins, carotenoid biosynthesis, and ascorbate and aldarate metabolism in the control group (CK) (Figure 8). Moreover, pathways such as metabolism, beta-lactam resistance, flagellar assembly, and bacterial chemotaxis exhibited a significantly higher abundance in CK compared to the Lactobacillus buchneri (LAB) and the combination of LAB and cellulase (LC) groups. Contrastingly, amino acid metabolism was found to be less pronounced in CK and cellulase (CE) groups than in LAB and LC treatments, as indicated by further ANOVA analysis (Figure 9). However, no significant difference was observed between CE and CK groups in this regard.
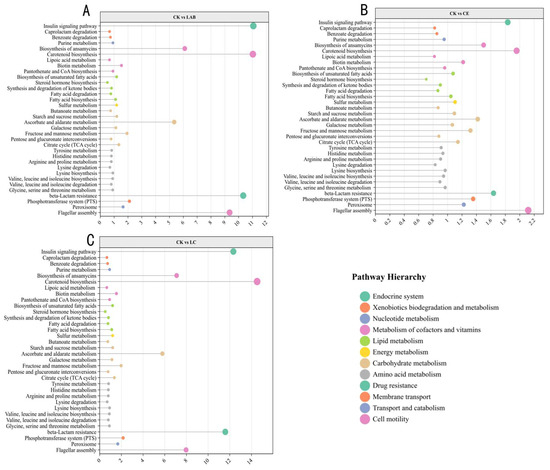
Figure 8.
KEGG pathway enrichment analysis of differential metabolites in Sorghum bicolor × S. propinquum silage. The differential abundance scores of the KEGG pathway between (A) CK and LAB, (B) CK and CE, and (C) CK and LC silage groups. The size of the pathway corresponds to different metabolite counts. A differential abundance score greater than 1 indicates a multiple less than CK, and a difference abundance score less than 1 indicates a multiple greater than CK.
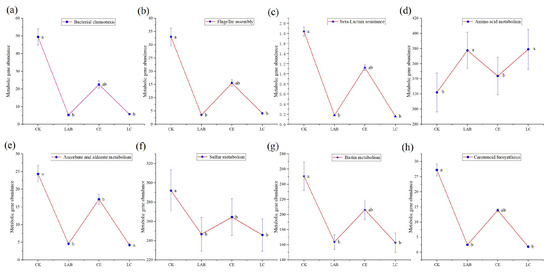
Figure 9.
Abundance of specific metabolic pathways in the silage groups. The data shown are the mean values, the error bars represent the standard deviations, and the different letters at the top indicate significant differences (test, p < 0.05). CK, control; LAB, Lactobacillus brucei; CE, cellulase; LC, Lactobacillus brucei + cellulase.
Correlations between specific bacterial genera and metabolic pathways were further explored (Figure 10), revealing negative correlations between Lentilactobacillus and several metabolic pathways, including Insulin signaling, biosynthesis of ansamycins, and carotenoid biosynthesis. Conversely, Levilactobacillus and Lactiplantibacillus showed opposite trends. Moreover, amino acid metabolism was negatively correlated with Levilactobacillus and Lactiplantibacillus but positively correlated with Lentilactobacillus.
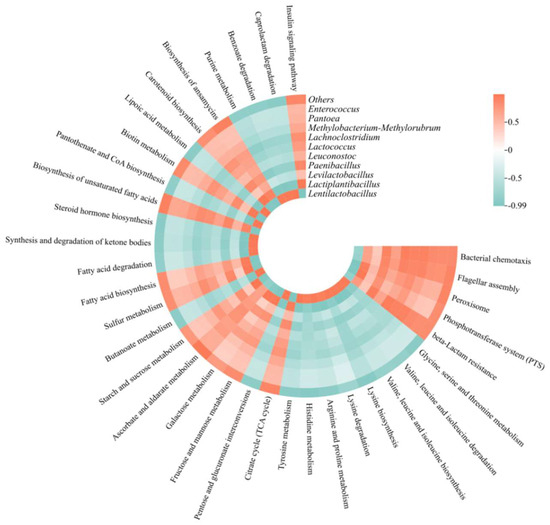
Figure 10.
Correlation between bacterial genera and metabolic pathways in different additive silage groups. Heat map based on Pearson correlation analysis. Green indicates a negative correlation, and red indicates a positive correlation.
