

Phosphorus/Sulfur-Enriched Reduced Graphene Oxide Papers Obtained from Recycled Graphite: Solid-State NMR Characterization and Electrochemical Performance for Energy Storage

 , and
, and
Abstract
1. Introduction
2. Experimental Methods
2.1. Samples Preparation
Materials
2.2. Characterization
2.2.1. X-ray Diffraction (XRD)
2.2.2. Solid-State 13C and 31P NMR
2.2.3. Fourier-Transform Infrared (FTIR) Spectroscopy
2.2.4. X-ray Fluorescence (XRF)
2.2.5. Electrochemical Characterization
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Brodie, B.C. On the Atomic Weight of Graphite. Philos. Trans. R. Soc. Lond. 1859, 149, 249–259. [Google Scholar]
- Pei, S.; Cheng, H.M. The Reduction of Graphene Oxide. Carbon 2012, 50, 3210–3228. [Google Scholar] [CrossRef]
- Huang, H.; Shi, H.; Das, P.; Qin, J.; Li, Y.; Wang, X.; Su, F.; Wen, P.; Li, S.; Lu, P.; et al. The Chemistry and Promising Applications of Graphene and Porous Graphene Materials. Adv. Funct. Mater. 2020, 30, 1909035. [Google Scholar] [CrossRef]
- Dreyer, D.R.; Park, S.; Bielawski, C.W.; Ruoff, R.S. The chemistry of graphene oxide. Chem. Soc. Rev. 2009, 39, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Brisebois, P.P.; Siaj, M. Harvesting Graphene Oxide—Years 1859 to 2019: A Review of Its Structure, Synthesis, Properties and Exfoliation. J. Mater. Chem. C 2020, 8, 1517–1547. [Google Scholar] [CrossRef]
- Park, S.; An, J.; Potts, J.R.; Velamakanni, A.; Murali, S.; Ruoff, R.S. Hydrazine-Reduction of Graphite-and Graphene Oxide. Carbon 2011, 49, 3019–3023. [Google Scholar] [CrossRef]
- De Silva, K.K.H.; Huang, H.H.; Joshi, R.K.; Yoshimura, M. Chemical Reduction of Graphene Oxide Using Green Reductants. Carbon 2017, 119, 190–199. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhao, Z.; Chen, Y.; Hu, H.; Qiu, J. Low Temperature Plasma-Mediated Synthesis of Graphene Nanosheets for Supercapacitor Electrodes. J. Mater. Chem. 2012, 22, 6061–6066. [Google Scholar] [CrossRef]
- Ghadim, E.E.; Rashidi, N.; Kimiagar, S.; Akhavan, O.; Manouchehri, F.; Ghaderi, E. Pulsed Laser Irradiation for Environment Friendly Reduction of Graphene Oxide Suspensions. Appl. Surf. Sci. 2014, 301, 183–188. [Google Scholar] [CrossRef]
