Abstract
In this study, the wild-type Rhodotorula mucilaginosa GDMCC 2.30 and its high carotenoid-producing mutant JH-R23, which was screened from the space mutation breeding treated wild type, were used as materials. Through whole-genome sequencing and resequencing analysis, the carotenoid metabolic pathway and mechanism of high carotenoid production in the mutant were explored. The R. mucilaginosa GDMCC 2.30 genome comprised 18 scaffolds and one circular mitochondrial genome with a total size of 20.31 Mb, a GC content of 60.52%, and encoding 7128 genes. The mitochondrial genome comprised 40,152 bp with a GC content of 40.59%. Based on functional annotations in the GO, KEGG, and other protein databases, nine candidate genes associated with carotenoid metabolic pathways, and candidate genes of the CrtS and CrtR homologous gene families were identified. The carotenoid metabolic pathway was inferred to start from sugar metabolism to the mevalonate pathway, as is common to most fungi, and the final product of the mevalonate pathway, geranylgeranyl diphosphate, is a precursor for various carotenoids, including β-carotene, lycopene, astaxanthin, and torularhodin, formed through the activity of crucial enzymes encoded by genes such as CrtI, CrtYB, CrtS, and CrtR. Resequencing analysis of the mutant JH-R23 detected mutations in the exons of four genes, including those encoding Gal83, 3-oxoacyl-reductase, p24 proteins, and GTPase. These mutations are interpreted to have an important impact on carotenoid synthesis by JH-R23.
1. Introduction
Carotenoids are fat-soluble pigments present in higher plants; fungi; algae; and bacteria [1]. These compounds are favored by the food (including health food) and cosmetics industries for their diverse physiological functions; such as antioxidant and antitumor activity and immunity enhancement [2,3]. The estimated global market value for carotenoids is projected to expand to USD 2 billion by 2026 with an annual growth rate of 4.2% [4]. Compared with chemical synthesis and biological extraction; fermentation is currently the main method used for commercial carotenoid production. To further improve the production efficiency of carotenoids; research on strains producing high carotenoid yields and their metabolic engineering has become an important focus for the industrial fermentation production of carotenoids Rhodotorula mucilaginosa is a red yeast rich in carotenoids and lipids and originates from seawater; sediments; glaciers; and other environments. Food waste; agricultural waste; and other substances can be used as culture media for the species for high density fermentation production of carotenoids and lipids [5,6,7]. Although wild-type red yeast strains are highly adaptable; carotenoid production is relatively low. Therefore; isolation of red yeast mutants producing high carotenoid yields and optimization of product metabolism regulation based on the clarity of the mutation mechanism are urgently required for enhanced industrial production [8]. Cutzu et al. applied ultraviolet mutagenesis to Rhodotorula glutinis and isolated a mutant with its β-carotene yield increased by 2.8-fold compared with that of the parental strain [9]. Nasrabadi et al. combined multiple physicochemical mutagenesis methods to obtain the mutant Rhodotorula acheniorum MRN and increased its carotenoid yield by 6.45-fold through culture medium optimization [10]. Zheng et al. used atmospheric and room temperature plasma mutagenesis and screened the mutant Rhodotorula toruloides M18; which showed a 14.68-fold increase in torularhodin production over that of the wild type [11]. In previous work; our research group loaded R. mucilaginosa GDMCC 2.30 onto the “New Generation Manned Spacecraft Test Ship” to obtain the mutant JH-R23 and increased its carotenoid production by 2.46-fold through optimization of the fermentation process [12]. However; little information is available on the mechanism of high carotenoid production in red yeast mutants; and only a few genome sketches assembled from Illumina sequencing data are available [13], still lacking high quality genomic studies; which largely hinders improvement of the product yield and scale of carotenoid production through metabolic regulation and optimization. In this study; we sequenced the complete genome of strain GDMCC 2.30 using PacBio and Illumina sequencing technologies and analyzed the carotenoid metabolic pathway by combining the present genomic information and previous research. The mutant JH-R23 was resequenced and its mutations were analyzed by comparative genomics to establish a theoretical basis for research on metabolic engineering for carotenoid production in R. mucilaginosa.
2. Materials and Methods
2.1. Strain Culture and Genomic DNA Extraction
The wild-type R. mucilaginosa GDMCC 2.30 was purchased from the Guangdong Microbial Culture Collection Center. The strain was cultured in 20 mL potato dextrose broth medium (20% [w/v] potato, 2% [w/v] glucose, and pH 7.0) at 28 °C and 200 rpm until the logarithmic phase (OD600 = 1.0). Cells were harvested by centrifugation at 13,400× g for 1 min and immediately flash frozen in liquid nitrogen for further extraction of genomic DNA and total RNA. Genomic DNA was extracted using the cetyltrimethylammonium bromide method [14]. Genomic DNA quality and integrity were assessed by agarose gel electrophoresis and comparison with appropriate size standards, while DNA yield and purity were measured using a NanoDrop™ 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and TM-380 fluorometer (Turner BioSystems, Inc., Sunnyvale, CA, USA). High quality DNA (OD260/280 = 1.8–2.0, >1 μg) was used for the study.
2.2. Genome Sequencing and Transcriptome Sequencing
The genomes of the wild-type GDMCC 2.30 and the mutant JH-R23 were sequenced by Shanghai Winnerbio Technology Co., Ltd. (Shanghai, China) using PacBio Sequel II platforms and Illumina NovaSeq 6000. For PacBio sequencing, fragments shorter than 500 bp were removed from the SMRTbell library before long-read sequencing. For Illumina sequencing, genomic DNA was fragmented using the E220 Focused-ultrasonicator (Covaris, Shelton Connecticut, MA, USA), and the library was prepared by end repair, A-tailing, adapter ligation, purification, and PCR amplification, followed by paired-end sequencing (2 × 150 bp).