4. Discussion
High-quality fermented silage is usually well preserved in terms of nutrient composition. After 45 days of silage, the dry matter content was reduced, and the dry matter loss was the largest in group CK. This phenomenon may be due to the fact that when there is insufficient soluble sugar in the environment, lactic acid bacteria will adjust their metabolic pathway in order to maintain their life activities and energy supply and change from homozygous fermentation to heterozygous fermentation [11]. The contents of CP and WSC in the CE and LC groups were higher than those in the CK group. The mechanism underlying this effect involves the decomposition of cellulose into sugars by cellulase, thereby augmenting the available substrate for fermentation [12]. Concurrently, the introduction of Lactobacillus buchneri (LAB) enhances the production of organic acids, facilitating the growth of beneficial micro-organisms. This process effectively breaks down cellulose and protein within the silage material, thereby optimizing the utilization of crude protein [13]. Additionally, cellulase contributes to the degradation of fiber and non-structural proteins, releasing plant cell structural proteins [14]. The CE group had the lowest NDF and ADF content, attributed to the conversion of cellulase-degradable fiber into soluble sugars [5]. Ash increases after silage, likely resulting from the transformation of organic matter present in the grass into inorganic matter throughout the silage process.
The pH and organic acids are also used to evaluate the quality of fermentation. Under each planting density, the pH value of the LAB, CE, and LC groups was lower than that of the CK group, and the organic acid concentration of LA was higher than that of CK. The application of Lactobacillus buchneri to the silage initiated the production of organic acids during fermentation, while cellulase activity led to the breakdown of cellulose into sugars, which were subsequently converted into organic acids, thereby contributing to a reduction in pH [15]. At the same time, this can be attributed to the ability of Lactobacillus species to outcompete other micro-organisms for nutrients during the fermentation process, thus efficiently utilizing the available carbon sources to enhance the production of lactic acid [16]. Supporting this, studies by Ren et al. and Hu et al. have indicated that the addition of cellulase can increase the relative abundance of lactic acid bacteria, further boosting the LA content in silage [17]. The concentration of AA in the treatment group with Lactobacillus buchneri was higher than that in the CK group. This is likely due to the fact that LAB, being of the Lactobacillus heterofermentative type, is capable of producing not only lactic acid but also a higher quantity of AA. These results indicated that in terms of nutrient composition and fermentation parameters, additives improved silage quality, and the combined use of Lactobacillus buchneri and cellulase was the best.
Furthermore, to understand the differences in bacterial community between treatments, we performed 16S rRNA high-throughput sequencing. The coverage index ranged from 0.997 to 0.999, confirming the reliability of the microbial community data and its suitability for bacterial community analysis. Typically, α-diversity explains bacterial communities’ species richness, evenness, and diversity. In this study, the Chao1, Shannon, Simpson, and Observed_species indices of the LAB and LC groups were lower than those of CK. This reduction in bacterial diversity following successful fermentation aligns with findings by Guan et al. [18], who observed a marked decrease in bacterial diversity post-fermentation. The incorporation of Lactobacillus boucheri, known to elevate levels of lactic acid (LA) and acetic acid (AA) while concurrently lowering pH, has been documented to inhibit the proliferation of competing bacterial species, thereby diminishing bacterial diversity [19]. This effect is indicative of the selective pressure imposed by the fermentation environment, which favors the growth of specific microbial populations while suppressing others. Additionally, the ß-diversity reflects the differences in bacterial communities among the treatments. The PCoA and NMDS maps showed significant separation between the CE and CK treatments and between the LAB and LC treatments, indicating differences in the bacterial communities. This observation also indicates that adding Lactobacillus buchneri reconfigured the bacterial community of silage.