- Dumée, L.F.; Feng, C.; He, L.; Allioux, F.M.; Yi, Z.; Gao, W.; Banos, C.; Davies, J.B.; Kong, L. Tuning the Grade of Graphene: Gamma Ray Irradiation of Free-Standing Graphene Oxide Films in Gaseous Phase. Appl. Surf. Sci. 2014, 322, 126–135. [Google Scholar] [CrossRef]
- Okhay, O.; Gonçalves, G.; Tkach, A.; Dias, C.; Ventura, J.; Ribeiro Da Silva, M.F.; Valente Gonçalves, L.M.; Titus, E. Thin Film versus Paper-like Reduced Graphene Oxide: Comparative Study of Structural, Electrical, and Thermoelectrical Properties. J. Appl. Phys. 2016, 120, 051706. [Google Scholar] [CrossRef]
- Khan, F.; Khan, S.; Kamal, S.; Arshad, M. Recent Advances in Graphene Oxide and Reduced Graphene Oxide Based Nanocomposites for the Photodegradation of Dyes. J. Mater. Chem. C 2020, 8, 15940–15955. [Google Scholar] [CrossRef]
- Zhu, Y.; Murali, S.; Cai, W.; Li, X.; Suk, J.W.; Potts, J.R.; Ruoff, R.S. Graphene and Graphene Oxide: Synthesis, Properties, and Applications. Adv. Mater. 2010, 22, 3906–3924. [Google Scholar] [CrossRef]
- Valentini, C.; Montes-Garcia, V.; Livio, P.A.; Chudziak, T.; Raya, J.; Ciesielski, A.; Samorì, P. Tuning the electrical properties of graphene oxide through low-temperature thermal annealing. Nanoscale 2023, 15, 5743–5755. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.A.; Baek, J. Doped Graphene Supercapacitors. Nanotechnology 2015, 26, 492001. [Google Scholar] [CrossRef]
- Yu, X.; Feng, L.; Park, H.S. Highly Flexible Pseudocapacitors of Phosphorus-Incorporated Porous Reduced Graphene Oxide Films. J. Power Sources 2018, 390, 93–99. [Google Scholar] [CrossRef]
- Qiao, X.; Liao, S.; You, C.; Chen, R. Phosphorus and Nitrogen Dual Doped and Simultaneously Reduced Graphene Oxide with High Surface Area as Efficient Metal-Free Electrocatalyst for Oxygen Reduction. Catalysts 2015, 5, 981–991. [Google Scholar] [CrossRef]
- Ghafuri, H.; Talebi, M. Water-Soluble Phosphated Graphene: Preparation, Characterization, Catalytic Reactivity, and Adsorption Property. Ind. Eng. Chem. Res. 2016, 55, 2970–2982. [Google Scholar] [CrossRef]
- Niu, F.; Tao, L.-M.; Deng, Y.-C.; Wang, Q.-H.; Song, W.-G. Phosphorus Doped Graphene Nanosheets for Room Temperature NH3 Sensing. N. J. Chem. 2014, 38, 2269. [Google Scholar] [CrossRef]
