

Terminal-Enhanced Polymerization in the Biosynthesis of Polysialic Acid
 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids and Bacterial Strains
2.2. Media, and Culture Conditions
2.3. Deletion of Chromosomal Genes Using CRISPR/Cas9 System
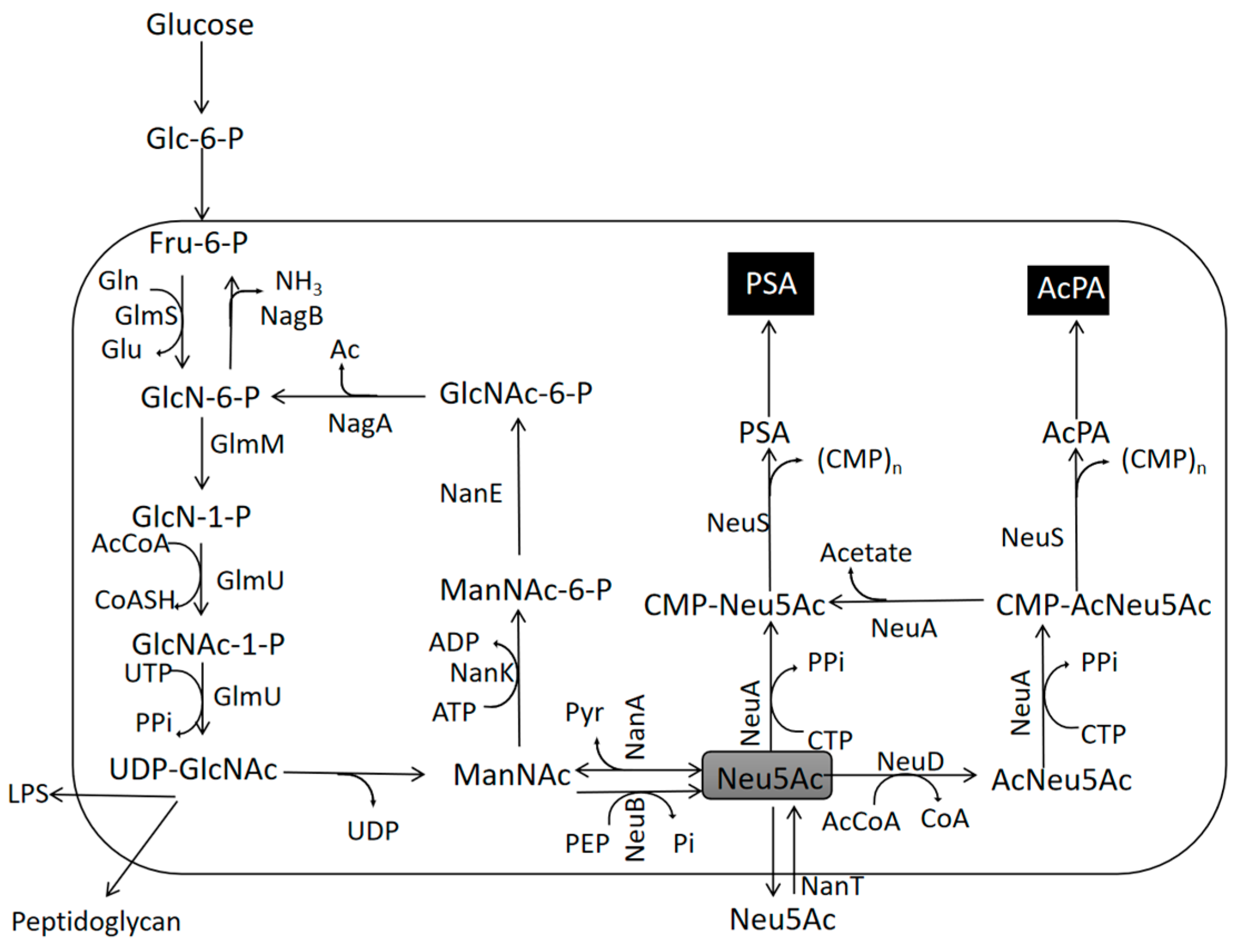
2.4. Terminal Polymerization Pathway Construction and Assembly
2.5. Quantitative Measurement of Polymerized PSA
2.5.1. Measurement of PSA Based on Resorcinol Colorimetric Assay
2.5.2. Measurement of Monomer Neu5Ac Based on HPLC
2.5.3. Calculation of Polymerization to Produce Polysialic Acid
2.6. Fluorescence Quantitative PCR Methods
2.7. Fermentation Culture Conditions
3. Results
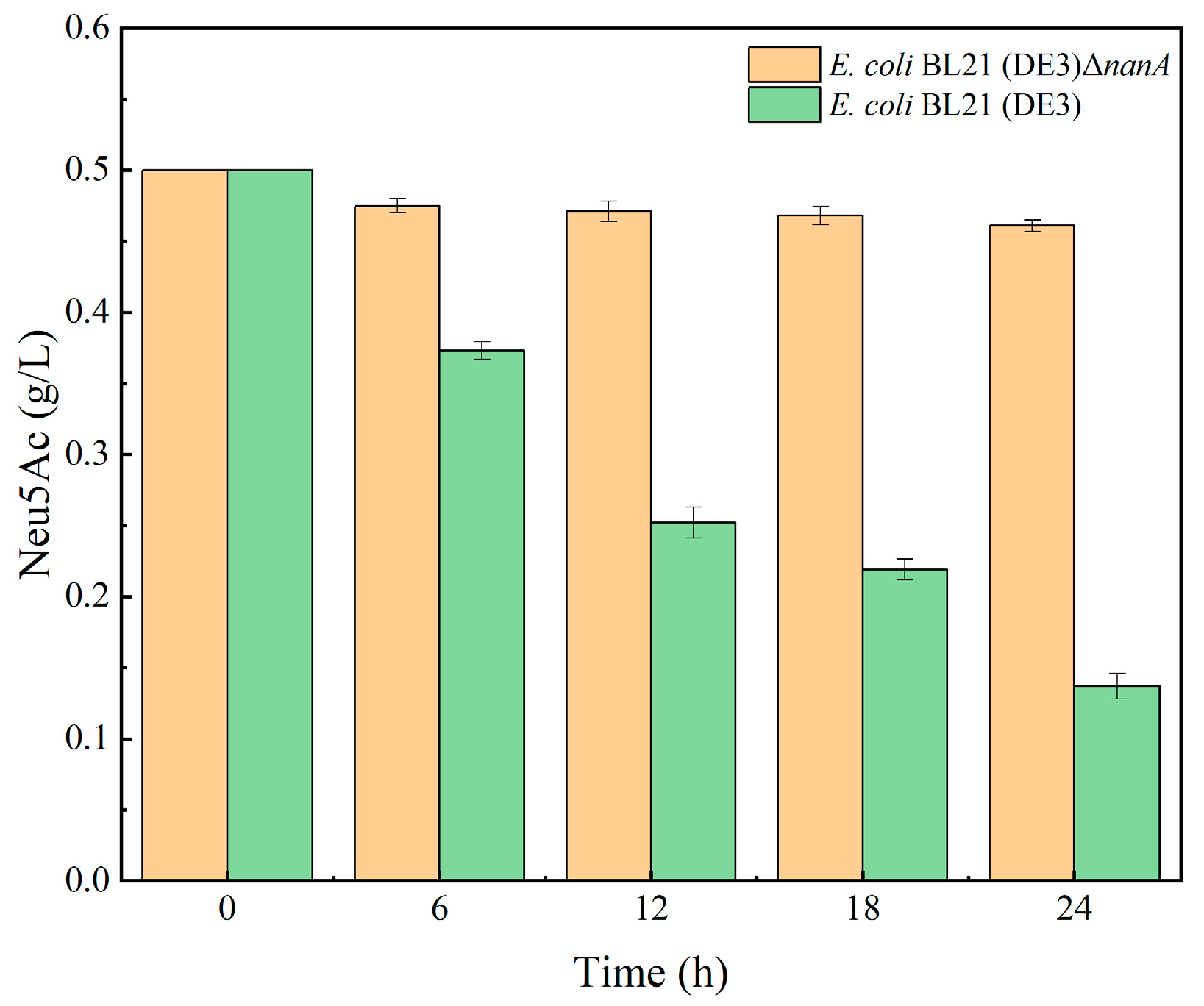
3.1. Phenotypic Verification of nanA Gene Knockout
3.2. Construction and Validation of Recombinant Bacteria
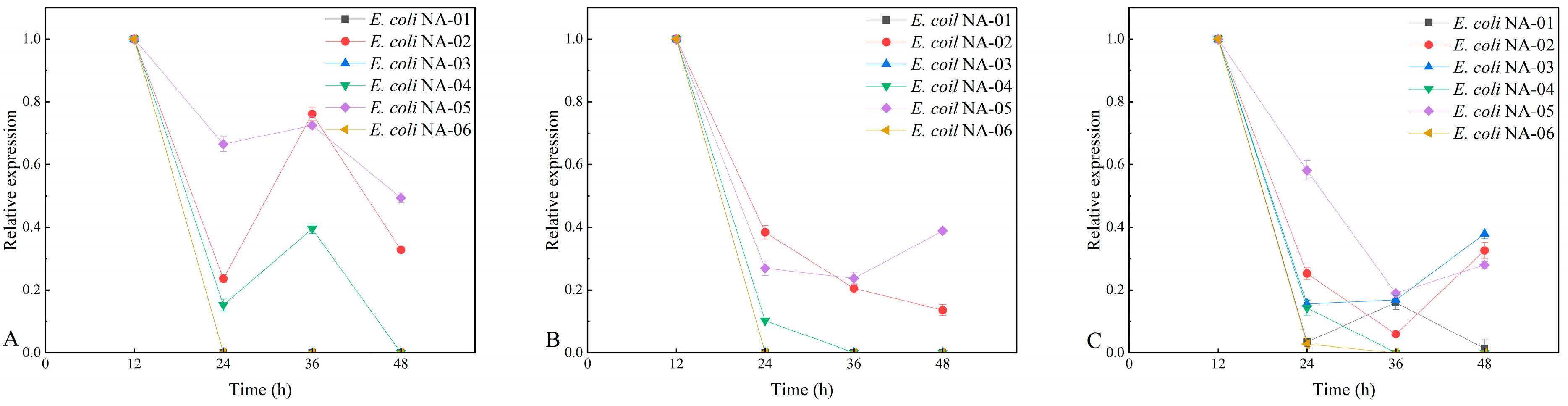
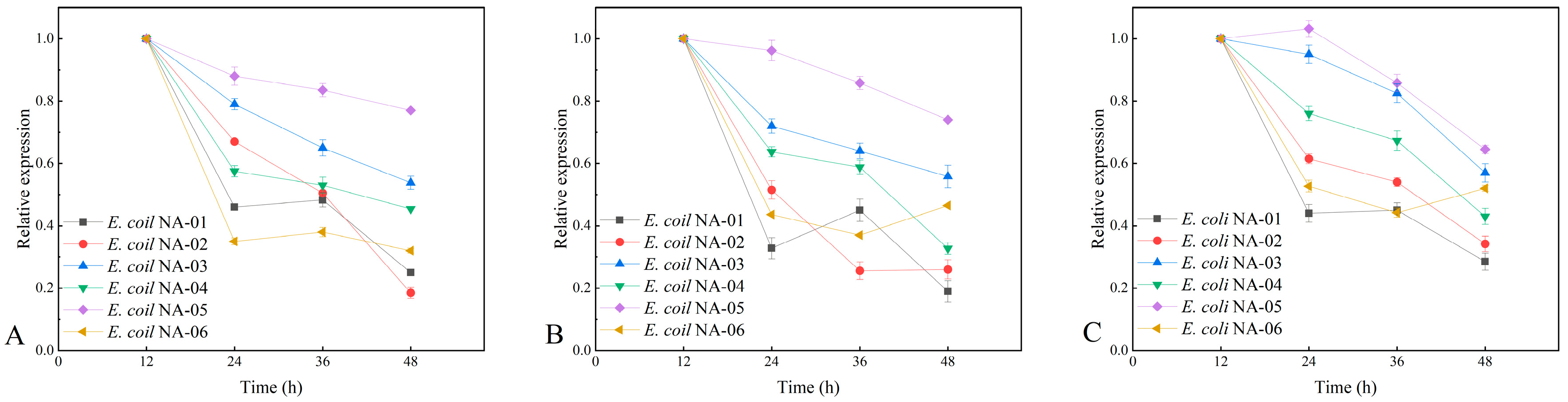
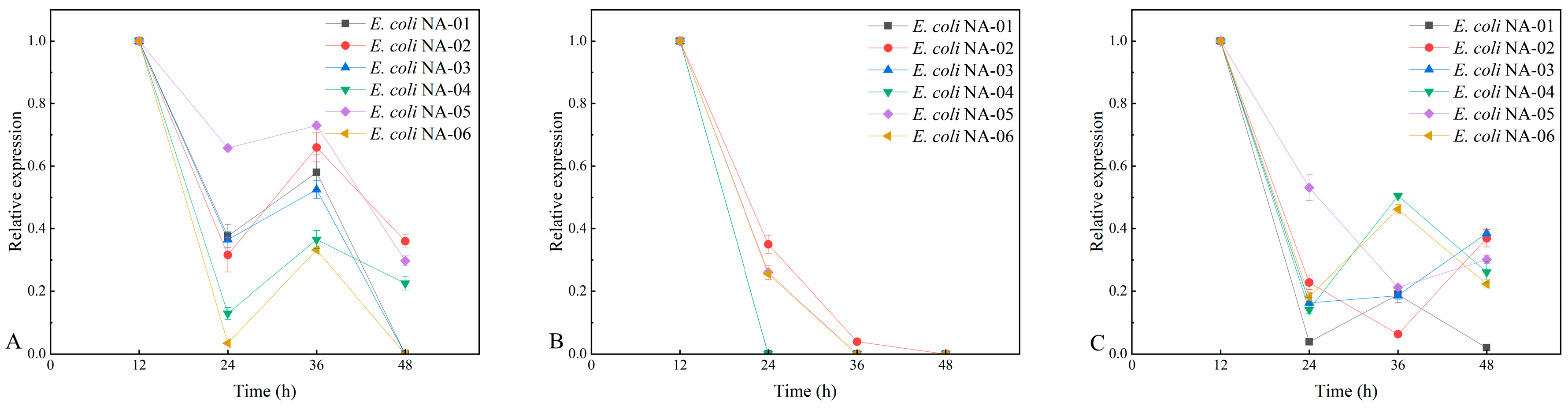
3.3. Plasmid Stability at Different Resistance Concentrations
3.3.1. Plasmid Stability at Standard Antibiotic Concentrations
3.3.2. Plasmid Stability at 50% Antibiotic Concentration
3.3.3. Plasmid Stability at Antibiotic-Free Concentrations
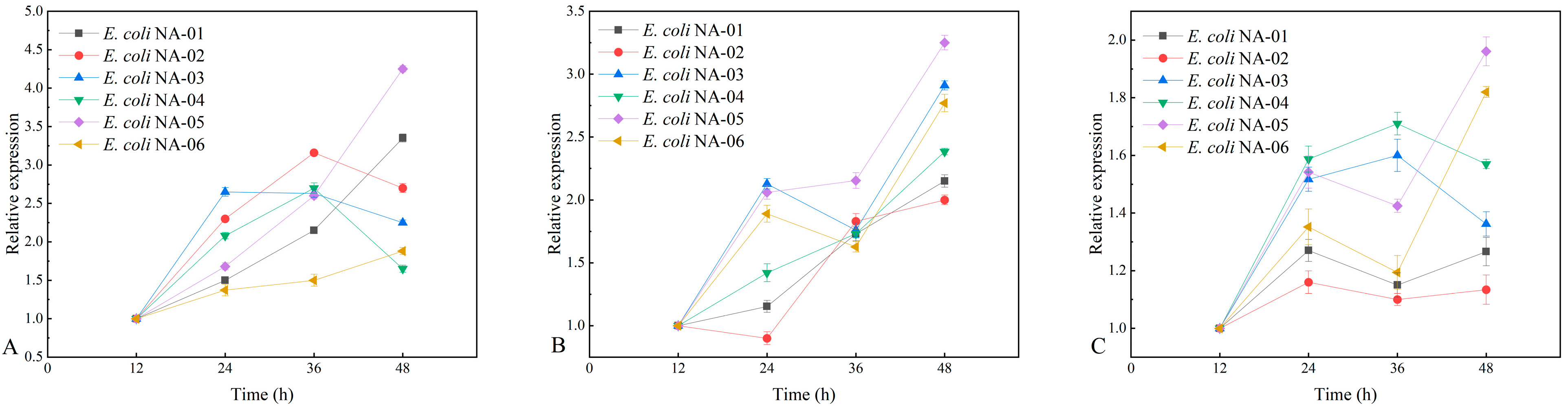