Total RNA was extracted from each strain using the TRIzol reagent kit (Sangon Biotech, Shanghai, China) following the manufacturer’s protocol. Transcriptome sequencing libraries were constructed using the NEBNext Ultra Directional RNA Library Prep Kit (New England Biolabs, Ipswich, MA, USA), and sequenced on an Illumina NovaSeq 6000 by Shanghai Winnerbio Technology Co., Ltd. (Shanghai, China). All genomic sequencing data and transcriptome sequencing data are available in the NCBI database under BioProject PRJNA1034680.
2.3. Genome Assembly and Prediction
The Illumina reads were further processed for quality control and filtered using fastp (v0.23.2) [15]. The k-mer count (k-mer = 19) was calculated using Jellyfish (v2.2.10) [16]. GenomeScope (v2.0) and Smudgeplot (v0.2.5) [17] were used to estimate the genome size, ploidy, repeat content, and heterozygosity. The PacBio data were corrected, trimmed, and assembled using Flye (v2.9.1) [18] and the genome sequence was polished using NextPolish (v1.4.0) [19]. The completeness of the assembly was evaluated using BUSCO (v5.4.5) [20] with the basidiomycota_odb10 database. Telomeric repeats (TTAGGG/CCCTAA) in the assembled genome were searched using Perl scripts.
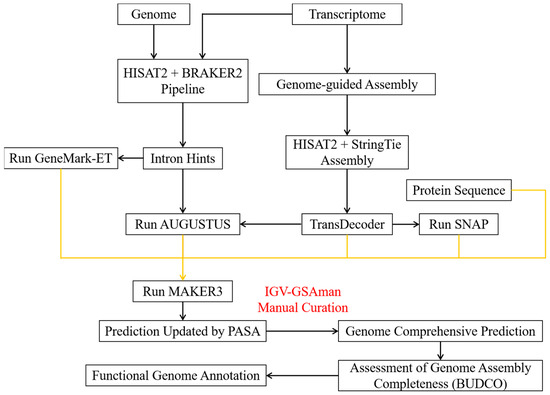
Figure 1.
Schematic illustration of the pipeline for the genome annotation of R. mucilaginosa GDMCC 2.30 with the supplements of transcriptome sequences. The orange lines indicate the import of transcripts in MAKER3 for the further prediction of gene models. The IGV-GSAman software correction schematic in Figure S1.
First, RNA clean reads were processed for quality control and filtering using fastp (v0.23.2) [15]. A reference genome index was built with HISAT2 (v2.2.1) [21] using default parameters, and clean reads were mapped to the R. mucilaginosa GDMCC 2.30 genome. The mapped reads were sorted and indexed using SAMtools (v1.16.1) [22]. The unmasked genome and mapped reads were entered into BRAKER2 (v2.1.6) [23] for ab initio prediction with default settings. After intron identification, preliminary training datasets for gene model prediction were generated using GeneMark-ET (v4.46) and AUGUSTUS (v3.4.0).
Transcripts were then assembled and merged by genome-guided assembly using StringTie (v2.2.1) [24] and HISAT2 (v2.2.1) [21] with default settings for the above mapped reads. The resulting transcriptome database was used to predict gene models with TransDecoder (v5.5.0) [25]. In parallel, the transcriptome database, R. mucilaginosa GDMCC 2.30 genome sequences, previously identified R. mucilaginosa GDMCC 2.30 repetitive sequences, and all available fungal homologous protein sequences in the genus Rhodotorula were used to generate an initial gene model prediction with the SNAP model implemented in MAKER3 (v3.01.03) [26]. This initial prediction, the transcriptome database, and preliminary training datasets from BRAKER2 (v2.1.6) were then incorporated into MAKER3 (v3.01.03) for the final gene model prediction.
After obtaining initial gene models, PASA was utilized to update and refine them. Additionally, gene structure annotation information was manually reviewed and corrected using IGV-GSAman (v0.7.14) (https://gitee.com/CJchen/IGV-sRNA, accessed on 13 February 2023). This allowed us to obtain the final gene model prediction. The genome annotation was submitted to the NCBI database under BioProject PRJNA1034680.
Repeats were identified by RepeatModeler (v2.0.3) [27] and RepeatMasker (v4.1.2) [28]. Barrnap (v0.9) (https://github.com/tseemann/barrnap, accessed on 14 May 2023) was used to predict rRNA genes, tRNAscan-SE (v2.0.11) [29] was used to predict tRNA genes, and snRNAs were identified by searching the Rfam database using CMsearch (v1.1.4) [30]. Functional annotation of genes in the genome (E-value ≤ 1 × 10−5) by searching against the Nr, SwissProt, KOG, and GO databases was performed using DIAMOND (v0.9.24) [31]. KEGG orthologs were assigned using KofamScan (v1.3.0) [32]. The mitochondrial genome was functionally annotated using GeSeq [33] online followed by manual curation and was visualized using Chloroplot [34].
2.4. Comparative Genomics and Genetic Variation Analysis
The Rhodotorula mucilaginosa JH-R23 genome and 13 representative red yeast genomes were used for a comparative genomic analysis, comprising Rhodotorula babjevae CBS 7808, Rhodotorula diobovata UCD-FST 08-225, Rhodotorula glutinis ZHK, Rhodotorula graminis WP1, Rhodotorula kratochvilovae YM25235, Rhodotorula mucilaginosa JY1105, Rhodotorula paludigena TL35-5, Rhodotorula sphaerocarpa GDMCC 60679, Rhodotorula taiwanensis MD1149, Rhodotorula toruloides NP11, Sporidiobolus pararoseus NGR, Sporidiobolus roseus SR19, and Phaffia rhodozyma CBS 6938 (Table S1). OrthoFinder (v2.5.4) [35] was used to identify and cluster gene families. Next, 1646 single-copy orthologs were globally aligned using MUSCLE (v5.1) [36] and subjected to phylogenetic analysis using the maximum likelihood method implemented in RAxML (v8.2.12) [37] with “-# 1000 -m PROTGAMMAAUTO”. Divergence times were estimated by MCMCTree (v4.9) [38] using fourfold degenerate sites of single-copy gene families with the parameter settings clock = 3 and model = 0. Expanded and contracted gene families were identified by CAFÉ5 [39]. Carotenoid gene clusters were identified by antiSMASH (v6.0.0) [40] using the ClusterBlast algorithm and visualized using the ChiPlot online Web Page (https://www.chiplot.online/, accessed on 18 May 2023). Candidate gene domains were predicted using CDD [41] and MEME [42] online. Phylogenetic trees were constructed and visualized using ChiPlot. Single-nucleotide polymorphisms (SNPs) and insertions/deletions (InDels) were identified using GATK4.0 and were annotated using Annovar [43].