Silage quality is also closely related to the composition of the bacterial community. The present study found mainly Firmicutes and Proteobacteria in the silage, and the abundance of Firmicutes was much higher than Proteobacteria. This dominance is likely attributable to the rich content of complex carbohydrates, such as cellulose, present in Sorghum bicolor × S. propinquum. These carbohydrates, which are challenging to degrade, are efficiently broken down by Firmicutes due to their potent cellulolytic capabilities. Consequently, Firmicutes can extract available nutrients from the complex carbohydrates found in forage silage materials, thereby securing a nutritional advantage over other bacterial populations and achieving a high relative abundance during the silage process [20]. Proteobacteria mainly existed in CK. However, minimal to no Proteobacteria were detected in the other additive treatments. This reduction can be explained by the rapid onset of anaerobic and acidic conditions induced by LAB and CE treatments, conditions that are unfavorable for the growth and reproduction of Proteobacteria during fermentation [21]. Additionally, Proteobacteria are known to degrade organic matter under aerobic conditions, which could lead to a higher loss of dry matter in the CK group [22]. These findings underscore the significant impact of fermentation additives on the taxonomic structure of bacterial communities within silage, particularly highlighting the role of specific bacterial phyla in the degradation of complex carbohydrates and the preservation of dry matter. The rapid establishment of anaerobic and acidic environments by cellulase and Lactobacillus buchneri treatments not only inhibits the growth of less desirable bacterial populations, such as Proteobacteria, but also enhances the nutritional quality and stability of the silage.
The abundance of Lentilactobacillus in the LAB and LC groups was higher than that in the CK group. This aligns well with the findings previously reported by Ogiy et al. [20]. Previous studies have demonstrated that the addition of specific micro-organisms to silage can effectively control the growth and proliferation of competing microbial entities within the silage [23]. In particular, the introduction of Lactobacillus buchneri has been shown to afford Lentilactobacillus a competitive edge. Furthermore, the breakdown of cellulose into more accessible carbon sources significantly benefits Lentilactobacillus. Lactiplantibacillus was abundant in CK and decreased with an increase in density. This pattern may be attributed to the natural prevalence of Lactiplantibacillus on the plant surface and the decrease due to the drop in ventilation and other conditions with an increase in planting density [24]. Levilactobacillus was mostly found in the CE group and was the lowest in M4 and M5. Levilactobacillus is known to perform glyco-based fermentation, and cellulase addition produces more soluble sugars. On the other hand, the low content of WSC in M4 and M5 resulted in a drop in their abundance. Additionally, adding Lactobacillus buchneri in silage decreased the abundance of harmful bacteria (Paenibacillus, Lactococcus, and Leuconostoc). There are typically outcompeted by more acid-tolerant bacteria, such as Lentilactobacillus and Lactiplantibacillus, under low-pH conditions. In brief, under varying planting density scenarios, harmful bacteria were detected only in CK, and the addition of additives significantly reduced their abundance, thereby improving the overall quality of the silage [25].
The LEfSe analysis was employed to delve deeper into the disparities in bacterial community composition between the additive treatment group and the control group during the silage process. In our study, Lactiplantibacillus was mainly enriched in CK, Lentilactobacillus in LAB and LC, and Levilactobacillus in CE. These results confirmed that using additives changed the bacterial community structure in silage, and Levilactobacillus was likely to be glycosylated. Paenibacillus, Pantoea, Lachnoclostrdium, Methylobacteria-methylorubrum, and Enterococcus were found only in CK and CE, suggesting that the use of LAB could improve the fermentation quality of silage. The investigation into the correlation between bacterial communities and silage quality fosters a comprehensive comprehension of the pivotal bacteria that are essential for optimizing silage fermentation quality. Additionally, we found that Lentilactobacillus and Levilactobacillus were positively correlated with LA and negatively correlated with pH, while Lactiplantibacillus showed an opposite trend. This finding is contrary to the observation made by Bai et al. [26]. In the groups treated with Lactobacillus buchneri (LAB) and the combination of LAB and cellulase (LC), the supplementation of Lactobacillus buchneri fortified the dominance of Lentilactobacillus over Lactiplantibacillus, thereby enhancing the production of LA by Lentilactobacillus. Meanwhile, this phenomenon can be attributed to the acidogenic properties of these bacteria, where Lentilactobacillus and Levilactobacillus contribute to acidification (lowering pH) through their metabolic activities, contrasting with Lactiplantibacillus which may have less impact on acid production.