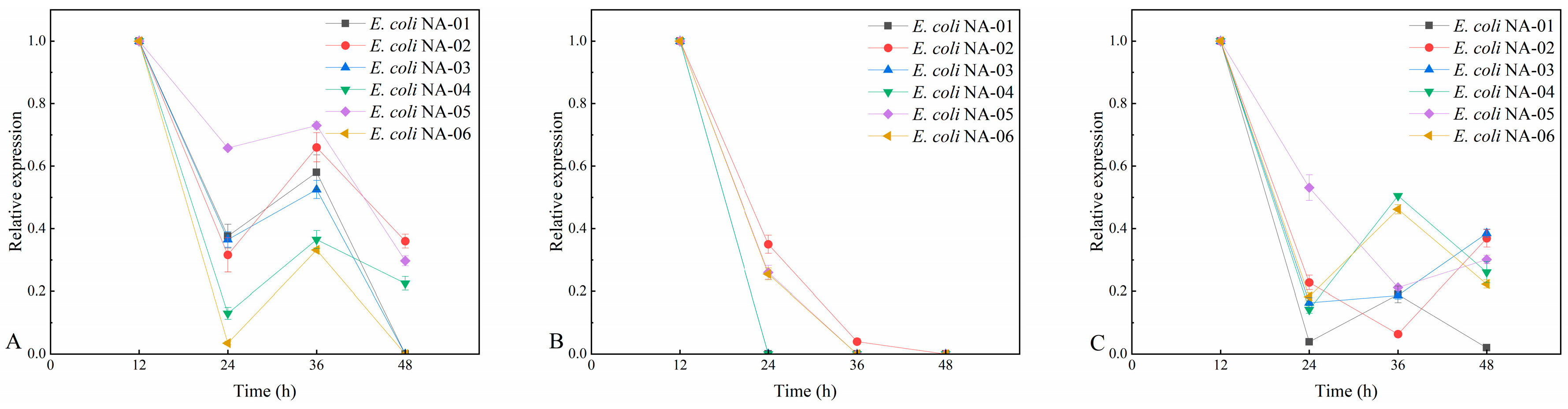
3.4. Association between Expression of Key Genes and Yield
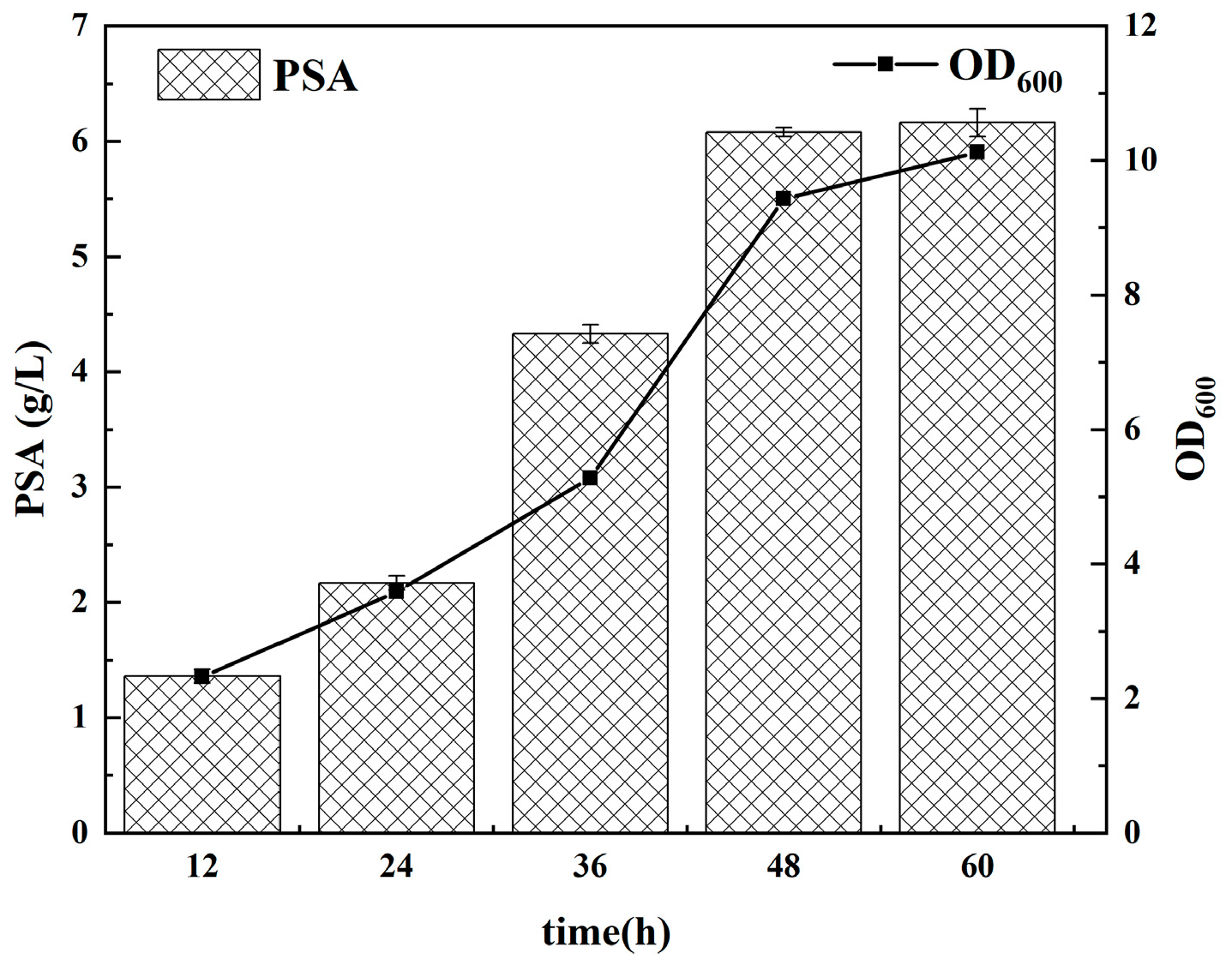
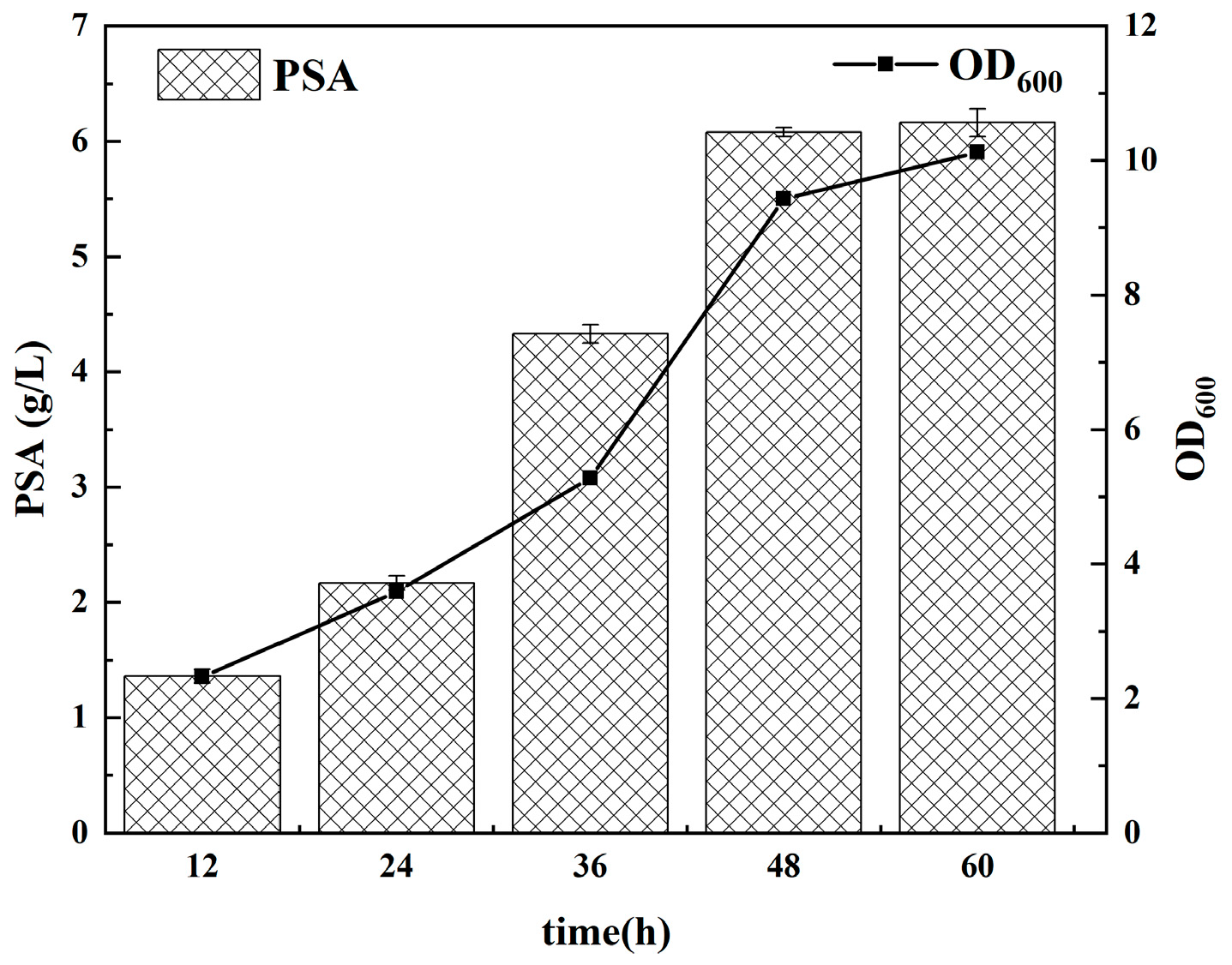
3.5. PSA Polymerization in Fermenter Level
4. Discussion
4.1. The Potential of Neu5Ac-Based PSA Synthesis and the Way Forward
4.2. Compatibility of Expression Vectors in Synthetic Biology
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McGowen, M.M.; Vionnet, J.; Vann, W.F. Elongation of alternating α2,8/2,9 polysialic acid by the Escherichia coli K92 polysialyltransferase. Glycobiology 2001, 11, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Azurmendi, H.F.; Vionnet, J.; Wrightson, L.; Trinh, L.B.; Shiloach, J.; Freedberg, D.I. Extracellular structure of polysialic acid explored by on cell solution NMR. Proc. Natl. Acad. Sci. USA 2007, 104, 11557–11561. [Google Scholar] [CrossRef]
- Brusés, J.L.; Rutishauser, U. Roles, regulation, and mechanism of polysialic acid function during neural development. Biochimie 2001, 83, 635–643. [Google Scholar] [CrossRef] [PubMed]
- El Maarouf, A.; Petridis, A.K.; Rutishauser, U. Use of polysialic acid in repair of the central nervous system. Proc. Natl. Acad. Sci. USA 2006, 103, 16989–16994. [Google Scholar] [CrossRef]
