
Surface Functionalization of (Pyrolytic) Carbon—An Overview



Abstract
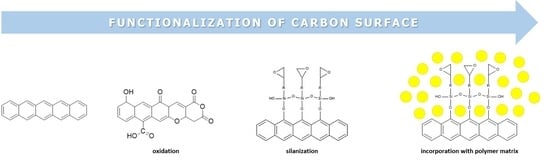
1. Introduction
2. Properties and Characteristics of (Pyrolytic) Carbon
3. Functionalization of (Pyrolytic) Carbon
3.1. Oxidation of Carbon Surface
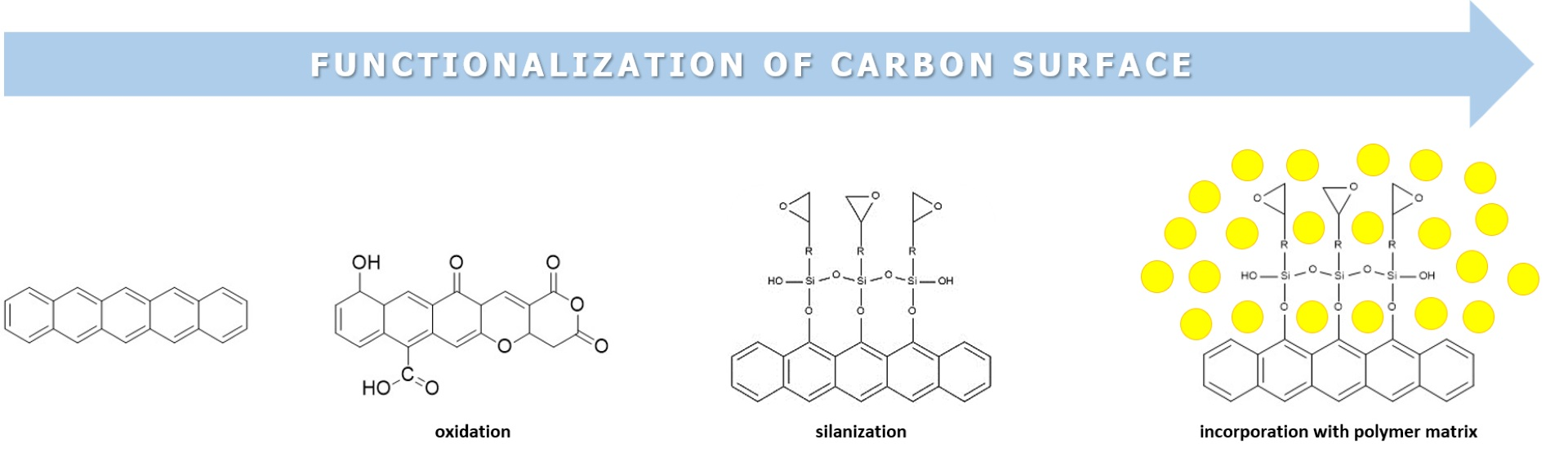
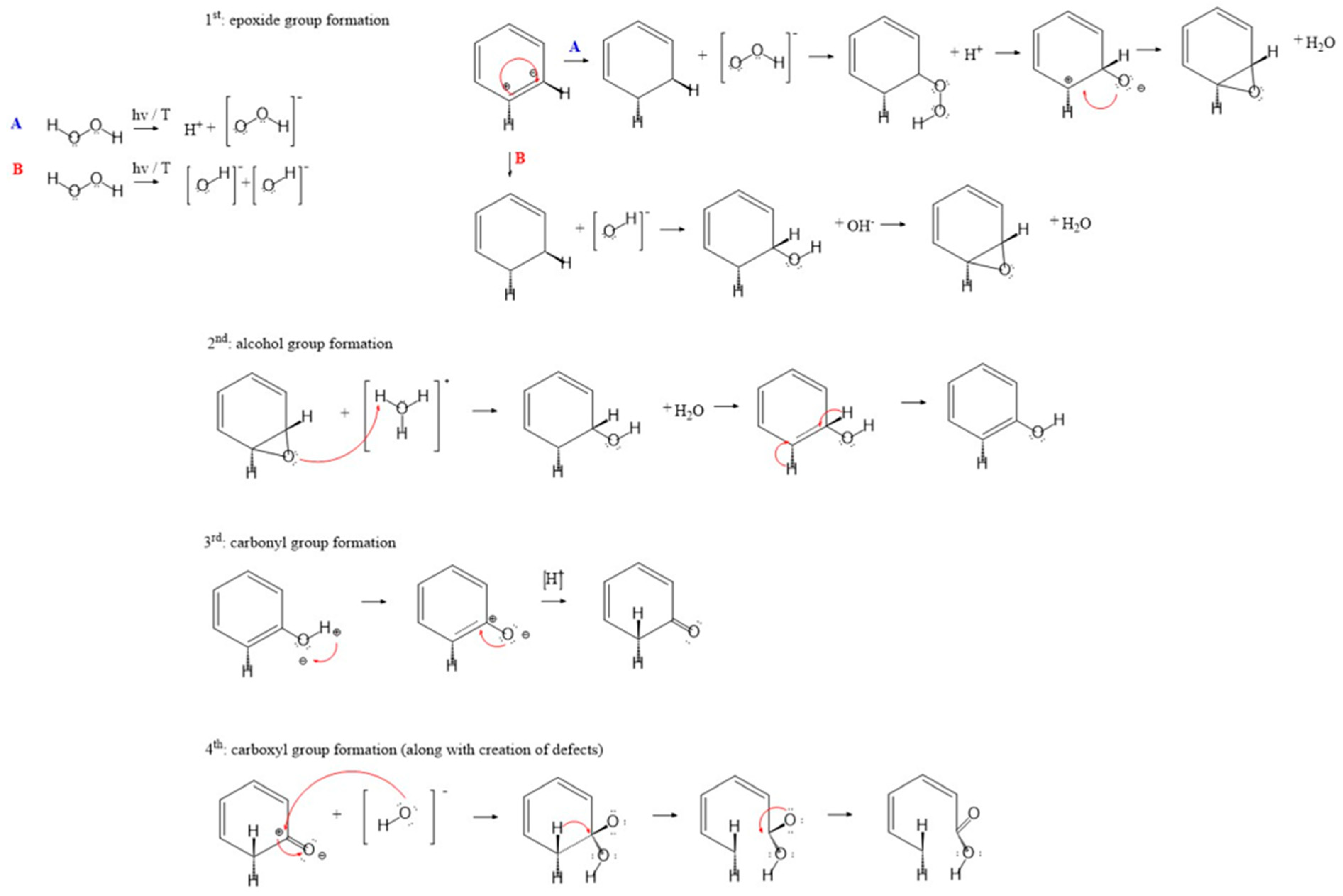
3.1.1. Structural Evolution of Surface Oxygen Groups
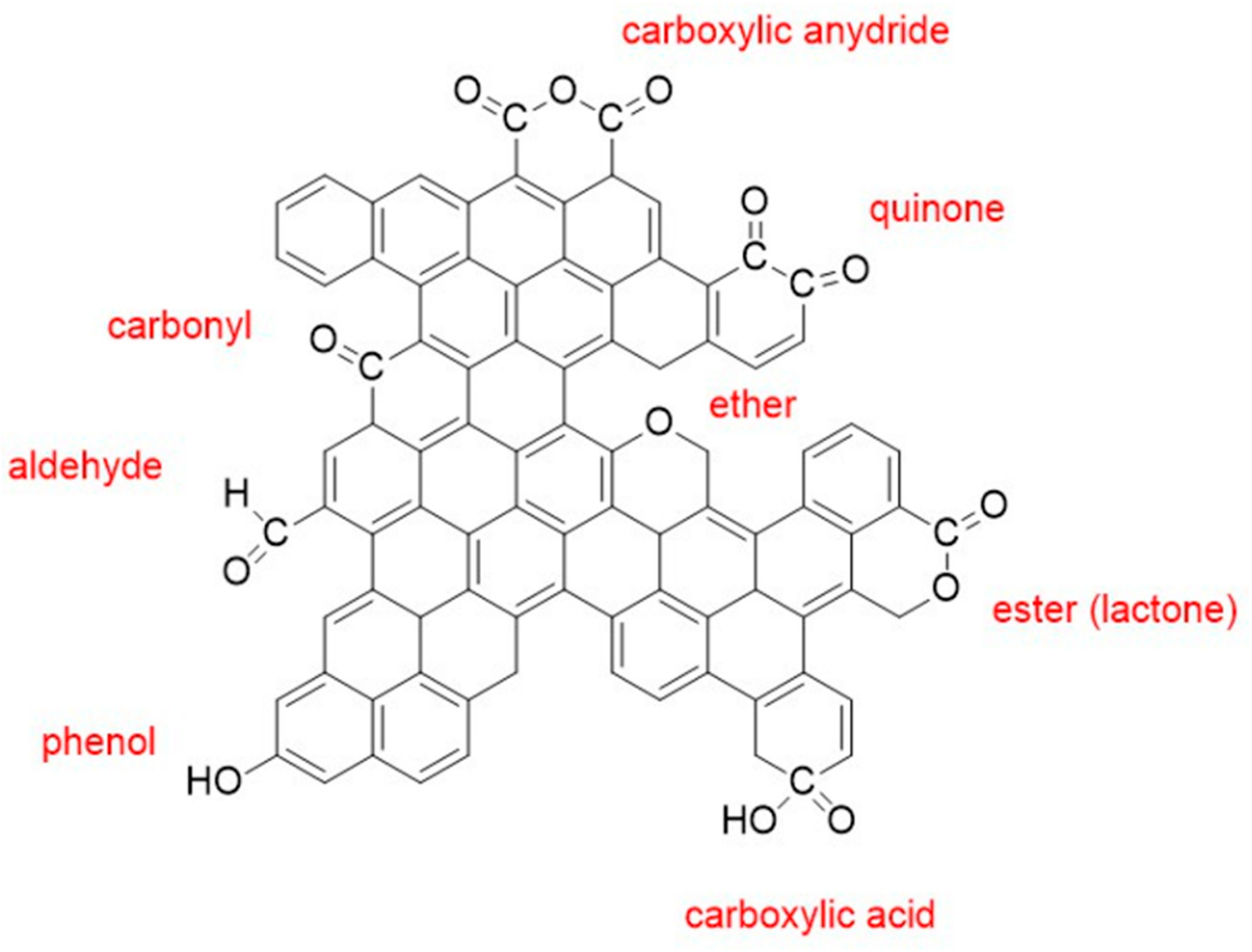