2.5. Space Breeding Mutation
For pre-flight preparation, R. mucilaginosa GDMCC 2.30 cells were cultured in tubes containing potato dextrose agar (HuanKai, Guangzhou, China) at 28 °C for 48 h. These tubes were shipped from Shenzhou Biotechnology Co. (Inner Mongolia, China) to the “New Generation Manned Spacecraft Test Ship”, which flew for 67 h in space under special conditions of microgravity (10−6–10−3 g), vacuum (101.325 kPa), temperature (17–23 °C), and cosmic ionizing radiation (0.146 Gy/y); the tubes were returned to the laboratory after the successful landing of the return capsule.
After the space mission, the mutants were gently scraped from the potato dextrose agar tubes, resuspended in 10 mL of sterile water and diluted to 10−5, 10−6, and 10−7 with sterile water, and plated on potato dextrose agar at 28 °C for 48 h to screen for the highest carotenoid-producing strain. The strain with the highest carotenoid production was screened on potato dextrose agar plates and named JH-R23. The wild-type and mutant strains were inoculated into 20 mL of potato dextrose broth medium at 28 °C and 200 rpm for 144 h, respectively. Carotenoid production and dry cell weight were determined every 24 h to compare their carotenoid differences. Meanwhile, they were plated on potato dextrose agar plates and incubated at 28 °C for 120 h to be stored for preservation.
The internal transcribed spacer (ITS) gene region of rDNA was amplified by polymerase chain reaction (PCR) using ITS1 (5′-TCCGTAGGTGAACCTGCGG-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) primers purchased from Sangon Biotech (Shanghai, China) [44]. The PCR products were analyzed by electrophoresis before being sent to Sangon Biotech (Shanghai, China) for sequencing. The obtained ITS sequences were available in the NCBI GenBank database under OR976268-OR976269, and then blasted against the NCBI database for identification.
2.6. Carotenoid Yield and Biomass Determination
Extraction of carotenoid was performed as described by Tian et al. [45] with some modifications as follows. After fermentation, cells were disrupted using the DMSO cell wall breaking method. Fermentation broth (5 mL) was centrifuged at 7576× g for 5 min to obtain wet cells, which were washed twice with triple distilled water, resuspended once in 5 mL of 99% anhydrous ethanol, and centrifuged at 7576× g for 5 min. The supernatant was discarded, 2 mL DMSO was added to resuspend the cells, the cell suspension was incubated in a 65 °C water bath for 1.5 h, then 6 mL acetone was added and shaken for 10 min until the cells were colorless. After centrifugation at 7576× g for 5 min, the supernatant was collected to measure the absorbance at 480 nm. Total carotenoid yield was calculated according to previous studies [46]. Carotenoid yield was calculated using the following formula:
where D is the dilution factor, V is the volume of organic solvent used for extraction (mL), W is the dry weight of cells used for extraction (g), and 0.16 is the extinction coefficient of the organic solvent.
Carotenoid yield per unit cell dry weight (μg/g) = A480 × D × V/(0.16 × W),
For biomass determination [47], 5 mL fermentation broth was centrifuged at 7576× g for 5 min. The supernatant was discarded, the wet cells were washed twice with 5 mL triple distilled water, and then centrifuged to obtain the wet cells. The wet cells were dried to constant weight in a 105 °C oven and the dry weight was measured as the biomass.
2.7. Statistical Analysis
The data are presented as the mean ± SD of at least three independent experiments. All data were statistically analyzed using SPSS software (v26.0). Significant differences were assessed using two-sided t-tests (p < 0.05).
3. Results and Discussion
3.1. Rhodotorula mucilaginosa GDMCC 2.30 Genome Sequencing, Assembly, and Evaluation
To assemble a high quality genome of R. mucilaginosa GDMCC 2.30, PacBio Sequel II and Illumina NovaSeq 6000 were used to sequence the complete genome. The results showed that a total of 6.94 Gb Illumina short reads and 6.42 Gb PacBio long reads were obtained, approximately 300× high quality subreads covered the 21.27 Mb genome (Table S2). Based on a k-mer analysis, the genome heterozygosity was 0.0141%, suggesting that the strain was a homozygous diploid (Figure S2). The genome contained 18 scaffolds and a circular mitochondrial genome with a total size of 20.31 Mb and a GC content of 60.52%. The circular mitochondrial genome comprised 40,152 bp with a GC content of 40.59% (Figure 2). A total of 1714 BUSCO genes were identified (97.1%) and telomere sequences existed at both ends of most scaffolds (Table S3), indicating the genome data were of high quality. Given the high-depth sequencing, novel sequencing technologies, and suitable assembly methods, the assembly for wild-type GDMCC 2.30 was more complete, accurate, and near chromosome level compared with assemblies reported for other conspecific strains (Table 1). This laid a sound foundation for functional genomic and mutation analysis of this strain.
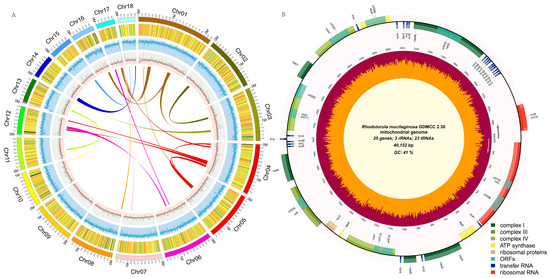
Figure 2.
(A) Circular maps of the Rhodotorula mucilaginosa GDMCC 2.30 nuclear genome; Tracks from the outermost to innermost circles represent chromosome information, gene expression level, GC content, sequencing depth, and collinearity, respectively. (B) mitochondrial genome; Genes within the circle are transcribed clockwise, while those outside are transcribed counter-clockwise. Genes are color-coded based on their functional groups. The inner circle’s orange represents the GC content, while the maroon represents the AT content.