- Mühlenhoff, M.; Eckhardt, M.; Gerardy-Schahn, R. Polysialic acid: Three-dimensional structure, biosynthesis and function. Curr. Opin. Struct. Biol. 1998, 8, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.-X.; Qiao, Y.; Shi, B.; Tao, Y. Polysialic acid biosynthesis and production in Escherichia coli: Current state and perspectives. Appl. Microbiol. Biotechnol. 2015, 100, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.Y.; Wang, S.Z.; Li, G.S.; Zhan, X.B.; Lin, C.C.; Wu, J.R.; Zhu, L. A new polysialic acid production process based on dual-stage pH control and fed-batch fermentation for higher yield and resulting high molecular weight product. Appl. Microbiol. Biotechnol. 2013, 97, 2405–2412. [Google Scholar] [CrossRef]
- Ferrero, M.A.; Aparicio, L.R. Biosynthesis and production of polysialic acids in bacteria. Appl. Microbiol. Biotechnol. 2010, 86, 1621–1635. [Google Scholar] [CrossRef]
- Ishikawa, M.; Koizumi, S. Microbial production of N-acetylneuraminic acid by genetically engineered Escherichia coli. Carbohydr. Res. 2010, 345, 2605–2609. [Google Scholar] [CrossRef]
- Roberts, I.S. The biochemistry and genetics of capsular polysaccharide production in bacteria. Annu. Rev. Microbiol. 1996, 50, 285–315. [Google Scholar] [CrossRef]