3.1.2. Chemical (Wet) Oxidation and Overview of Reaction Mechanisms
Oxidation with HNO3, H2O2 and (NH4)2S2O8
Reaction Mechanisms of Carbon Oxidation by HNO3 and H2O2
Hydrothermal and Solvothermal Oxidation Methods
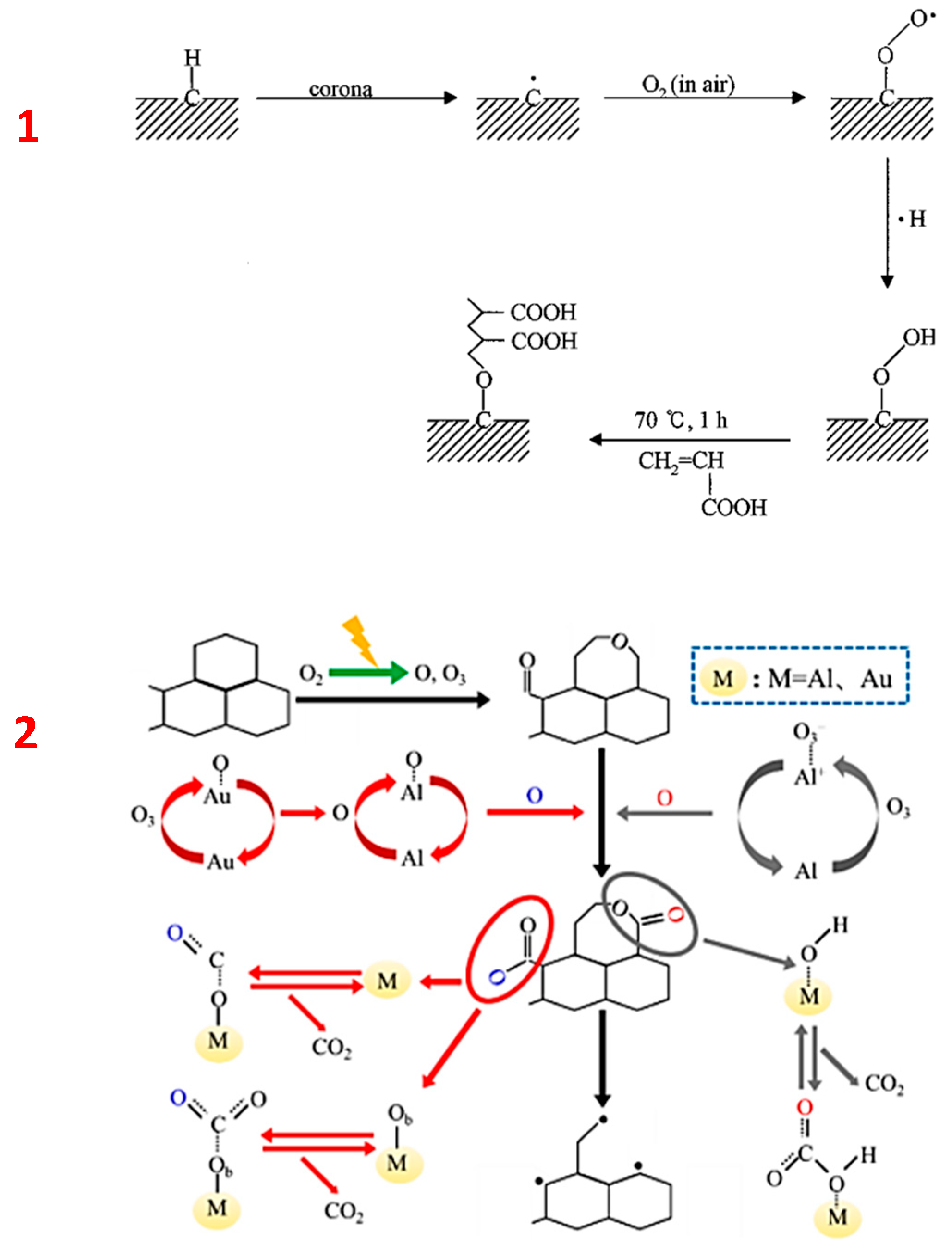
3.1.3. Physical (Dry) Oxidation and Overview of Reaction Mechanisms
Hydrophobic Recovery
- Overturn of the polar groups at the (polymer) surface, i.e., the created hydrophilic groups re-orientate away from the surface;
- Migration of the created polar moieties from the surface to the bulk (outside-in);
- Migration of the untreated moieties through the bulk matrix to the surface (inside out);
- The loss of volatile, e.g., oxygen rich species (and other polar functionalities) to the atmosphere;
- A change in surface roughness.
Reaction Mechanisms of Plasma-Assisted Carbon Oxidation
- O2 is excited and decomposed into two O atoms where one directly reacts with the catalyst surface and forms M-O, and the other reacts with O2 and forms O3;
- O3 reacts with the catalyst and forms another M-O unit, O2 and/or intermediate species: M-O3 and/or M+-O3;
- Since the aluminium catalyst is present in the system, it is the source of residual H2O (M-H2O) which can form M-OH and a bicarbonate complex M-O(CO2);
- The bicarbonate complex reacts with the oxygenated catalyst M-O and forms M-O(CO2).