- Sun, X.; Cheng, P.; Wang, H.; Xu, H.; Dang, L.; Liu, Z.; Lei, Z. Activation of Graphene Aerogel with Phosphoric Acid for Enhanced Electrocapacitive Performance. Carbon 2015, 92, 1–10. [Google Scholar] [CrossRef]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved Synthesis of Graphene Oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef] [PubMed]
- Hummers, W.S.; Offeman, R.E. Preparation of Graphitic Oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Chua, C.K.; Sofer, Z.; Pumera, M. Graphite Oxides: Effects of Permanganate and Chlorate Oxidants on the Oxygen Composition. Chem.-A Eur. J. 2012, 18, 13453–13459. [Google Scholar] [CrossRef] [PubMed]
- Costa, T.L.G.; Vieira, M.A.; Gonçalves, G.R.; Cipriano, D.F.; Lacerda Jr., V.; Gonçalves, A.S.; Scopel, W.L.; de Siervo, A.; Freitas, J.C.C. Combined computational and experimental study about the incorporation of phosphorus into the structure of graphene oxide. Phys. Chem. Chem. Phys. 2023, 25, 6927–6943. [Google Scholar] [CrossRef]
- Cory, D.G.; Ritchey, W.M. Suppression of Signals from the Probe in Bloch Decay Spectra. J. Magn. Reson. 1988, 32, 128–132. [Google Scholar] [CrossRef]
- Sanderson, K. Carbon Makes Super-Tough Paper. Nature 2007. [Google Scholar] [CrossRef]
- Eigler, S.; Dotzer, C.; Hof, F.; Bauer, W.; Hirsch, A. Sulfur Species in Graphene Oxide. Chem.-A Eur. J. 2013, 19, 9490–9496. [Google Scholar] [CrossRef]
- Farjadian, F.; Abbaspour, S.; Sadatlu, M.A.A.; Mirkiani, S.; Ghasemi, A.; Hoseini-Ghahfarokhi, M.; Mozaffari, N.; Karimi, M.; Hamblin, M.R. Recent Developments in Graphene and Graphene Oxide: Properties, Synthesis, and Modifications: A Review. ChemistrySelect 2020, 5, 10200–10219. [Google Scholar] [CrossRef]
- Moreno-Fernández, G.; Gómes-Urbano, J.L.; Enterría, M.; Cid, R.; López del Amo, J.M.; Mysyk, R.; Carriazo, D. Understanding enhanced charge storage of phosphorus-functionalized graphene in aqueous acidic electrolytes. Electrochim. Acta 2020, 361, 136985. [Google Scholar] [CrossRef]
- Dreyer, D.R.; Todd, A.D.; Bielawski, C.W. Harnessing the Chemistry of Graphene Oxide. Chem. Soc. Rev. 2014, 43, 5288–5301. [Google Scholar] [CrossRef]
- Aliyev, E.; Filiz, V.; Khan, M.M.; Lee, Y.J.; Abetz, C.; Abetz, V. Structural Characterization of Graphene Oxide: Surface Functional Groups and Fractionated Oxidative Debris. Nanomaterials 2019, 9, 1180. [Google Scholar] [CrossRef]