- Whitfield, C.; Roberts, I.S. Structure, assembly and regulation of expression of capsules in Escherichia coli. Mol. Microbiol. 1999, 31, 1307–1319. [Google Scholar] [CrossRef]
- Daines, D.A.; Silver, R.P. Evidence for Multimerization of Neu Proteins Involved in Polysialic Acid Synthesis in Escherichia coli K1 Using Improved LexA-Based Vectors. J. Bacteriol. 2000, 182, 5267–5270. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Tian, R.Z.; Shen, Q.Y.; Liu, Y.F.; Liu, L.; Li, J.H.; Du, G.C. Pathway Engineering of Bacillus subtilis for Enhanced N-Acetylneuraminic Acid Production via Whole-Cell Biocatalysis. Biotechnol. J. 2019, 14, 8. [Google Scholar] [CrossRef]
- Lundgren, B.R.; Boddy, C.N. Sialic acid and N-acyl sialic acid analog production by fermentation of metabolically and genetically engineered Escherichia coli. Org. Biomol. Chem. 2007, 5, 1903–1909. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Gu, P.F.; Wang, Y.; Li, Y.K.; Yang, F.; Wang, Q.; Qi, Q.S. Engineering of an N-acetylneuraminic acid synthetic pathway in Escherichia coli. Metab. Eng. 2012, 14, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Yu, H.; Cao, H.Z.; Lau, K.; Muthana, S.; Tiwari, V.K.; Son, B.; Chen, X. Pasteurella multocida sialic acid aldolase: A promising biocatalyst. Appl. Microbiol. Biotechnol. 