
Exploring the Intestinal Microbial Community of Lantang Pigs through Metagenome-Assembled Genomes and Carbohydrate Degradation Genes

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. Genomic DNA Extraction and Metagenomic Sequencing
2.3. Bioinformatics
3. Results
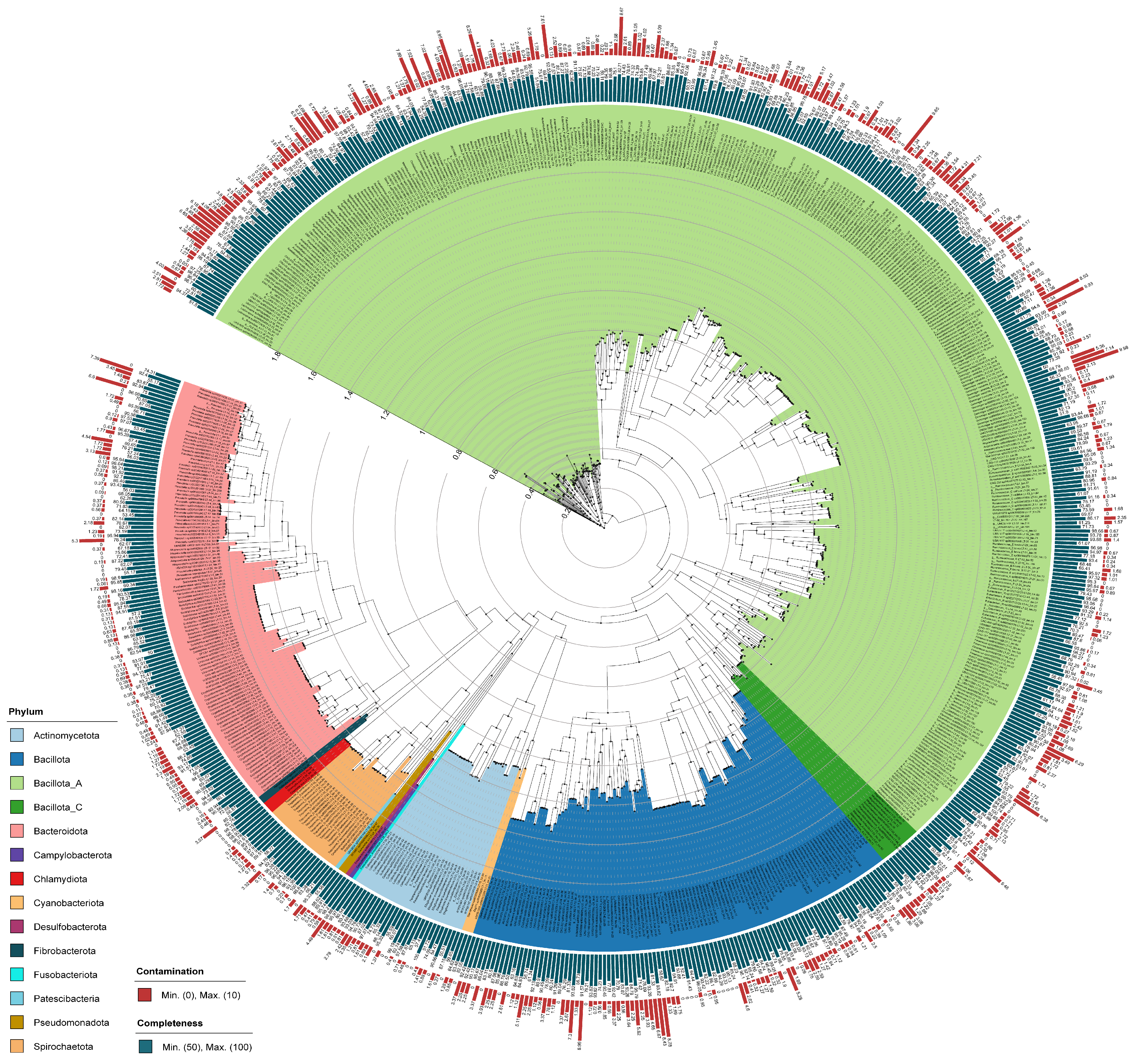
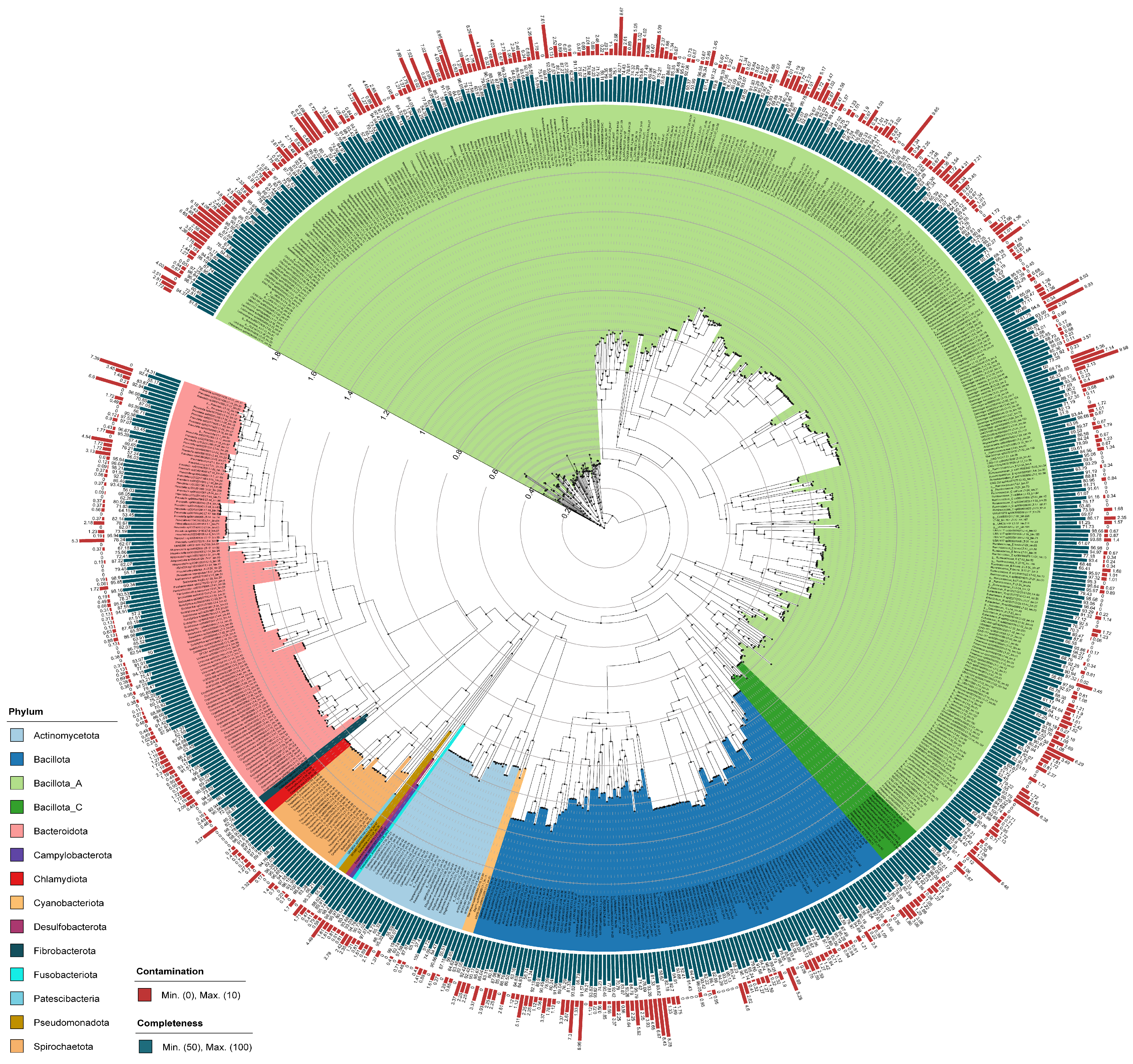
3.1. Metagenome-Assembled Genomes
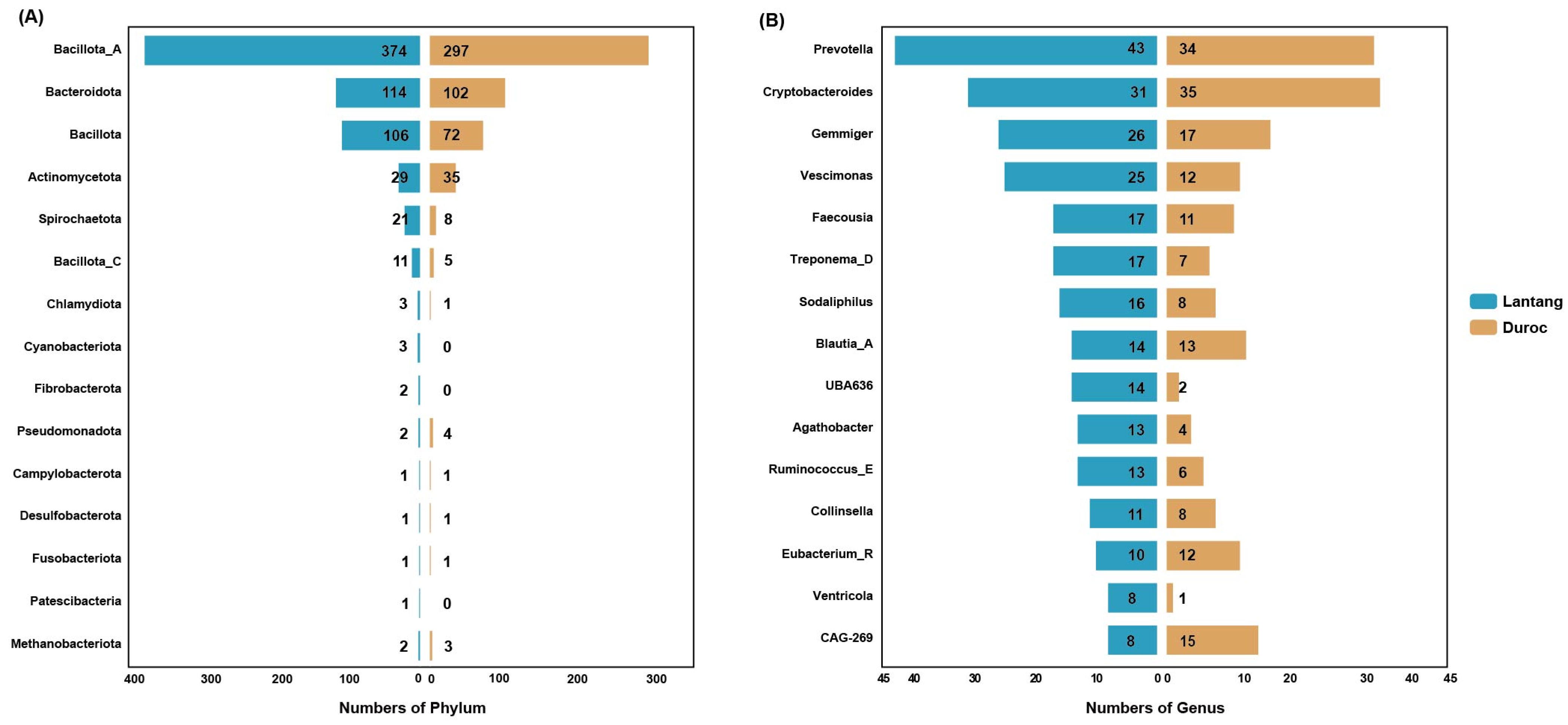
3.2. Classification of Genomes Based on Taxonomy
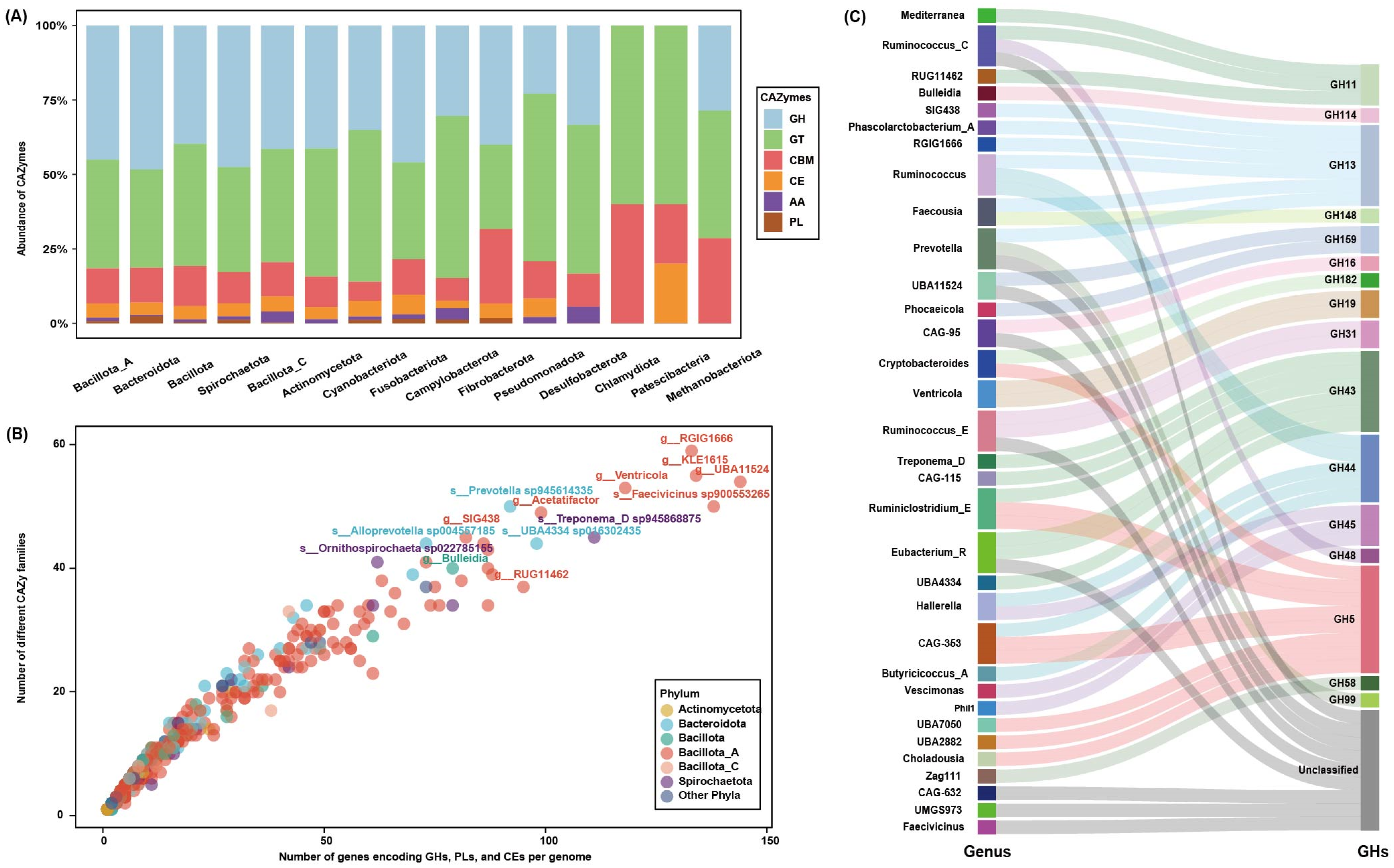
3.3. Overview of Carbohydrate Genes in the Microbiome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Song, B.; Zheng, C.; Zheng, J.; Zhang, S.; Zhong, Y.; Guo, Q.; Li, F.; Long, C.; Xu, K.; Duan, Y. Comparisons of carcass traits, meat quality, and serum metabolome between Shaziling and Yorkshire pigs. Anim. Nutr. 2022, 8, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, X.; Mu, L.; Ding, Z. Immune responses in pigs induced by recombinant DNA vaccine co-expressing swine IL-18 and membrane protein of porcine reproductive and respiratory syndrome virus. Int. J. Mol. Sci. 2012, 13, 5715–5728. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, S.; Luo, J.; Chen, T.; Xi, Q.; Zhang, Y.; Sun, J. The difference of intestinal microbiota composition between Lantang and Landrace newborn piglets. BMC Vet. Res. 2023, 19, 174. [Google Scholar] [CrossRef] [PubMed]
- Wassie, T.; Cheng, B.; Zhou, T.; Gao, L.; Lu, Z.; Xie, C.; Wu, X. Microbiome-metabolome analysis reveals alterations in the composition and metabolism of caecal microbiota and metabolites with dietary Enteromorpha polysaccharide and Yeast glycoprotein in chickens. Front. Immunol. 2022, 13, 996897. [Google Scholar] [CrossRef]
- Wardman, J.F.; Bains, R.K.; Rahfeld, P.; Withers, S.G. Carbohydrate-active enzymes (CAZymes) in the gut microbiome. Nat. Rev. Microbiol. 2022, 20, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Kaddouch, E.; Cleveland, M.E.; Navarro, D.; Grisel, S.; Haon, M.; Brumer, H.; Lafond, M.; Berrin, J.-G.; Bissaro, B. A simple and direct ionic chromatography method to monitor galactose oxidase activity. RSC Adv. 2022, 12, 26042–26050. [Google Scholar] [CrossRef]