- Skákalová, V.; Kotrusz, P.; Jergel, M.; Susi, T.; Mittelberger, A.; Vretenár, V.; Šiffalovič, P.; Kotakoski, J.; Meyer, J.C.; Hulman, M. Chemical Oxidation of Graphite: Evolution of the Structure and Properties. J. Phys. Chem. C 2018, 122, 929–935. [Google Scholar] [CrossRef]
- Gao, W.; Alemany, L.B.; Ci, L.; Ajayan, P.M. New Insights into the Structure and Reduction of Graphite Oxide. Nat. Chem. 2009, 1, 403–408. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Riedl, T.; Lerf, A.; Klinowski, J. Solid-State NMR Studies of the Structure of Graphite Oxide. J. Phys. Chem. 1996, 100, 19954–19958. [Google Scholar] [CrossRef]
- Rawal, A.; Man, S.H.C.; Agarwal, V.; Yao, Y.; Thickett, S.C.; Zetterlund, P.B. Structural Complexity of Graphene Oxide: The Kirigami Model. ACS Appl. Mater. Interfaces 2021, 13, 18255–18263. [Google Scholar] [CrossRef]
- Freitas, J.C.C.; Emmerich, F.G.; Cernicchiaro, G.R.C.; Sampaio, L.C.; Bonagamba, T.J. Magnetic Susceptibility Effects on 13C MAS NMR Spectra of Carbon Materials and Graphite. Solid State Nucl. Magn. Reson. 2001, 20, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.A.; Gonçalves, G.R.; Cipriano, D.F.; Schettino, M.A.; Silva Filho, E.A.; Cunha, A.G.; Emmerich, F.G.; Freitas, J.C.C. Synthesis of Graphite Oxide from Milled Graphite Studied by Solid-State 13C Nuclear Magnetic Resonance. Carbon 2016, 98, 496–503. [Google Scholar] [CrossRef]
- Puziy, A.M.; Poddubnaya, O.I.; Socha, R.P.; Gurgul, J.; Wisniewski, M. XPS and NMR Studies of Phosphoric Acid Activated Carbons. Carbon 2008, 46, 2113–2123. [Google Scholar] [CrossRef]
- Wang, Y.; Zuo, S.; Yang, J.; Yoon, S.H. Evolution of Phosphorus-Containing Groups on Activated Carbons during Heat Treatment. Langmuir 2017, 33, 3112–3122. [Google Scholar] [CrossRef] [PubMed]
- Lopes, T.R.; Cipriano, D.F.; Gonçalves, G.R.; Honorato, H.A.; Schettino, M.A.; Cunha, A.G.; Emmerich, F.G.; Freitas, J.C.C. Multinuclear Magnetic Resonance Study on the Occurrence of Phosphorus in Activated Carbons Prepared by Chemical Activation of Lignocellulosic Residues from the Babassu Production. J. Environ. Chem. Eng. 2017, 5, 6016–6029. [Google Scholar] [CrossRef]