Further analyses revealed that specific bacteria are associated with distinct fermentation pathways: Lactiplantibacillus was negatively correlated with crude protein (CP), suggesting a lesser role in nitrogen-based fermentation, whereas Lentilactobacillus exhibited a negative correlation with water-soluble carbohydrates (WSC), indicating a potential preference for glycosyl-based fermentation pathways. The increased abundance of Lentilactobacillus in the CE group, compared to CK, supports this hypothesis. Further analyses revealed that specific bacteria are associated with distinct fermentation pathways: Lentilactobacillus was positively correlated with crude protein (CP), indicating that nitrogen-based fermentation may have been occurring, whereas Levilactobacillus exhibited a positive correlation with water-soluble carbohydrates (WSC), indicating a potential preference for glycosyl-based fermentation pathways. The increased abundance of Levilactobacillus in the CE group, compared to CK, supports this hypothesis. The aerobic nature of bacteria such as Paenibacillus, Pantoea, Lachnoclostridium, Methylobacteria-methylotrophus, and Enterococcus [27] and their positive correlation with pH further support the notion that the presence and activity of aerobic bacteria contribute to higher pH levels in silage. The lower relative abundance of these bacteria in additive-treated groups (Figure 6B and Figure 7) emphasizes the effectiveness of the additives in creating an anaerobic environment conducive to lowering the pH and enhancing silage quality through the selective promotion of beneficial bacterial populations.
Predicting the functional alterations within bacterial communities is instrumental in assessing their influence on the shifting quality of silage. As found in this study, the significant enrichment of the Insulin signaling pathway in CK was comparable to LAB and LC groups, while amino acid metabolism was notably enriched in the LAB and LC groups. These alterations suggest a higher decomposition and metabolism rate of amino acids in the CK group, potentially reducing the protein content, as detailed in Figure 2 [28]. Typically, bacterial response to antibiotic pressure manifests through an increase in beta-lactam resistance. The addition of LAB appeared to mitigate this pressure, resulting in a markedly lower abundance of lactam in LAB and LC groups compared to CK. Additionally, the energy-intensive process of flagellar assembly, rather than being allocated towards fermentation and acid production [29], likely contributed to a higher pH in CK than in LAB-treated silage. Excessive carotenoid biosynthesis and activity of ascorbate and aldarate metabolism could negatively impact fermentation and feed quality, possibly due to substrate competition and the accumulation of metabolites such as uronic acid, adversely affecting nutritional balance and animal health [30]. The presence of ansamycins might exhibit specificity for Lactiplantibacillus in CK, accounting for its higher abundance compared to LAB-treated silage. The specific bacterial genera and metabolic pathways suggest that Lentilactobacillus may play a crucial role in inhibiting certain metabolic pathways. In the meantime, Lentilactobacillus may stimulate amino acid metabolism, leading to the production of more beneficial peptides, thus improving the quality of the silage.
5. Conclusions
This study revealed that the combined use of Lactobacillus buchneri and cellulase optimized the microbial community and improved the quality of Sorghum bicolor × S. propinquum silage. Specifically, the combination promoted the proliferation of beneficial bacteria, such as Lentilactobacillus and Levilactobacillus, and reduced pH, effectively curbing harmful micro-organisms, such as Paenibacillus. Compared with the addition of Lactobacillus buchneri or cellulose alone, combined treatment increased the content of water-soluble carbohydrates and the content of crude protein, lactic acid, and acetic acid, significantly improving feeding value. The combination of M2 and LC demonstrated the best effect among the various treatments. This research provides a scientific basis for planning strategies for efficiently storing and utilizing Sorghum bicolor × S. propinquum silage. However, the actual mechanisms underlying the fermentation process need to be explored. Future studies should analyze the metabolic process and reveal the mechanism of Sorghum bicolor × S. propinquum silage preparation.
Author Contributions
X.Y., S.Z. and Q.D. designed the experiments and revised the manuscript. Q.D., B.L. and Y.T. performed the experiments. Q.D. and X.Y. wrote the manuscript. Q.D. and P.W. performed the data analysis. X.Y., S.Z., X.D., L.Z., C.G. and P.W. contributed to the grammar and language revision of the paper and contributed to this experiment. All authors have read and agreed to the published version of the manuscript.
Funding
This project was supported by the Guizhou Province Key Research and Development Program of China (Qiankehezhicheng[2021]Yiban143) and Guizhou high-level innovative talents project (Qiankehepingtairencai-GCC[2022]022-1).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Our raw sequence data are available under the NCBI project PRJNA1119948 with the accession number SRP511592.