Table 1.
Published genomic data for Rhodotorula mucilaginosa (as of November 2023).
3.2. Rhodotorula mucilaginosa GDMCC 2.30 Gene Annotation and Carotenoid Biosynthesis Pathway Prediction
A Repeats accounted for 1.71% of the R. mucilaginosa GDMCC 2.30 genome, including LTR transposons, which accounted for 1.03%. The MAKER annotation pipeline predicted 7128 protein-coding genes, 118 tRNAs, 23 rRNAs, and 7 snRNAs. Integrated annotations from the Nr, SwissProt, KOG, GO, and KEGG databases revealed 7015 genes (98.41%) with predictable functions (Figure S3, Table S4), with an average gene sequence length of 1717.9 bp and an average protein sequence length of 571.7 aa; the average number of exons per gene was 6.39 with an average length of 268.8 bp, and the average number of introns per gene was 5.39 with an average length of 89 bp. The mitochondrial genome contained 23 tRNA genes, 3 rRNA genes, and 25 protein-coding genes (Figure 2B). Compared with the reported R. mucilaginosa RIT389 (NC_036340.1) mitochondrial genome [48], the GC content and genome size were basically consistent, with sequence similarity of 99% (Figure S4).
Given that annotations of the R. mucilaginosa GDMCC 2.30 genome included six candidate genes associated with the mevalonate pathway and five carotenoid biosynthesis candidate genes in total (Table S5), it was inferred that, with acetyl coenzyme A (acetyl-CoA) as the initial substrate, geranylgeranyl diphosphate (GGPP) could be synthesized through the synergistic activities of multiple crucial enzymes to provide precursors for the biosynthesis of C40 carotenoids [54]. In addition, the genome contained numerous genes and gene clusters encoding carotenoid biosynthetic enzymes, such as lycopene cyclase/phytoene synthase (CrtYB), carotenoid oxygenase (CCD1), phytoene desaturase (CrtI), and geranylgeranyl pyrophosphate synthase (CrtE) (Figure 3A). These enzymes collaboratively catalyze the conversion of GGPP into various carotenoids, including lycopene, β-carotene, and torulene [55,56].
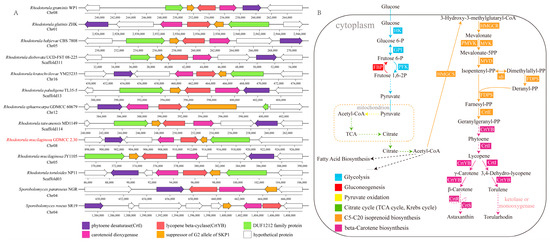
Figure 3.
Analysis of the carotenoid metabolic pathway in Rhodotorula mucilaginosa GDMCC 2.30. (A) Comparison of carotenoid biosynthetic gene clusters among 13 red yeast species. Homologous genes are indicated by the same color; (B) Putative carotenoid metabolic pathway map. All protein-coding genes are annotated from the KEGG database. The full gene names are listed in Table S5.
Currently, the metabolic process of astaxanthin and torularhodin synthesis in Rhodotorula yeasts remains controversial. Previous studies indicate that, in P. rhodozyma, CrtS and CrtR collaboratively catalyze the ketolation and hydroxylation of β-carotene into astaxanthin [57,58,59]. By constructing phylogenetic trees for the Ascomycota CrtS (OG00000414) and the CrtR (OG00004469) gene families to predict their domain structures (Figure S5), we observed that both gene families had similar domain compositions and motif constitutions (Table S6). Motif_7a showed species specificity, whereas motif_7b showed copy number specificity. The CYP_FUM15-like structural domain is highly conserved in the CrtS gene family, while the CYPOR and Flavodoxin_1 structural domains are conserved in the CrtR gene family. Therefore, although R. mucilaginosa is phylogenetically distant from P. rhodozyma, analysis of the amino acid sequence similarity and domain structure suggested a strong similarity in adaptive evolution. It is inferred that the enzymes encoded by these gene families in R. mucilaginosa have similar ketolation or hydroxylation functions as those in P. rhodozyma and participate in the synthesis of carotenoids such as astaxanthin and torularhodin. However, the exact synthesis pathways for these carotenoids in R. mucilaginosa GDMCC 2.30 require further verification by performing heterologous expression experiments. In summary, based on the present genomic information and previous research, the carotenoid biosynthetic pathway in R. mucilaginosa GDMCC 2.30 was resolved from a KEGG pathway analysis (Figure 3B), which lays the foundation for future understanding of carotenoid biosynthesis, as well as providing new insights into the study of carotenoid metabolic pathways.
3.3. Analysis of the Mechanism of High Carotenoid Production in the Mutant JH-R23
Through space mutation breeding treatment of R. mucilaginosa GDMCC 2.30, we diluted the yeast cells and plated them on potato dextrose agar plates to screen for a high carotenoid-producing mutant strain. Pigment intensity analysis was performed on 46 colonies grown on potato dextrose agar plates (Figure 4A, left) and we obtained a deep red mutant strain, JH-R23, which was molecularly identified, fermentation verified, and strain conserved (Figure 4A, right). A 617 bp fragment of the rDNA-ITS gene was amplified and sequenced. The sequence obtained was compared with sequences in the NCBI database and showed 100% similarity to the R. mucilaginosa strain (OR976269.1), confirming the homology of the isolated strain. Before 24 h of cultivation, the carotenoid production of JH-R23 and wild-type strains was similar, with no significant difference. However, after 48 h of cultivation, the carotenoid production per 24 h of the JH-R23 strain was 1.98, 1.94, 2.01, 2.39, and 2.46 times higher than that of the wild-type strain, respectively, and reached a peak after 144 h of cultivation. The carotenoid production of the wild-type strain was 151.39 μg/g and that of the JH-R23 mutant strain was 372.84 μg/g (Figure 4B). Mutagenesis has been used to enhance carotenoid production in R. mucilaginosa to varying degrees. However, current research has mainly focused on UV mutagenesis. Issa et al. mutagenized R. mucilaginosa A734 using UV light at 254 nm, resulting in a 1.12-fold increase in total carotenoids [60]. However, the observed increase was much lower than that of JH-R23 in this study (2.46-fold), and this is the first time that such a significant increase in carotenoids has been reported for R. mucilaginosa by space mutagenesis.