- Zhou, Y.; Ma, R.; Candelaria, S.L.; Wang, J.; Liu, Q.; Uchaker, E.; Li, P.; Chen, Y.; Cao, G. Phosphorus/Sulfur Co-Doped Porous Carbon with Enhanced Specific Capacitance for Supercapacitor and Improved Catalytic Activity for Oxygen Reduction Reaction. J. Power Sources 2016, 314, 39–48. [Google Scholar] [CrossRef]
- Yu, X.; Kang, Y.; Park, H.S. Sulfur and Phosphorus Co-Doping of Hierarchically Porous Graphene Aerogels for Enhancing Supercapacitor Performance. Carbon 2016, 101, 49–56. [Google Scholar] [CrossRef]
- Ying, K.; Tian, R.; Zhou, J.; Li, H.; Dugnani, R.; Lu, Y.; Duan, H.; Guo, Y.; Liu, H. A Three Dimensional Sulfur/Reduced Graphene Oxide with Embedded Carbon Nanotubes Composite as a Binder-Free, Free-Standing Cathode for Lithium-Sulfur Batteries. RSC Adv. 2017, 7, 43483–43490. [Google Scholar] [CrossRef]
- Thomas, H.R.; Marsden, A.J.; Walker, M.; Wilson, N.R.; Rourke, J.P. Sulfur-Functionalized Graphene Oxide by Epoxide Ring-Opening. Angew. Chem. Int. Ed. 2014, 53, 7613–7618. [Google Scholar] [CrossRef]
- Bi, Z.; Huo, L.; Kong, Q.; Li, F.; Chen, J.; Ahmad, A.; Wei, X.; Xie, L.; Chen, C. Structural Evolution of Phosphorus Species on Graphene with a Stabilized Electrochemical Interface. Appl. Mater. Interfaces 2019, 11, 11421–11430. [Google Scholar] [CrossRef]
- Al, M.; Oh, P.O.; Frost, R.L.; Scholz, R.; López, A.; Xi, Y. A Vibrational Spectroscopic Study of the Phosphate Mineral Whiteite CaMn(++)Mg2Al2(PO4)4(OH)2·8(H2O). Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 124, 243–248. [Google Scholar]
- Gurusamy, L.; Anandan, S.; Liu, N.; Wu, J.J. Synthesis of a Novel Hybrid Anode Nanoarchitecture of Bi2O3/Porous-RGO Nanosheets for High-Performance Asymmetric Supercapacitor. J. Electroanal. Chem. 2020, 856, 113489. [Google Scholar] [CrossRef]
- Zhao, X.; Li, W.; Kong, F.; Chen, H.; Wang, Z.; Liu, S.; Jin, C. Carbon Spheres Derived from Biomass Residue via Ultrasonic Spray Pyrolysis for Supercapacitors. Mater. Chem. Phys. 2018, 219, 461–467. [Google Scholar] [CrossRef]
- Pandolfo, A.G.; Hollenkamp, A.F. Carbon Properties and Their Role in Supercapacitors. J. Power Sources 2006, 157, 11–27. [Google Scholar] [CrossRef]