Acknowledgments
We thank the Personal Biotechnology Co., LTD (Shanghai, China) for providing technical support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Qu, H.; Liu, X.B.; Dong, C.F.; Lu, X.Y.; Shen, Y.X. Field performance and nutritive value of sweet sorghum in eastern China. Field Crops Res. 2014, 157, 84–88. [Google Scholar] [CrossRef]
- Muck, R.E. Silage microbiology and its control through additives. Rev. Bras Zootec. 2010, 39, 183–191. [Google Scholar] [CrossRef]
- Huang, Z.; Wang, M.; Ke, W.; Guo, X. Screening of high 1, 2-propanediol production by Lactobacillus buchneri strains and their effects on fermentation characteristics and aerobic stability of whole-plant corn silage. Agriculture 2021, 11, 590. [Google Scholar] [CrossRef]
- Si, H.; Liu, H.; Li, Z.; Nan, W.; Sui, Y.; Li, G. Effect of Lactobacillus plantarum and Lactobacillus buchneri addition on fermentation, bacterial community and aerobic stability in lucerne silage. Anim. Prod. Sci. 2018, 59, 1528–1536. [Google Scholar] [CrossRef]
- Auerbach, H.; Theobald, P. Additive type affects fermentation, aerobic stability and mycotoxin formation during air exposure of early-cut rye (Secale cereale L.) Silage. Agronomy 2020, 10, 1432. [Google Scholar] [CrossRef]
- Liu, W.; Si, Q.; Sun, L.; Wang, Z.; Liu, M.; Du, S.; Ge, G.; Jia, Y. Effects of cellulase and xylanase addition on fermentation quality, aerobic stability, and bacteria composition of low water-soluble carbohydrates oat silage. Fermentation 2023, 9, 638. [Google Scholar] [CrossRef]
- Li, L.; Wu, H.K.; Xie, X.G.; Zhao, G.Q.; He, J.J.; Hu, Z.C.; Yang, X.J.; Wu, H. Effects of addition of cellulase and starch on fermentation quality of silage. Chin. J. Anim. Nutr. 2019, 33, 5025–5035. (In Chinese) [Google Scholar] [CrossRef]
- He, L.; Zhou, W.; Wang, C.; Yang, F.; Chen, X.; Zhang, Q. Effect of cellulase and Lactobacillus casei on ensiling characteristics, chemical composition, antioxidant activity, and digestibility of mulberry leaf silage. J. Dairy Sci. 2019, 102, 9919–9931. [Google Scholar] [CrossRef]
- Contreras-Govea, F.E.; Muck, R.E.; Armstrong, K.L.; Albrecht, K.A. Fermentability of corn–lablab bean mixtures from different planting densities. Anim. Feed Sci. Technol. 2009, 149, 298–306. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, B.; Nishino, N.; Wang, X.; Yu, Z. Fermentation and microbial population dynamics during the ensiling of native grass and subsequent exposure to air. Anim. Sci. J. 2016, 87, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yuan, X.; Desta, S.T.; Dong, Z.; Mugabe, W.; Shao, T. Characterization of Enterococcus faecalis JF85 and Enterococcus faecium Y83 isolated from Tibetan yak (Bos grunniens) for ensiling Pennisetum sinese. Bioresour. Technol. 2018, 257, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Geren, H.; Yaşar Tuncer, K. Effect of different plant densities on the yield and some silage quality characteristics of giant king grass (Pennisetum hybridum) under mediterranean climatic conditions. Turk. J. Field Crops. 2015, 20, 85–91. [Google Scholar] [CrossRef]
- Oladosu, Y.; Rafii, M.Y.; Abdullah, N.; Magaji, U.; Hussin, G.; Ramli, A.; Miah, G. Fermentation quality and additives: A case of rice straw silage. Biomed. Res. Int. 2016, 1–14. [Google Scholar] [CrossRef]
- Liu, W.; Du, S.; Sun, L.; Wang, Z.; Ge, G.; Jia, Y. Study on dynamic fermentation of oat silage assisted by exogenous fibrolytic enzymes. Plants 2024, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; He, H.; Zhang, S.; Kong, J. Effects of inoculants Lactobacillus brevis and Lactobacillus parafarraginis on the fermentation characteristics and microbial communities of corn stover silage. Sci. Rep. 2017, 7, 13614. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Ran, Q.; Li, H.; Zhang, X. Succession of microbial communities of corn silag inoculated with heterofermentative lactic acid bacteria from ensiling to aerobic exposure. Fermentation 2021, 7, 258. [Google Scholar] [CrossRef]