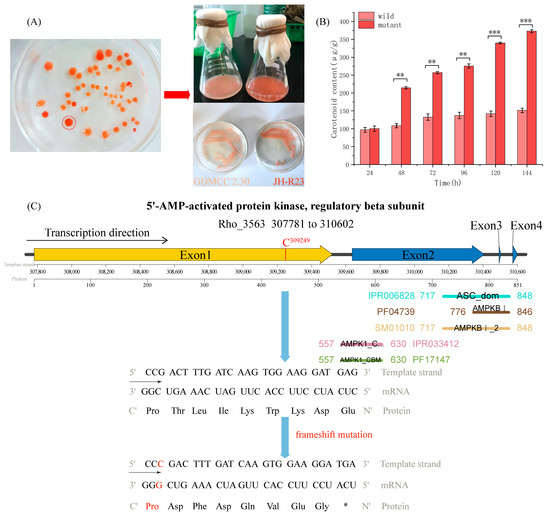
Figure 4.
Analysis of the mechanism of high carotenoid production of Rhodotorula mucilaginosa JH-R23. (A) Screening plate, and comparison of wild-type strain (GDMCC 2.30) and mutant strain (JH-R23) cultures; (B) Carotenoid yield of the wild-type GDMCC 2.30 and mutant JH-R23; ** denotes significant differences (p < 0.01), *** denotes highly significant differences (p < 0.001). (C) Predicted insertion mutation of a cytosine nucleotide in the Gal83 gene and domain analysis of JH-R23; * denotes a stop codon (Ter).
Genome resequencing analysis of the genetic variation in JH-R23 detected 38 SNPs and 58 InDels (Table S7). The majority of mutations were located in intergenic regions, but mutations were detected in the exons of four genes encoding Gal83, 3-oxoacyl-reductase, p24 family protein, and GTPase.
The Gal83 protein is the β subunit of the Snf1 protein kinase complex, forming a trimer complex with the α subunit of Snf1 and γ subunit of Snf4 [61]. As an important intracellular energy sensor, this complex is activated under glucose starvation and participates in relieving the inhibition of glucose catabolism products. For example, the Snf1 protein kinase complex promotes expression of the hexose transporter genes HXT2 and HXT4 and phosphorylates the Mig1 transcriptional repressor to relieve the inhibition of Gal gene transcription regulated by galactose induction [62,63]. The insertion of a cytosine nucleotide in the Gal83 coding sequence of JH-R23 caused a frameshift mutation, shortening the protein length from 851 to 540 amino acids (Figure 4C), resulting in the loss of glycogen-binding capability. However, Gal83 contains a highly conserved glycogen-binding domain that is homologous to that of AMPK family proteins. The mutation that eliminated Gal83–glycogen binding would also affect the activity of the Snf1 protein kinase complex, thus weakening the feedback inhibition effect of glucose catabolism products. This would lead to sustained expression of high-affinity transport proteins, thereby enhancing the co-consumption capabilities of glucose and xylose to provide substrates and energy for carotenoid synthesis [64]. In addition, this mutation may influence the conformation of adjacent domains or interactions with unknown signaling molecules, and thus the specific mechanism requires further study [64]. As shown by Wang et al. [54], transcriptome comparison between a Gal83 knockout strain and the wild type revealed upregulation in the acetyl-CoA and CoA biosynthesis pathways, but downregulation in the sugar lipid metabolism and ether lipid metabolism pathways. This finding highlighted the regulatory role of Snf1 in carbon source utilization, sporulation, trap formation, oxidative stress response, and other metabolic activities, providing substrates and energy for the downstream synthesis of metabolic products [65]. In addition, 3-oxoacyl-reductase plays an important role in the primary stage of lipid synthesis and multiple synonymous mutations may reduce the expression of lipid synthetic enzymes by changing the codon usage preference, thereby reducing the competition for carotenoid precursors. Mutation of GTPase can increase the sensitivity of a strain to osmotic pressure and oxidative stress, stimulating elevated carotenoid production for antioxidation [66]. In summary, the Gal83 mutation may be the major reason for the change in carotenoid yield in the JH-R23 mutant, but the mutations in the genes encoding 3-oxoacyl-reductase and GTPase also may increase carotenoid yield to some extent.
Comparative analysis of 13 red yeast genomes revealed 4084 orthologous gene families and 1646 single-copy orthologous gene families (Table S8). Based on the gene family analysis, a phylogenetic tree for 14 red yeast species was constructed (Figure 5). A GO enrichment analysis indicated that JH-R23-specific gene families were mainly enriched in purine nucleotide biosynthesis and metabolism, cytoplasmic ribosomes, pantothenate biosynthesis and metabolism, and amide biosynthesis and metabolism (Table S9). Pantothenate is a precursor of CoA, which can promote the synthesis of acetyl-CoA and stimulate energy metabolism, thereby increasing the supply of carotenoid precursors and energy, and enabling the potential capability for high carotenoid production in JH-R23 [67,68]. Comparative evolutionary analysis showed that 88 gene families were significantly expanded, and 69 gene families were significantly contracted in JH-R23 (p < 0.05; Figure 5). Furthermore, GO and KEGG enrichment analyses indicated that these gene families were associated with ABC transporters, ribosomes, and other pathways (Table S10). These findings suggested that the mutant JH-R23 exhibited higher metabolic activity, unique pantothenate synthesis, and stronger transmembrane transport capabilities, thus enabling the high carotenoid production.