- Cuña, A.; Ortega Vega, M.R.; da Silva, E.L.; Tancredi, N.; Radtke, C.; Malfatti, C.F. Nitric Acid Functionalization of Carbon Monoliths for Supercapacitors: Effect on the Electrochemical Properties. Int. J. Hydrogen Energy 2016, 41, 12127–12135. [Google Scholar] [CrossRef]
- Liu, Y.; Chang, Z.; Yao, L.; Yan, S.; Lin, J.; Chen, J.; Lian, J.; Lin, H. Nitrogen/Sulfur Dual-Doped Sponge-like Porous Carbon Materials Derived from Pomelo Peel Synthesized at Comparatively Low Temperatures for Superior-Performance Supercapacitors. J. Electroanal. Chem. 2019, 847, 113111. [Google Scholar] [CrossRef]
- Qiang, Z.; Dan, D.; Wang, M.; Xiong, C.; Ying, X.; Jie, Z. Sulfur Modification of Carbon Materials as Well as the Redox Additive of Na2S for Largely Improving Capacitive Performance of Supercapacitors. J. Electroanal. Chem. 2020, 856, 113678. [Google Scholar]
- Béguin, F.; Frackowiak, E. Supercapacitors: Materials, Systems, and Applications; Wiley-VCH: Hoboken, NJ, USA, 2013. [Google Scholar]
- Mishra, A.K.; Ramaprabhu, S. Functionalized Graphene-Based Nanocomposites for Supercapacitor Application. J. Phys. Chem. C 2011, 115, 14006–14013. [Google Scholar] [CrossRef]
- Cuña, A.; Tancredi, N.; Bussi, J.; Deiana, A.C.; Sardella, M.F.; Barranco, V.; Rojo, J.M.E. Grandis as a Biocarbons Precursor for Supercapacitor Electrode Application. Waste Biomass Valorization 2014, 5, 305–313. [Google Scholar] [CrossRef]
- Vieira, M.A.; Frasson, C.M.R.; Costa, T.L.G.; Cipriano, D.F.; Schettino, M.A.; Cunha, A.G.; Freitas, J.C.C. Solid state 13C NMR study on the synthesis of graphite oxide from different graphitic precursors. Quim. Nova 2017, 40, 1164–1171. [Google Scholar]
- Jeong, H.K.; Jin, M.H.; So, K.P.; Lim, S.C.; Lee, Y.H. Tailoring the characteristics of graphite oxides by different oxidation times. J. Phys. D Appl. Phys. 2009, 42, 065418. [Google Scholar] [CrossRef]
- Wang, H.; Hu, Y.H. Effect of oxygen content on structures of graphite oxides. Ind. Eng. Chem. Res. 2011, 50, 6132–6137. [Google Scholar] [CrossRef]
- Mu, S.J.; Su, Y.C.; Xiao, L.H.; Liu, S.D.; Hu, T.; Tang, H.B. X-ray Difraction Pattern of Graphite Oxide. Chin. Phys. Lett. 2013, 30, 096101. [Google Scholar] [CrossRef]