- Kaoutari, A.E.; Armougom, F.; Gordon, J.I.; Raoult, D.; Henrissat, B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat. Rev. Microbiol. 2013, 11, 497–504. [Google Scholar] [CrossRef]
- Kassan, M.; Kwon, Y.; Munkhsaikhan, U.; Sahyoun, A.M.; Ishrat, T.; Galán, M.; Gonzalez, A.A.; Abidi, A.H.; Kassan, A.; Ait-Aissa, K. Protective Role of Short-Chain Fatty Acids against Ang-II-Induced Mitochondrial Dysfunction in Brain Endothelial Cells: A Potential Role of Heme Oxygenase 2. Antioxidants 2023, 12, 160. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Tao, S.; Song, T.; Lyu, W.; Li, Y.; Wang, W.; Shen, Q.; Ni, Y.; Zhu, J.; Zhao, J. Clostridium butyricum and carbohydrate active enzymes contribute to the reduced fat deposition in pigs. iMeta 2024, 3, e160. [Google Scholar] [CrossRef]
- Wang, Y.; LaPointe, G. Arabinogalactan Utilization by Bifidobacterium longum subsp. longum NCC 2705 and Bacteroides caccae ATCC 43185 in Monoculture and Coculture. Microorganisms 2020, 8, 1703. [Google Scholar] [CrossRef]
- Cockburn, D.W.; Koropatkin, N.M. Polysaccharide degradation by the intestinal microbiota and its influence on human health and disease. J. Mol. Biol. 2016, 428, 3230–3252. [Google Scholar] [CrossRef]
- Nieter, A.; Haase-Aschoff, P.; Kelle, S.; Linke, D.; Krings, U.; Popper, L.; Berger, R.G. A chlorogenic acid esterase with a unique substrate specificity from Ustilago maydis. Appl. Environ. Microbiol. 2015, 81, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Cavalier, D.M.; Lerouxel, O.; Neumetzler, L.; Yamauchi, K.; Reinecke, A.; Freshour, G.; Zabotina, O.A.; Hahn, M.G.; Burgert, I.; Pauly, M. Disrupting two Arabidopsis thaliana xylosyltransferase genes results in plants deficient in xyloglucan, a major primary cell wall component. Plant Cell 2008, 20, 1519–1537. [Google Scholar] [CrossRef] [PubMed]