- Yan, Y.H.; Li, X.M.; Guan, H.; Huang, L.K.; Ma, X.; Peng, Y.; Li, Z.; Nie, G.; Zhou, J.Q.; Yang, W.Y.; et al. Microbial community and fermentation characteristic of Italian ryegrass silage prepared with corn stover and lactic acid bacteria. Bioresour. Technol. 2019, 279, 166–173. [Google Scholar] [CrossRef]
- Ren, H.; Wang, L.; Sun, Y.; Zhao, Q.; Sun, Y.; Li, J.; Zhang, B. Enhancing the co-ensiling performance of corn stover and cabbage waste via the addition of cellulase. BioResources 2021, 6342–6362. [Google Scholar] [CrossRef]
- Guan, H.; Yan, Y.; Li, X.; Li, X.; Shuai, Y.; Feng, G.; Ran, Q.; Cai, Y.; Li, Y.; Zhang, X. Microbial communities and natural fermentation of corn silages prepared with farm bunker-silo in Southwest China. Bioresour. Technol. 2018, 265, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Ogiy, S.; Chen, Y.; Pasvolsky, R.; Weinberg, Z.G.; Shemesh, M. High resolution melt analysis to confirm the establishment of Lactobacillus plantarum and Enterococcus faecium from silage inoculants during ensiling of wheat. Grassl. Sci. 2016, 62, 29–36. [Google Scholar] [CrossRef]
- McAllister, T.A.; Dunière, L.; Drouin, P.; Xu, S.; Wang, Y.; Munns, K.; Zaheer, R. Silage review: Using molecular approaches to define the microbial ecology of silage. J. Dairy Sci. 2018, 101, 4060–4074. [Google Scholar] [CrossRef]
- Wang, Y.; He, L.W.; Xing, Y.Q.; Zhou, W.; Pian, R.Q.; Yang, F.Y.; Chen, X.Y.; Zhang, Q. Bacterial diversity and fermentation quality of Moringa oleifera leaves silage prepared with lactic acid bacteria inoculants and stored at different temperatures. Bioresour. Technol. 2019, 284, 349. [Google Scholar] [CrossRef]
- Yuan, X.; Dong, Z.; Li, J.; Shao, T. Microbial community dynamics and their contributions to organic acid production during the early stage of the ensiling of Napier grass (Pennisetum purpureum). Grass Forage Sci. 2020, 75, 37–44. [Google Scholar] [CrossRef]
- Wang, X.; Haruta, S.; Wang, P.; Ishii, M.; Igarashi, Y.; Cui, Z. Diversity of a stable enrichment culture which is useful for silage inoculant and its succession in alfalfa silage. FEMS Microbiol. Ecol. 2006, 57, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Hardoim, P.R.; Overbeek, L.S.; Berg, G.; Pirttilä, A.M.; Compant, S.; Campisano, A.; Döring, M.; Sessitsch, A. The hidden world within plants: Ecological and evolutionary considerations for defining functioning of microbial endophytes. Microbiol. Mol. Biol. Rev. 2015, 79, 293–320. [Google Scholar] [CrossRef]
- Wu, B.; Hu, Z.; Wei, M.; Yong, M.; Niu, H. Effects of inoculation of Lactiplantibacillus plantarum and Lentilactobacillus buchneri on fermentation quality, aerobic stability, and microbial community dynamics of wilted Leymus chinensis silage. Front. Microbiol. 2022, 13, 928731. [Google Scholar] [CrossRef]
- Bai, B.; Qiu, R.; Wang, Z.; Liu, Y.; Bao, J.; Sun, L.; Liu, T.; Ge, G.; Jia, Y. Effects of cellulase and lactic acid bacteria on ensiling performance and bacterial community of Caragana korshinskii Silage. Microorganisms 2023, 11, 337. [Google Scholar] [CrossRef]
- Wang, F.; Shi, H.; Wang, S.; Wang, Y.; Cao, Z.; Li, S. Amino acid metabolism in dairy cows and their regulation in milk synthesis. Curr. Drug Metab. 2019, 20, 36–45. [Google Scholar] [CrossRef]
- Macnab, R.M. How bacteria assemble flagella. Annu. Rev. Microbiol. 2003, 57, 77–100. [Google Scholar] [CrossRef] [PubMed]
- Dixon, D.P.; Lapthorn, A.; Edwards, R. Plant glutathione transferases. Genome Biol. 2002, 3, 1–10. [Google Scholar] [CrossRef][Green Version]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).