- Rattana, T.; Chaiyakun, S.; Witit-Anun, N.; Nuntawong, N.; Chindaudom, P.; Oaew, S.; Kedkeaw, C.; Limsuwan, P. Preparation and characterization of graphene oxide nanosheets. Procedia Eng. 2012, 32, 759–764. [Google Scholar] [CrossRef]
- Huh, S.H. Thermal Reduction of Graphene Oxide; Mikhailov, S., Ed.; InTech: London, UK, 2011; Volume 19, pp. 73–90. [Google Scholar]
- Stankovich, S.; Dikin, D.A.; Piner, R.D.; Kohlhaas, K.A.; Kleinhammes, A.; Jia, Y.; Wu, Y.; Nguyen, S.T.; Ruoff, R.S. Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon 2007, 45, 1558–1565. [Google Scholar] [CrossRef]
- Krishnamoorthy, K.; Veerapandian, M.; Yun, K.; Kim, S.J. The chemical and structural analysis of graphene oxide with different degrees of oxidation. Carbon 2013, 53, 38–49. [Google Scholar] [CrossRef]
- Tegou, E.; Pseiropoulos, G.; Filippidou, M.K.; Chatzandroulis, S. Low-temperature thermal reduction of graphene oxide films in ambient atmosphere: Infra-red spectroscopic studies and gas sensing applications. Microelectron. Eng. 2016, 159, 146–150. [Google Scholar] [CrossRef]
- Hontoria-Lucas, C.; López-Peinado, A.J.; López-González, J.D.; Rojas-Cervantes, M.L.; Martín-Aranda, R.M. Study of oxygen-containing groups in a series of graphite oxides: Physical and chemical characterization. Carbon 1995, 33, 1585–1592. [Google Scholar] [CrossRef]
- Cai, W.; Piner, R.D.; Stadermann, F.J.; Park, S.; Shaibat, M.A.; Ishii, Y.; Yang, D.; Velamakanni, A.; An, S.J.; Stoller, M.; et al. Synthesis and Solid-State NMR Structural Characterization of 13C-Labeled Graphite Oxide. Science 2008, 321, 1815–1817. [Google Scholar] [CrossRef] [PubMed]
- Lerf, A.; He, H.; Forster, M.; Klinowski, J. Structure of Graphite Oxide Revisited. J. Phys. Chem. B 1998, 102, 4477–4482. [Google Scholar] [CrossRef]
- Dimiev, A.M.; Eigler, S. Graphene Oxide: Fundamentals and Applications, 1st ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2016. [Google Scholar]
- Sorokina, N.E.; Shornikova, O.N.; Avdeev, V.V. Stability limits of graphite intercalation compounds in the systems graphite-HNO3(H2SO4)-H2O-KMnO4. Inorg. Mater. 2007, 43, 822–826. [Google Scholar] [CrossRef]