- Plakys, G.; Gasparavičiūtė, R.; Vaitekūnas, J.; Rutkienė, R.; Meškys, R. Characterization of Paenibacillus sp. GKG endo-β-1, 3-glucanase, a member of family 81 glycoside hydrolases. Microorganisms 2022, 10, 1930. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Madupu, R.; Durkin, A.S.; Ekborg, N.A.; Pedamallu, C.S.; Hostetler, J.B.; Radune, D.; Toms, B.S.; Henrissat, B.; Coutinho, P.M. The complete genome of Teredinibacter turnerae T7901: An intracellular endosymbiont of marine wood-boring bivalves (shipworms). PLoS ONE 2009, 4, e6085. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, C.; Kirstein, I.V.; Meunier, C.L.; Rick, J.; Fofonova, V.; Wiltshire, K.H.; Steinke, N.; Vidal-Melgosa, S.; Hehemann, J.-H.; Huettel, B. Dissolved storage glycans shaped the community composition of abundant bacterioplankton clades during a North Sea spring phytoplankton bloom. Microbiome 2023, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Andaleeb, H.; Ullah, N.; Falke, S.; Perbandt, M.; Brognaro, H.; Betzel, C. High-resolution crystal structure and biochemical characterization of a GH11 endoxylanase from Nectria haematococca. Sci. Rep. 2020, 10, 15658. [Google Scholar] [CrossRef] [PubMed]
- Schultz-Johansen, M.; Bech, P.K.; Hennessy, R.C.; Glaring, M.A.; Barbeyron, T.; Czjzek, M.; Stougaard, P. A novel enzyme portfolio for red algal polysaccharide degradation in the marine bacterium Paraglaciecola hydrolytica S66T encoded in a sizeable polysaccharide utilization locus. Front. Microbiol. 2018, 9, 839. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wu, Q.; Dai, J.; Zhang, S.; Wei, F. Evidence of cellulose metabolism by the giant panda gut microbiome. Proc. Natl. Acad. Sci. USA 2011, 108, 17714–17719. [Google Scholar] [CrossRef]
- Borbón-García, A.; Reyes, A.; Vives-Flórez, M.; Caballero, S. Captivity shapes the gut microbiota of Andean bears: Insights into health surveillance. Front. Microbiol. 2017, 8, 1316. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, Y.; Yang, Z.; Li, C.; Liang, H.; Wu, Z.; Pu, W. Yeast probiotics shape the gut microbiome and improve the health of early-weaned piglets. Front. Microbiol. 2018, 9, 2011. [Google Scholar] [CrossRef]