2008, 79, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.X.; Zhang, Z.J.; Liu, W.F.; Dong, Z.Y.; Tao, Y. Enhanced production of N-acetyl-D-neuraminic acid by multi-approach whole-cell biocatalyst. Appl. Microbiol. Biotechnol. 2013, 97, 4775–4784. [Google Scholar] [CrossRef] [PubMed]
- Song, L.L.; Zhou, H.; Cai, X.H.; Li, C.Y.; Liang, J.N.; Jin, C. NeuA O-acetylesterase activity is specific for CMP-activated O-acetyl sialic acid in Streptococcus suis serotype 2. Biochem. Biophys. Res. Commun. 2011, 410, 212–217. [Google Scholar] [CrossRef]
- Zapata, G.; Vann, W.F.; Aaronson, W.; Lewis, M.S.; Moos, M. Sequence of the Cloned Escherichia coli K1 CMP-N-acetylneuraminic Acid Synthetase Gene. J. Biol. Chem. 1989, 264, 14769–14774. [Google Scholar] [CrossRef]
- Steenbergen, S.M.; Lee, Y.C.; Vann, W.F.; Vionnet, J.; Wright, L.F.; Vimr, E.R. Separate pathways for O acetylation of polymeric and monomeric sialic acidsand identification of sialyl O-acetyl esterase in Escherichia coli K1. J. Bacteriol. 2006, 188, 6195–6206. [Google Scholar] [CrossRef]