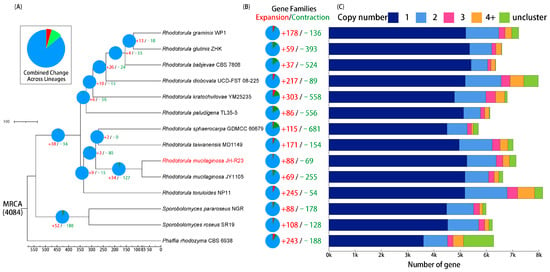
Figure 5.
Comparative genomic analysis of Rhodotorula mucilaginosa JH-R23 and other red yeast species. (A) Phylogenetic tree and divergence time estimation for 13 representative red yeast species; (B) Expanded (red) and contracted (green) gene families; (C) Distribution of single-copy, multi-copy, and unmatched gene families among 14 red yeast species.
In summary, an exon mutation disrupted the glycogen-binding domain of Gal83, which would affect the activity of the Snf1 protein kinase complex and weaken its feedback inhibition of glucose. This would lead to sustained expression of high-affinity transport proteins, thereby increasing sugar consumption to provide substrates and energy for carotenoid synthesis.
4. Conclusions
In this study, we generated a near chromosome-level genome assembly of R. mucilaginosa GDMCC 2.30 using PacBio Sequel II and Illumina NovaSeq 6000 sequencing data. This genome contained 18 scaffolds and a circular mitochondrial genome with a total size of 20.31 Mb and a GC content of 60.52%. The mitochondrial genome comprised 40,152 bp with a GC content of 40.59%. A total of 1714 BUSCO genes (97.1%) were identified, and most scaffold ends contained telomere sequences, indicating the data quality was high, and enabling further analysis of the biological evolution and functional genomics of the strain. Through in-depth analysis and sorting of carotenoid-related gene clusters and families, the carotenoid biosynthesis pathway in the strain was inferred to start from sugar metabolism to mevalonate metabolism, and the final product of mevalonate metabolism (GGPP) is a precursor for various carotenoids formed through the activity of crucial enzymes encoded by genes such as CrtI, CrtYB, CrtS, and CrtR. The mutant JH-R23, screened after loading GDMCC 2.30 on the “New Generation Manned Spacecraft Test Ship”, was resequenced and comparative genomic analysis showed that an exon mutation disrupted the glycogen-binding domain of Gal83, thereby affecting the activity of the Snf1 protein kinase complex and weakening the feedback inhibition of glucose catabolism. These changes would lead to sustained expression of high-affinity transport proteins, thereby enhancing sugar consumption to provide substrates and energy for carotenoid synthesis. Multiple synonymous mutations of 3-oxoacyl-reductase and GTPase were additional important factors that would contribute to the increase in carotenoid production, establishing the genetic foundation for elevated carotenoid production.
Supplementary Materials
The following supporting information can be downloaded at: https://doi.org/10.5281/zenodo.10431767, Figure S1. IGV-GSAman software correction schematic. Figure S2: Evaluation of R. mucilaginosa genome size and ploidy by k-mer analysis. Figure S3: R. mucilaginosa GDMCC 2.30 gene annotation status. Figure S4: R. mucilaginosa GDMCC 2.30 mitochondrion alignment status. Figure S5: Phylogenetic relationships, protein structural domains, and motif structures of specific gene families. Table S1: Table of Species in Comparative Genomic Analysis. Table S2: Summary of sequencing data of the R. mucilaginosa GDMCC 2.30 genome. Table S3: Summary of BUSCO’s completeness analysis of R. mucilaginosa GDMCC 2.30 genome. Table S4: Annotation Statistics of the R. mucilaginosa GDMCC 2.30 genome. Table S5: List of Genes used in Figure 2B and its TPM. Table S6: Amino acid sequences for the CrtS gene family (OG00000414) and the CrtR gene family (OG00004469) for each species. Table S7: SNP/Indel analysis results in GDMCC 2.30 vs. JH-R23. Table S8: Statistics for gene family inference and gene counts for each family and each species. Table S9: GO enrichment results of species-specific genes of the R. mucilaginosa JH-R23. Table S10: GO/KEGG enrichment results of significantly expanded genes in R. mucilaginosa JH-R23.
Author Contributions
Conceptualization, methodology, visualization, and writing—original draft preparation, J.H.; conceptualization, methodology, and formal analysis, S.Y.; supervision, writing—review and editing, funding acquisition, project administration, and resources, H.J. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the Key-Area Research and Development Program of Guangdong Province 2018B020206001.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
This project has been deposited at NCBI under the accession PRJNA1034680.