- Fevrier, C.; Bourdon, D.; Aumaitre, A. Effects of level of dietary fibre from wheat bran on digestibility of nutrients, digestive enzymes and performance in the European Large White and Chinese Mei Shan pig. J. Anim. Physiol. Anim. Nutr. 1992, 68, 60–72. [Google Scholar] [CrossRef]
- Lei, L.; Wang, Z.; Li, J.; Yang, H.; Yin, Y.; Tan, B.; Chen, J. Comparative microbial profiles of colonic digesta between ningxiang pig and large white pig. Animals 2021, 11, 1862. [Google Scholar] [CrossRef]
- Rios-Covian, D.; Salazar, N.; Gueimonde, M.; de Los Reyes-Gavilan, C.G. Shaping the metabolism of intestinal Bacteroides population through diet to improve human health. Front. Microbiol. 2017, 8, 376. [Google Scholar] [CrossRef] [PubMed]
- Pinnell, L.J.; Reyes, A.A.; Wolfe, C.A.; Weinroth, M.D.; Metcalf, J.L.; Delmore, R.J.; Belk, K.E.; Morley, P.S.; Engle, T.E. Bacteroidetes and firmicutes drive differing microbial diversity and community composition among micro-environments in the bovine rumen. Front. Vet. Sci. 2022, 9, 897996. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Shu, G.; Zhu, X.; Gao, P.; Wang, S.; Wang, L.; Xi, Q.; Zhang, S.; Zhang, Y.; Li, Y. Fatty acid and transcriptome profiling of longissimus dorsi muscles between pig breeds differing in meat quality. Int. J. Biol. Sci. 2013, 9, 108. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langdon, W.B. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData Min. 2015, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Warr, A.; Affara, N.; Aken, B.; Beiki, H.; Bickhart, D.M.; Billis, K.; Chow, W.; Eory, L.; Finlayson, H.A.; Flicek, P. An improved pig reference genome sequence to enable pig genetics and genomics research. Gigascience 2020, 9, giaa051. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.; Han, Y.; Huang, Y.; Li, D.; Chai, J.; Deng, L.; Wei, M.; Wu, K.; Zhao, H.; Yang, G. A comprehensive analysis of antibiotic resistance genes in the giant panda gut. iMeta 2024, e171. [Google Scholar] [CrossRef]