- Zhu, Y.Y.; Wan, L.; Meng, J.W.; Luo, G.C.; Chen, G.; Wu, H.; Zhang, W.L.; Mu, W.M. Metabolic Engineering of Escherichia coli for Lacto-N-triose II Production with High Productivity. J. Agric. Food Chem. 2021, 69, 3702–3711. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chen, B.; Duan, C.L.; Sun, B.B.; Yang, J.J.; Yang, S. Multigene Editing in the Escherichia coli Genome via the CRISPR-Cas9 System. Appl. Environ. Microbiol. 2015, 81, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Svennerholm, L. Quantitive estimation of sialic acids: II. A colorimetric resorcinol-hydrochloric acid method. Biochim. Et Biophys. Acta 1957, 24, 604–611. [Google Scholar] [CrossRef]
- Vimr, E.R.; Troy, F.A. Regulation of sialic acid metabolism in Escherichia coli: Role of N-acylneuraminate pyruvate-lyase. J. Bacteriol. 1985, 164, 854–860. [Google Scholar] [CrossRef]
- Ajikumar, P.K.; Xiao, W.H.; Tyo, K.E.J.; Wang, Y.; Simeon, F.; Leonard, E.; Mucha, O.; Phon, T.H.; Pfeifer, B.; Stephanopoulos, G. Isoprenoid Pathway Optimization for Taxol Precursor Overproduction in Escherichia coli. Science 2010, 330, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Simon, E.S.; Bednarski, M.D.; Whitesides, G.M. Synthesis of CMP-NeuAc from N-acetylglucosamine—Generation of CTP from CMP Using Adenylate Kinase. J. Am. Chem. Soc. 1988, 110, 7159–7163. [Google Scholar] [CrossRef]