Acknowledgments
We gratefully acknowledge the WinnerBio Bioinformatics Platform for assistance with the bioinformatics and for helpful discussions. We thank Robert McKenzie from Liwen Bianji (Edanz) (www.liwenbianji.cn, accessed on 31 October 2023) for editing a draft of this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Rodriguez-Concepcion, M.; Avalos, J.; Bonet, M.L.; Boronat, A.; Gomez-Gomez, L.; Hornero-Mendez, D.; Limon, M.C.; Melendez-Martinez, A.J.; Olmedilla-Alonso, B.; Palou, A.; et al. A global perspective on carotenoids: Metabolism, biotechnology, and benefits for nutrition and health. Prog. Lipid Res. 2018, 70, 62–93. [Google Scholar] [CrossRef] [PubMed]
- Frengova, G.I.; Beshkova, D.M. Carotenoids from Rhodotorula and Phaffia: Yeasts of biotechnological importance. J. Ind. Microbiol. Biotechnol. 2009, 36, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Kot, A.M.; Blazejak, S.; Gientka, I.; Kieliszek, M.; Brys, J. Torulene and torularhodin: “new” fungal carotenoids for industry? Microb. Cell Fact. 2018, 17, 49. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.G.; Otero, P.; Echave, J.; Carreira-Casais, A.; Chamorro, F.; Collazo, N.; Jaboui, A.; Lourenco-Lopes, C.; Simal-Gandara, J.; Prieto, M.A. Xanthophylls from the Sea: Algae as Source of Bioactive Carotenoids. Mar. Drugs 2021, 19, 188. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-T.; Yang, C.-F. Using strain Rhodotorula mucilaginosa to produce carotenoids using food wastes. J. Taiwan Inst. Chem. Eng. 2016, 61, 270–275. [Google Scholar] [CrossRef]
- Kot, A.M.; Blazejak, S.; Kieliszek, M.; Gientka, I.; Brys, J. Simultaneous Production of Lipids and Carotenoids by the Red Yeast Rhodotorula from Waste Glycerol Fraction and Potato Wastewater. Appl. Biochem. Biotechnol. 2019, 189, 589–607. [Google Scholar] [CrossRef] [PubMed]
- Dias Rodrigues, T.V.; Amore, T.D.; Teixeira, E.C.; de Medeiros Burkert, J.F. Carotenoid Production by Rhodotorula mucilaginosa in Batch and Fed-Batch Fermentation Using Agroindustrial Byproducts. Food Technol. Biotechnol. 2019, 57, 388–398. [Google Scholar] [CrossRef]
- Li, Z.; Li, C.; Cheng, P.; Yu, G. Rhodotorula mucilaginosa-alternative sources of natural carotenoids, lipids, and enzymes for industrial use. Heliyon 2022, 8, e11505. [Google Scholar] [CrossRef]
- Cutzu, R.; Coi, A.; Rosso, F.; Bardi, L.; Ciani, M.; Budroni, M.; Zara, G.; Zara, S.; Mannazzu, I. From crude glycerol to carotenoids by using a Rhodotorula glutinis mutant. World J. Microbiol. Biotechnol. 2013, 29, 1009–1017. [Google Scholar] [CrossRef]
- Nasrabadi, M.R.N.; Razavi, S.H. Optimization of β-carotene production by a mutant of the lactose-positive yeast Rhodotorula acheniorum from whey ultrafiltrate. Food Sci. Biotechnol. 2011, 20, 445–454. [Google Scholar] [CrossRef]
- Elfeky, N.; Elmahmoudy, M.; Zhang, Y.; Guo, J.; Bao, Y. Lipid and Carotenoid Production by Rhodotorula glutinis with a Combined Cultivation Mode of Nitrogen, Sulfur, and Aluminium Stress. Appl. Sci. 2019, 9, 2444. [Google Scholar] [CrossRef]
- Yang, S.; Jian, H.; Wei, F. Optimization of culture conditions for astaxanthin production by space mutation strain JH-R23. Food Sci. Technol. 2021, 46, 1–8. [Google Scholar]
- Daudu, R.; Parker, C.W.; Singh, N.K.; Wood, J.M.; Debieu, M.; O’Hara, N.B.; Mason, C.E.; Venkateswaran, K. Draft Genome Sequences of Rhodotorula mucilaginosa Strains Isolated from the International Space Station. Microbiol. Resour. Announc. 2020, 9, e00570-20. [Google Scholar] [CrossRef] [PubMed]
- Pilo, P.; Tiley, A.M.M.; Lawless, C.; Karki, S.J.; Burke, J.; Feechan, A. A rapid fungal DNA extraction method suitable for PCR screening fungal mutants, infected plant tissue and spore trap samples. Physiol. Mol. Plant Pathol. 2022, 117, 101758. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Marcais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Hu, J.; Fan, J.; Sun, Z.; Liu, S. NextPolish: A fast and efficient genome polishing tool for long-read assembly. Bioinformatics 2020, 36, 2253–2255. [Google Scholar] [CrossRef]
- Shen, Z.-Q.; Jiang, J.-H.; Li, C.-T.; Li, Y.; Zhou, L.-W. Genome Re-Annotation and Transcriptome Analyses of Sanghuangporus sanghuang. J. Fungi 2023, 9, 505. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Brůna, T.; Hoff, K.J.; Lomsadze, A.; Stanke, M.; Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 2021, 3, lqaa108. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 25, 4.10.1–4.10.14. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Cui, X.; Lu, Z.; Wang, S.; Jing-Yan Wang, J.; Gao, X. CMsearch: Simultaneous exploration of protein sequence space and structure space improves not only protein homology detection but also protein structure prediction. Bioinformatics 2016, 32, i332–i340. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Poczai, P.; Hyvonen, J.; Tang, J.; Amiryousefi, A. Chloroplot: An Online Program for the Versatile Plotting of Organelle Genomes. Front. Genet. 2020, 11, 576124. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- De Bie, T.; Cristianini, N.; Demuth, J.P.; Hahn, M.W. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Augustijn, H.E.; Reitz, Z.L.; Biermann, F.; Alanjary, M.; Fetter, A.; Terlouw, B.R.; Metcalf, W.W.; Helfrich, E.J.N.; et al. antiSMASH 7.0: New and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 2023, 51, W46–W50. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Bolchacova, E.; Voigt, K.; Crous, P.W.; et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.Q.; Xu, X.; Jiang, L.; Zhang, Z.D.; Huang, H. Optimization of fermentation conditions for carotenoid production in the radiation-resistant strain Deinococcus xibeiensis R13. Bioprocess Biosyst. Eng. 2019, 42, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, J.C. Carotenoids as Colorants and Vitamin A Precursors; Academic Press: Cambridge, MA, USA, 1981. [Google Scholar]