- Deng, F.; Wang, C.; Li, D.; Peng, Y.; Deng, L.; Zhao, Y.; Zhang, Z.; Wei, M.; Wu, K.; Zhao, J. The unique gut microbiome of giant pandas involved in protein metabolism contributes to the host’s dietary adaption to bamboo. Microbiome 2023, 11, 180. [Google Scholar] [CrossRef]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. dRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, R.; Peng, Y.; Chai, J.; Li, Y.; Deng, F. The role of gut archaea in the pig gut microbiome: A mini-review. Front. Microbiol. 2023, 14, 1284603. [Google Scholar] [CrossRef]
- Chen, T.; Long, W.; Zhang, C.; Liu, S.; Zhao, L.; Hamaker, B.R. Fiber-utilizing capacity varies in Prevotella-versus Bacteroides-dominated gut microbiota. Sci. Rep. 2017, 7, 2594. [Google Scholar] [CrossRef] [PubMed]
- Gorvitovskaia, A.; Holmes, S.P.; Huse, S.M. Interpreting Prevotella and Bacteroides as biomarkers of diet and lifestyle. Microbiome 2016, 4, 15. [Google Scholar] [CrossRef]
- Flint, H.J.; Bayer, E.A.; Rincon, M.T.; Lamed, R.; White, B.A. Polysaccharide utilization by gut bacteria: Potential for new insights from genomic analysis. Nat. Rev. Microbiol. 2008, 6, 121–131. [Google Scholar] [CrossRef]
- Artzi, L.; Bayer, E.A.; Moraïs, S. Cellulosomes: Bacterial nanomachines for dismantling plant polysaccharides. Nat. Rev. Microbiol. 2017, 15, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Zhuang, Y.; Cui, K.; Bi, Y.; Zhang, N. Metagenomics reveals the temporal dynamics of the rumen resistome and microbiome in goat kids. Microbiome 2024, 12, 14. [Google Scholar] [CrossRef]
- Helbert, W.; Poulet, L.; Drouillard, S.; Mathieu, S.; Loiodice, M.; Couturier, M.; Lombard, V.; Terrapon, N.; Turchetto, J.; Vincentelli, R. Discovery of novel carbohydrate-active enzymes through the rational exploration of the protein sequences space. Proc. Natl. Acad. Sci. USA 2019, 116, 6063–6068. [Google Scholar] [CrossRef]
- Li, H.; Ma, L.; Li, Z.; Yin, J.; Tan, B.; Chen, J.; Jiang, Q.; Ma, X. Evolution of the gut microbiota and its fermentation characteristics of Ningxiang pigs at the young stage. Animals 2021, 11, 638. [Google Scholar] [CrossRef]