- Wu, J.J.; Du, G.C.; Zhou, J.W.; Chen, J. Metabolic engineering of Escherichia coli for (2S)-pinocembrin production from glucose by a modular metabolic strategy. Metab. Eng. 2013, 16, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Tao, Y.; Jin, C.; Xu, Y.; Lin, B.X. Enhanced production of polysialic acid by metabolic engineering of Escherichia coli. Appl. Microbiol. Biotechnol. 2015, 99, 2603–2611. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Characteristics |
|---|---|
| E. coli DH5α | F-φ80, lac ZΔM15, Δ(lacZYA-arg F), U169, endA1, recA1, hsdR17(rk−,mk+), supE44λ-, thi-1, gyrA96, relA1, phoA |
| E. coli BL21 (DE3) | F-, ompT, hsdSB(rB-mB-), dcm, gal,(DE3) |
| E. coli BL21 (DE3)ΔnanA | E. coli BL21 (DE3) with nanA knocked out |
| E. coli NA-01 | As E. coli BL21 (DE3)ΔnanA with plasmids pRSF-UA, pACYC-UD, and pET-US |
| E. coli NA-02 | As E. coli BL21 (DE3)ΔnanA with plasmids pRSF-UA, pACYC-US, and pET-UD |
| E. coli NA-03 | As E. coli BL21 (DE3)ΔnanA with plasmids pRSF-UD, pACYC-UA, and pET-US |
| E. coli NA-04 | As E. coli BL21 (DE3)ΔnanA with plasmids pRSF-UD, pACYC-US, and pET-UA |