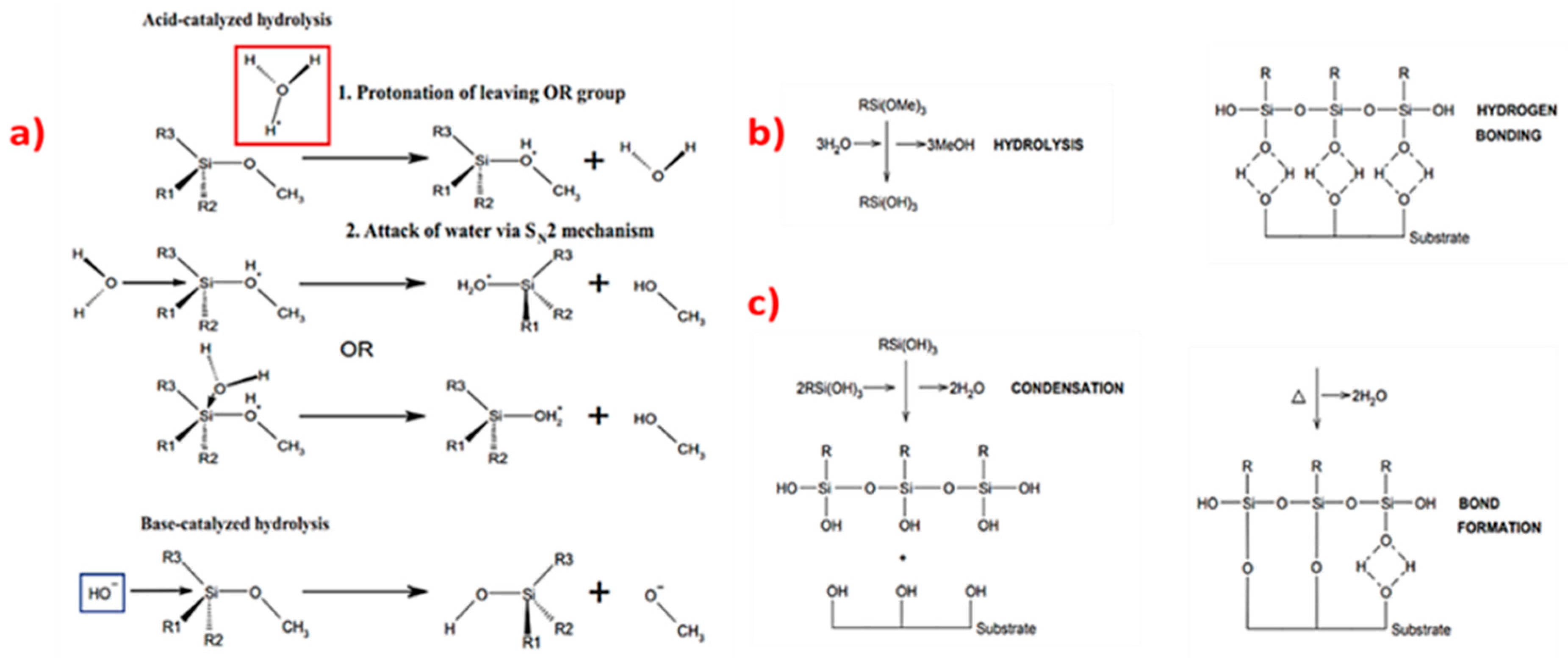
3.2. Silane Coupling to Carbon Surfaces
3.3. Other Chemical Functionalization Methods
- Addition of free radicals such as aryl radicals (Ar•) to benzenoid structures;
- Cycloaddition [1+2] of nitrene species to C=C bonds;
- Cycloaddition [1+2] of carbene species to C=C bonds;
- Cycloadditions [3+2] of, e.g., azomethine ylide to C=C bonds;
- Cycloaddition [4+2] of dienes to C=C bonds (Diels–Alder reaction);
- Reactions with superficial OH groups with of Meldrum’s acid, acid chlorides and anhydrides;
- Producing a hydrogen terminated carbon surface (C–H) followed by generation of radicals (C•) and “grafting-from” reactions;
- Reaction with amines.