- Xu, Q.; Yuan, X.; Gu, T.; Li, Y.; Dai, W.; Shen, X.; Song, Y.; Zhang, Y.; Zhao, W.; Chang, G. Comparative characterization of bacterial communities in geese fed all-grass or high-grain diets. PLoS ONE 2017, 12, e0185590. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Liu, J.; Chen, W.; He, Y.; Kahaer, M.; Li, R.; Tian, T.; Liu, Y.; Bai, B.; Cui, Y. Diagnostic model for predicting hyperuricemia based on alterations of the gut microbiome in individuals with different serum uric acid levels. Front. Endocrinol. 2022, 13, 925119. [Google Scholar] [CrossRef] [PubMed]
- Jiminez, J.A.; Uwiera, T.C.; Abbott, D.W.; Uwiera, R.R.; Inglis, G.D. Impacts of resistant starch and wheat bran consumption on enteric inflammation in relation to colonic bacterial community structures and short-chain fatty acid concentrations in mice. Gut Pathog. 2016, 8, 67. [Google Scholar] [CrossRef] [PubMed]
- Wielgosz-Grochowska, J.P.; Domanski, N.; Drywień, M.E. Efficacy of an irritable bowel syndrome diet in the treatment of small intestinal bacterial overgrowth: A narrative review. Nutrients 2022, 14, 3382. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Zhang, H.; Wei, Q.; Zhao, C.; Yang, X.; Wu, X.; Xia, T.; Liu, G.; Zhang, L.; Gao, Y. Comparative analyses of fecal microbiota in European mouflon (Ovis orientalis musimon) and blue sheep (Pseudois nayaur) living at low or high altitudes. Front. Microbiol. 2019, 10, 1735. [Google Scholar] [CrossRef] [PubMed]
- Bevins, C.L.; Salzman, N.H. The potter’s wheel: The host’s role in sculpting its microbiota. Cell. Mol. Life Sci. 2011, 68, 3675–3685. [Google Scholar] [CrossRef]
- Xiao, K.; Liang, X.; Lu, H.; Li, X.; Zhang, Z.; Lu, X.; Wang, H.; Meng, Y.; Roy, A.; Luo, W. Adaptation of gut microbiome and host metabolic systems to lignocellulosic degradation in bamboo rats. ISME J. 2022, 16, 1980–1992. [Google Scholar] [CrossRef] [PubMed]
- Pu, G.; Li, P.; Du, T.; Niu, Q.; Fan, L.; Wang, H.; Liu, H.; Li, K.; Niu, P.; Wu, C. Adding appropriate fiber in diet increases diversity and metabolic capacity of distal gut microbiota without altering fiber digestibility and growth rate of finishing pig. Front. Microbiol. 2020, 11, 533. [Google Scholar] [CrossRef]