| E. coli NA-05 | As E. coli BL21 (DE3)ΔnanA with plasmids pRSF-US, pACYC-UA, and pET-UD |
| E. coli NA-06 | As E. coli BL21 (DE3)ΔnanA with plasmids pRSF-US, pACYC-UD, and pET-UA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Chang, H.; Liu, X.; Zhao, H.; Guo, J.; Peng, S.; Wang, W.; Zhu, D.; Liu, X. Terminal-Enhanced Polymerization in the Biosynthesis of Polysialic Acid. Fermentation 2024, 10, 64. https://doi.org/10.3390/fermentation10010064
Wang C, Chang H, Liu X, Zhao H, Guo J, Peng S, Wang W, Zhu D, Liu X. Terminal-Enhanced Polymerization in the Biosynthesis of Polysialic Acid. Fermentation. 2024; 10(1):64. https://doi.org/10.3390/fermentation10010064
Chicago/Turabian StyleWang, Chongchuan, Huanan Chang, Xiaomeng Liu, Haiyang Zhao, Jianing Guo, Shuo Peng, Wenhao Wang, Deqiang Zhu, and Xinli Liu. 2024. "Terminal-Enhanced Polymerization in the Biosynthesis of Polysialic Acid" Fermentation 10, no. 1: 64. https://doi.org/10.3390/fermentation10010064
APA StyleWang, C., Chang, H., Liu, X., Zhao, H., Guo, J., Peng, S., Wang, W., Zhu, D., & Liu, X. (2024). Terminal-Enhanced Polymerization in the Biosynthesis of Polysialic Acid. Fermentation, 10(1), 64. https://doi.org/10.3390/fermentation10010064