- Dimiev, A.M.; Polson, T.A. Contesting the two-component structural model of graphene oxide and reexamining the chemistry of graphene oxide in basic media. Carbon 2015, 93, 544–554. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Ouyang, Z.; Lei, Y.; Chen, Y.; Zhang, Z.; Jiang, Z.; Hu, J. Preparation and Specific Capacitance Properties of Sulfur, Nitrogen Co-Doped Graphene Quantum Dots. Nanoscale Res. Lett. 2019, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
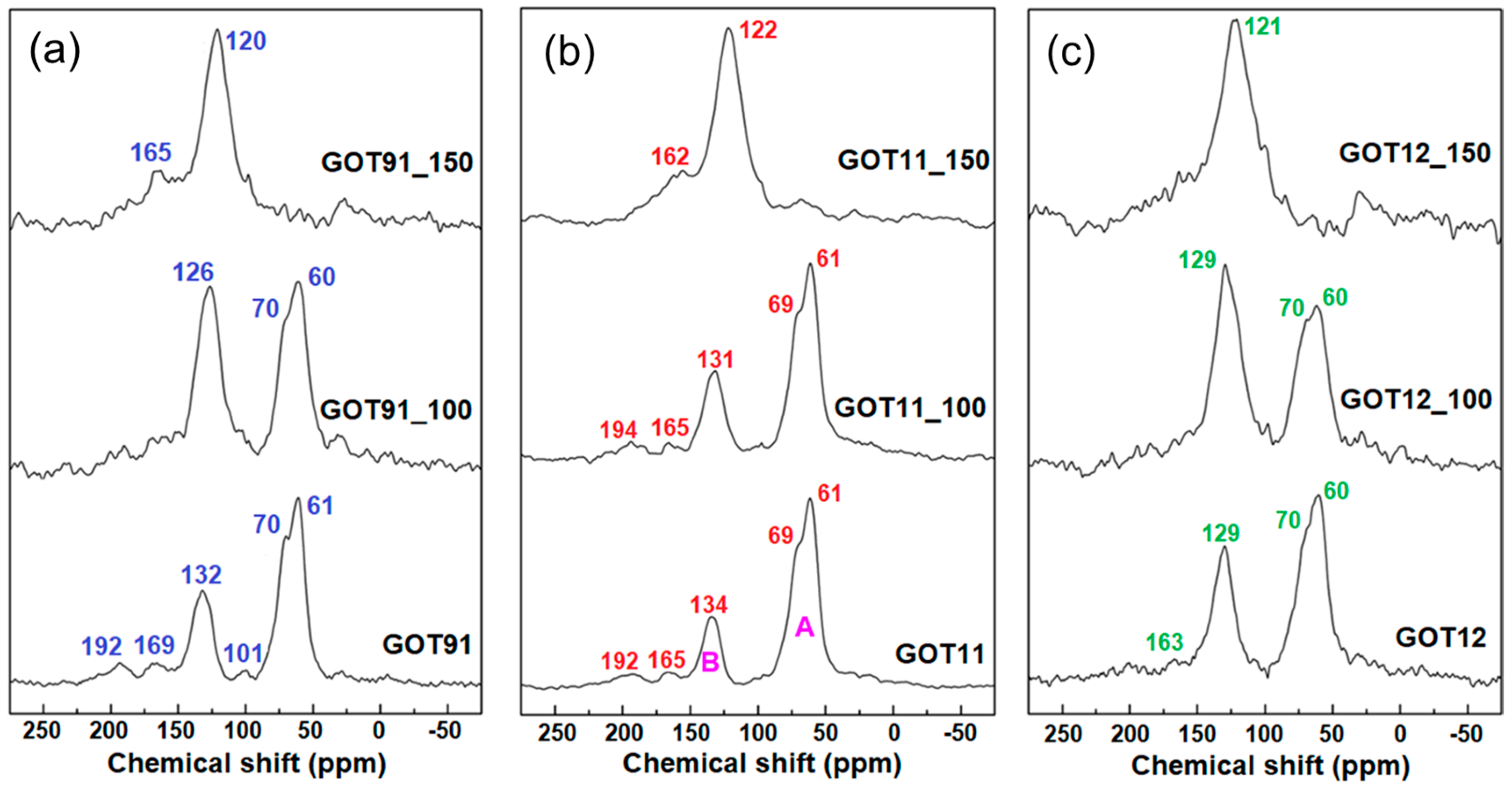{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
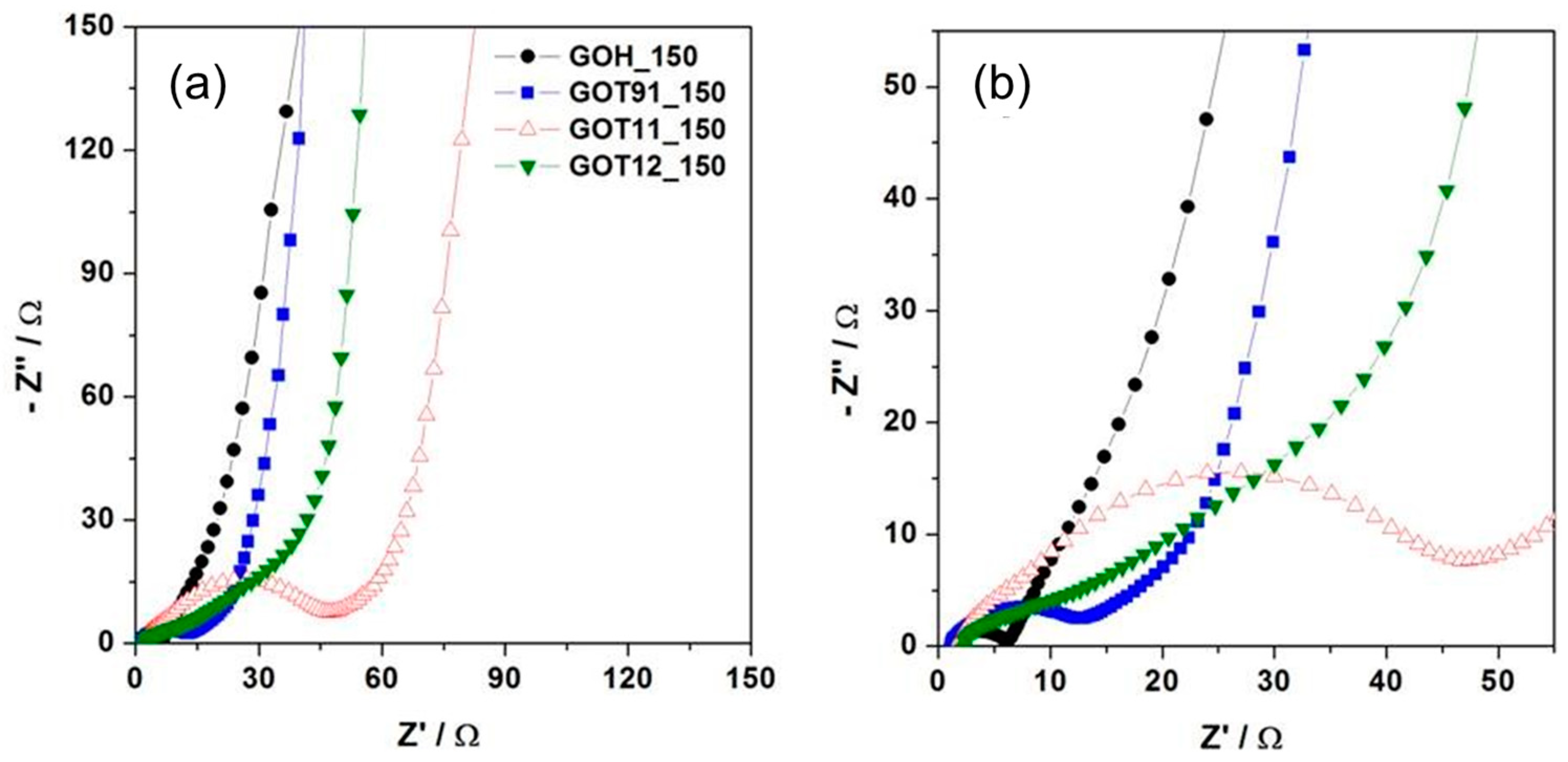{kind=link}
{kind=link}
| Sample | P Content (wt.%) | S Content (wt.%) |
|---|---|---|
| GOT91 | 0.26 ± 0.01 | 3.6 ± 0.2 |
| GOT91_150 | 0.27 ± 0.01 | 6.8 ± 0.3 |
| GOT11 | 1.4 ± 0.1 | 1.1 ± 0.1 |
| GOT11_150 | 2.3 ± 0.1 | 2.7 ± 0.1 |
| GOT12 | 2.8 ± 0.1 | 1.4 ± 0.1 |
| GOT12_150 | 5.4 ± 0.3 | 2.1 ± 0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vieira, M.A.; Costa, T.L.G.; Gonçalves, G.R.; Cipriano, D.F.; Schettino, M.A., Jr.; da Silva, E.L.; Cuña, A.; Freitas, J.C.C. Phosphorus/Sulfur-Enriched Reduced Graphene Oxide Papers Obtained from Recycled Graphite: Solid-State NMR Characterization and Electrochemical Performance for Energy Storage. C 2023, 9, 60. https://doi.org/10.3390/c9020060
Vieira MA, Costa TLG, Gonçalves GR, Cipriano DF, Schettino MA Jr., da Silva EL, Cuña A, Freitas JCC. Phosphorus/Sulfur-Enriched Reduced Graphene Oxide Papers Obtained from Recycled Graphite: Solid-State NMR Characterization and Electrochemical Performance for Energy Storage. C. 2023; 9(2):60. https://doi.org/10.3390/c9020060
Chicago/Turabian StyleVieira, Mariana A., Tainara L. G. Costa, Gustavo R. Gonçalves, Daniel F. Cipriano, Miguel A. Schettino, Jr., Elen L. da Silva, Andrés Cuña, and Jair C. C. Freitas. 2023. "Phosphorus/Sulfur-Enriched Reduced Graphene Oxide Papers Obtained from Recycled Graphite: Solid-State NMR Characterization and Electrochemical Performance for Energy Storage" C 9, no. 2: 60. https://doi.org/10.3390/c9020060
APA StyleVieira, M. A., Costa, T. L. G., Gonçalves, G. R., Cipriano, D. F., Schettino, M. A., Jr., da Silva, E. L., Cuña, A., & Freitas, J. C. C. (2023). Phosphorus/Sulfur-Enriched Reduced Graphene Oxide Papers Obtained from Recycled Graphite: Solid-State NMR Characterization and Electrochemical Performance for Energy Storage. C, 9(2), 60. https://doi.org/10.3390/c9020060