- Meng, X.; Zheng, J.; Wang, F.; Zheng, J.; Yang, D. Dietary fiber chemical structure determined gut microbiota dynamics. iMeta 2022, 1, e64. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Delgado Palacio, S.; Arboleya Montes, S.; Mancabelli, L. The first microbial colonizers of the human gut: Composition, activities, and health implications of the infant gut microbiota. Microbiol. Mol. Biol. Rev. 2017, 81, e00036-17. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, P.; Wang, P.; Hu, X.; Chen, F. Gut microbiota promotes production of aromatic metabolites through degradation of barley leaf fiber. J. Nutr. Biochem. 2018, 58, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Abdelsattar, M.M.; Wang, X.; Zhang, N.; Chai, J. In Vitro Modulation of Rumen Fermentation by Microbiota from the Recombination of Rumen Fluid and Solid Phases. Microbiol. Spectr. 2023, 11, e0338722. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Fahmi, M.; Tanaka, J.; Seki, K.; Kubota, Y.; Ito, M. Genome-wide analysis of whole human glycoside hydrolases by data-driven analysis in silico. Int. J. Mol. Sci. 2019, 20, 6290. [Google Scholar] [CrossRef] [PubMed]
- Agyekum, A.K.; Nyachoti, C.M. Nutritional and Metabolic Consequences of Feeding High-Fiber Diets to Swine: A Review. Engineering 2017, 3, 716–725. [Google Scholar] [CrossRef]
- Weimer, P.J. Degradation of Cellulose and Hemicellulose by Ruminal Microorganisms. Microorganisms 2022, 10, 2345. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Roux, S.; Seshadri, R.; Udwary, D.; Varghese, N.; Schulz, F.; Wu, D.; Paez-Espino, D.; Chen, I.-M.; Huntemann, M. A genomic catalog of Earth’s microbiomes. Nat. Biotechnol. 2021, 39, 499–509. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Phylum | MAGs | GHs | CEs | PLs | Total | ||||
|---|---|---|---|---|---|---|---|---|---|
| No. | Mean | No. | Mean | No. | Mean | No. | Mean | ||
| Actinomycetota | 27 | 121 | 4.5 | 12 | 0.4 | 0 | 0.0 | 133 | 4.9 |
| Bacillota | 99 | 585 | 5.9 | 66 | 0.7 | 5 | 0.1 | 656 | 6.6 |
| Bacillota_A | 356 | 5837 | 16.4 | 625 | 1.8 | 92 | 0.3 | 6554 | 18.4 |