- Watabe, Y.; Takahashi, S. Production of Enhanced Carotenoid-Producing Strains of the Yeast Rhodotorula gracilis Using the Antibiotic Zeocin. Appl. Biochem. Biotechnol. 2023, 195, 7889–7897. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.M.; Thomas, B.N.; Cavanaugh, N.T.; Morales, G.H.; Mayers, A.N.; Savka, M.A.; Hudson, A.O. Whole genome sequencing of Rhodotorula mucilaginosa isolated from the chewing stick (Distemonanthus benthamianus): Insights into Rhodotorula phylogeny, mitogenome dynamics and carotenoid biosynthesis. PeerJ 2017, 5, e4030. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.; Paul, K.; Saha, C.; Mukherjee, G.; Nag, M.; Ghosh, S.; Das, A.; Seal, A.; Tripathy, S. A unique life-strategy of an endophytic yeast Rhodotorula mucilaginosa JGTA-S1-a comparative genomics viewpoint. DNA Res. 2019, 26, 131–146. [Google Scholar] [CrossRef]
- Tang, W.; Wang, Y.; Cai, Y.; Liu, S.; Zhang, J.; He, Z. Genome Sequence of a Marine Carotenoid Producing Yeast Rhodotorula mucilaginosa CYJ03. J. Ocean. Univ. China 2020, 19, 466–472. [Google Scholar] [CrossRef]
- Deligios, M.; Fraumene, C.; Abbondio, M.; Mannazzu, I.; Tanca, A.; Addis, M.F.; Uzzau, S. Draft Genome Sequence of Rhodotorula mucilaginosa, an Emergent Opportunistic Pathogen. Genome Announc. 2015, 3, e00201-15. [Google Scholar] [CrossRef]
- Marcisauskas, S.; Kim, Y.; Blasche, S.; Patil, K.R.; Ji, B.; Nielsen, J. Draft Genome Sequences of Five Fungal Strains Isolated from Kefir. Microbiol. Resour. Announc. 2021, 10, e0019521. [Google Scholar] [CrossRef] [PubMed]
- Sundararaju, S.; Salah, H.; Ibrahim, E.B.; Perez-Lopez, A.; Abid, F.B.; Tsui, C.K.M. Draft Genome Sequence of Rhodotorula mucilaginosa from an Adult Patient in Qatar. Microbiol. Resour. Announc. 2021, 10, e0072521. [Google Scholar] [CrossRef] [PubMed]
- Sandmann, G. Carotenoids and Their Biosynthesis in Fungi. Molecules 2022, 27, 1431. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Li, C.; Zhang, N.; Li, B. Integrative analysis of genomic and metabolomic data reveals key metabolic pathways involved in lipid and carotenoid biosynthesis in oleaginous red yeast Rhodosporidiobolus odoratus XQR. Microbiol. Res. 2023, 270, 127339. [Google Scholar] [CrossRef] [PubMed]
- Bo, S.; Ni, X.; Guo, J.; Liu, Z.; Wang, X.; Sheng, Y.; Zhang, G.; Yang, J. Carotenoid Biosynthesis: Genome-Wide Profiling, Pathway Identification in Rhodotorula glutinis X-20, and High-Level Production. Front. Nutr. 2022, 9, 918240. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, V.; Rodriguez-Saiz, M.; de la Fuente, J.L.; Gudina, E.J.; Godio, R.P.; Martin, J.F.; Barredo, J.L. The crtS gene of Xanthophyllomyces dendrorhous encodes a novel cytochrome-P450 hydroxylase involved in the conversion of beta-carotene into astaxanthin and other xanthophylls. Fungal. Genet. Biol. 2006, 43, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Alcaino, J.; Barahona, S.; Carmona, M.; Lozano, C.; Marcoleta, A.; Niklitschek, M.; Sepulveda, D.; Baeza, M.; Cifuentes, V. Cloning of the cytochrome p450 reductase (crtR) gene and its involvement in the astaxanthin biosynthesis of Xanthophyllomyces dendrorhous. BMC Microbiol. 2008, 8, 169. [Google Scholar] [CrossRef]
- Ukibe, K.; Hashida, K.; Yoshida, N.; Takagi, H. Metabolic engineering of Saccharomyces cerevisiae for astaxanthin production and oxidative stress tolerance. Appl. Environ. Microbiol. 2009, 75, 7205–7211. [Google Scholar] [CrossRef]
- Issa, S.; Alhajali, A.; Alamir, L. Improving carotenoid pigments production in Rhodotorula mucilaginosa using UV irradiation. Int. Food Res. J. 2016, 23, 873–878. [Google Scholar]
- Vincent, O.; Townley, R.; Kuchin, S.; Carlson, M. Subcellular localization of the Snf1 kinase is regulated by specific beta subunits and a novel glucose signaling mechanism. Genes Dev. 2001, 15, 1104–1114. [Google Scholar] [CrossRef]
- Papapetridis, I.; Verhoeven, M.D.; Wiersma, S.J.; Goudriaan, M.; van Maris, A.J.A.; Pronk, J.T. Laboratory evolution for forced glucose-xylose co-consumption enables identification of mutations that improve mixed-sugar fermentation by xylose-fermenting Saccharomyces cerevisiae. FEMS Yeast Res. 2018, 18, foy056. [Google Scholar] [CrossRef] [PubMed]
- Vincent, O.; Carlson, M. Gal83 mediates the interaction of the Snf1 kinase complex with the transcription activator Sip4. EMBO J. 1999, 18, 6672–6681. [Google Scholar] [CrossRef] [PubMed]
- Wiatrowski, H.A.; Van Denderen, B.J.; Berkey, C.D.; Kemp, B.E.; Stapleton, D.; Carlson, M. Mutations in the gal83 glycogen-binding domain activate the snf1/gal83 kinase pathway by a glycogen-independent mechanism. Mol. Cell Biol. 2004, 24, 352–361. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, W.; Zhao, Y.; Bai, N.; Zhang, K.-Q.; Yang, J. AMPK is involved in regulating the utilization of carbon sources, conidiation, pathogenicity, and stress response of the nematode-trapping fungus Arthrobotrys oligospora. Microbiol. Spectr. 2022, 10, e02225-22. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Zhang, Y.; Wang, J.; Lv, C.; Chen, C. BcMtg2 is required for multiple stress tolerance, vegetative development and virulence in Botrytis cinerea. Sci. Rep. 2016, 6, 28673. [Google Scholar] [CrossRef] [PubMed]
- Pronk, J.T.; Yde Steensma, H.; Van Dijken, J.P. Pyruvate Metabolism in Saccharomyces cerevisiae. Yeast 1996, 12, 1607–1633. [Google Scholar] [CrossRef]
- Vadali, R.V.; Bennett, G.N.; San, K.Y. Cofactor engineering of intracellular CoA/acetyl-CoA and its effect on metabolic flux redistribution in Escherichia coli. Metab. Eng. 2004, 6, 133–139. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).