| Bacillota_C | 11 | 180 | 16.4 | 22 | 2.0 | 1 | 0.1 | 203 | 18.5 |
| Bacteroidota | 107 | 1229 | 11.5 | 106 | 1.0 | 62 | 0.6 | 1397 | 13.1 |
| Campylobacterota | 1 | 24 | 24.0 | 2 | 2.0 | 1 | 1.0 | 27 | 27.0 |
| Chlamydiota | 2 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 |
| Cyanobacteriota | 3 | 93 | 31.0 | 14 | 4.7 | 3 | 1.0 | 110 | 36.7 |
| Desulfobacterota | 1 | 6 | 6.0 | 0 | 0.0 | 0 | 0.0 | 6 | 6.0 |
| Fibrobacterota | 2 | 24 | 12.0 | 3 | 1.5 | 1 | 0.5 | 28 | 14.0 |
| Fusobacteriota | 1 | 62 | 62.0 | 9 | 9.0 | 2 | 2.0 | 73 | 73.0 |
| Methanobacteriota | 1 | 2 | 2.0 | 0 | 0.0 | 0 | 0.0 | 2 | 2.0 |
| Patescibacteria | 1 | 0 | 0.0 | 1 | 1.0 | 0 | 0.0 | 1 | 1.0 |
| Pseudomonadota | 2 | 11 | 5.5 | 3 | 1.5 | 0 | 0.0 | 14 | 7.0 |
| Spirochaetota | 20 | 447 | 22.4 | 41 | 2.1 | 11 | 0.6 | 499 | 25.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Fan, Y.; Jin, R.; Peng, Y.; Chai, J.; Wei, X.; Zhao, Y.; Deng, F.; Zhao, J.; Li, Y. Exploring the Intestinal Microbial Community of Lantang Pigs through Metagenome-Assembled Genomes and Carbohydrate Degradation Genes. Fermentation 2024, 10, 207. https://doi.org/10.3390/fermentation10040207
Yang J, Fan Y, Jin R, Peng Y, Chai J, Wei X, Zhao Y, Deng F, Zhao J, Li Y. Exploring the Intestinal Microbial Community of Lantang Pigs through Metagenome-Assembled Genomes and Carbohydrate Degradation Genes. Fermentation. 2024; 10(4):207. https://doi.org/10.3390/fermentation10040207
Chicago/Turabian StyleYang, Jianbo, Ying Fan, Rui Jin, Yunjuan Peng, Jianmin Chai, Xiaoyuan Wei, Yunxiang Zhao, Feilong Deng, Jiangchao Zhao, and Ying Li. 2024. "Exploring the Intestinal Microbial Community of Lantang Pigs through Metagenome-Assembled Genomes and Carbohydrate Degradation Genes" Fermentation 10, no. 4: 207. https://doi.org/10.3390/fermentation10040207
APA StyleYang, J., Fan, Y., Jin, R., Peng, Y., Chai, J., Wei, X., Zhao, Y., Deng, F., Zhao, J., & Li, Y. (2024). Exploring the Intestinal Microbial Community of Lantang Pigs through Metagenome-Assembled Genomes and Carbohydrate Degradation Genes. Fermentation, 10(4), 207. https://doi.org/10.3390/fermentation10040207