3.4. Polymer-Based Carbon Composites
3.4.1. Selected Matrix Resins for Carbon Composites
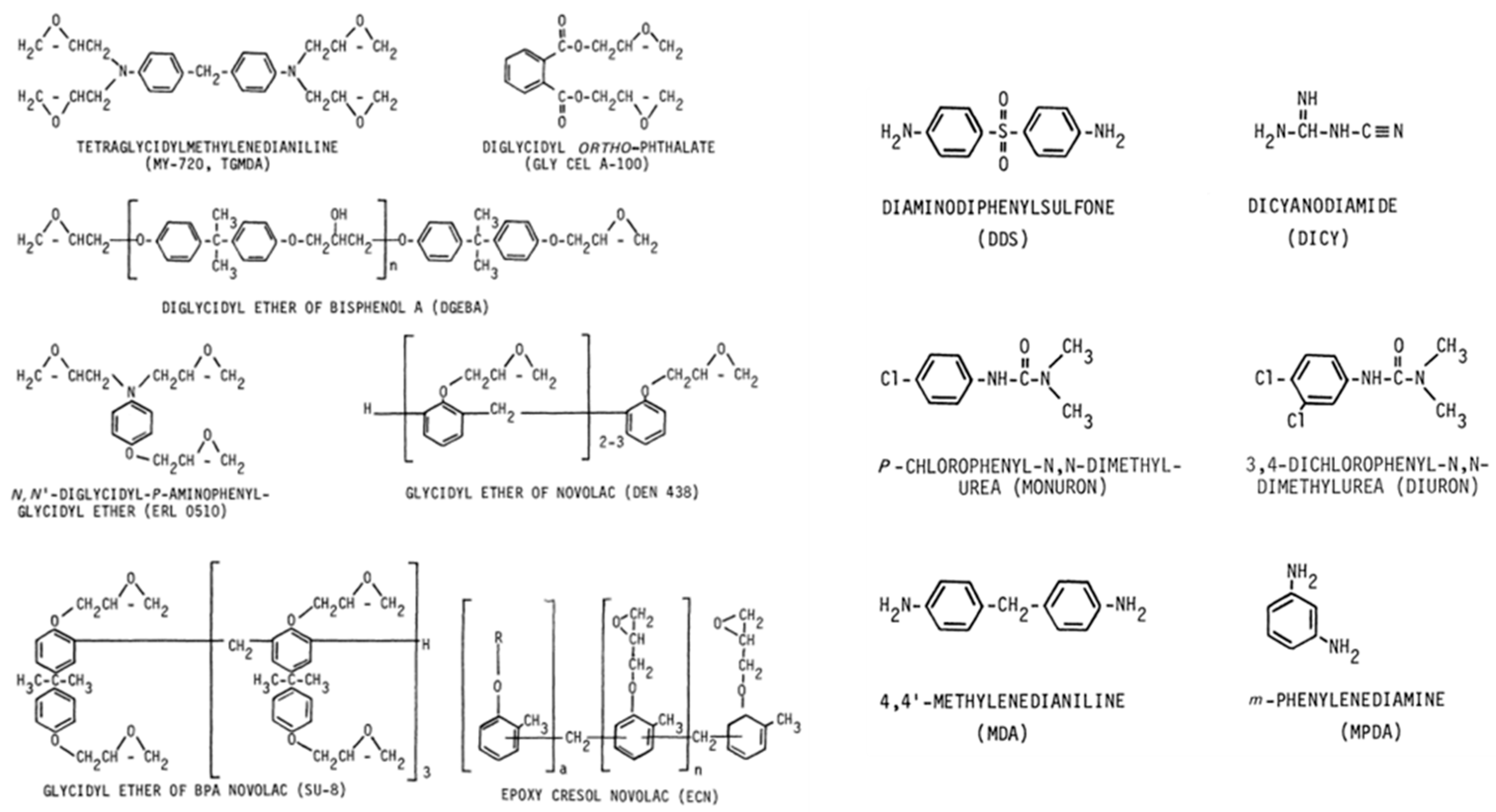
Epoxy Resins
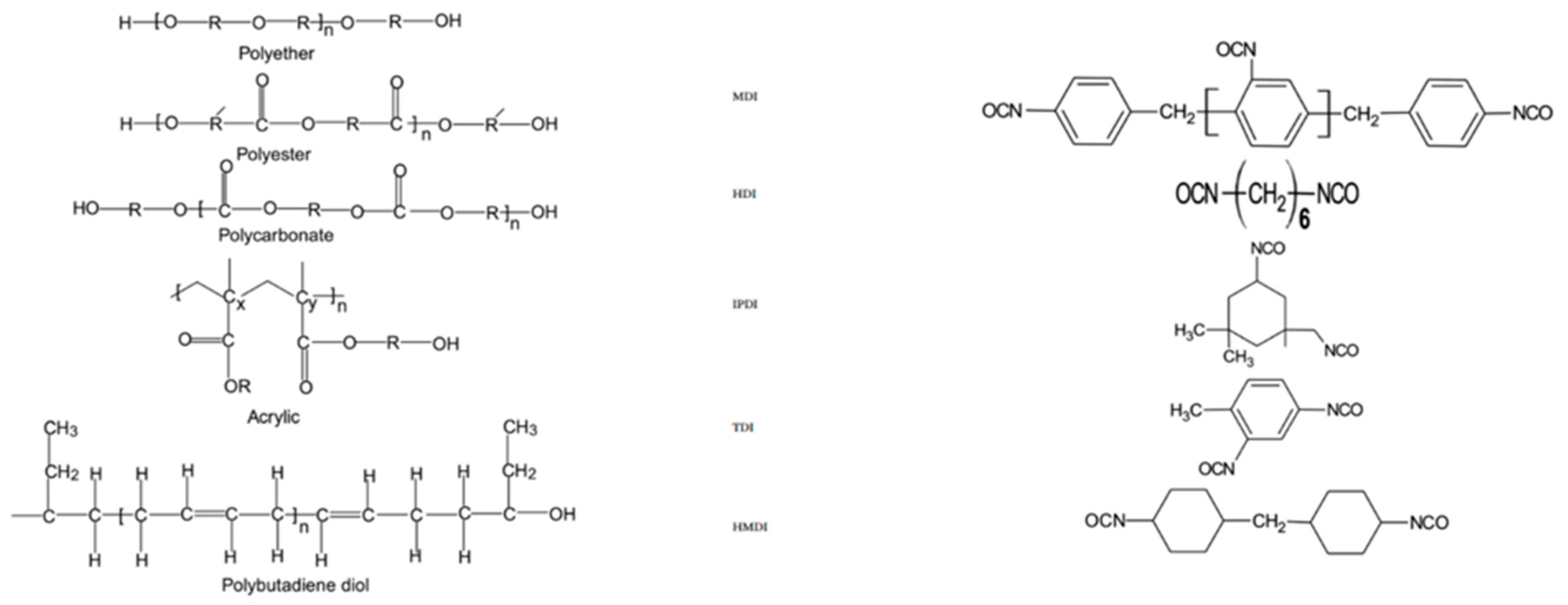
Polyurethane Resins
3.4.2. Characteristic Examples of Carbon Composites with Epoxy Resins and Polyurethanes
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andresen, J.; Lim, X.Y. Pyrolysis Processes and Technology for the Conversion of Hydrocarbons and Biomass. In Advances in Clean Hydrocarbon Fuel Processing; Elsevier: Amsterdam, The Netherlands, 2011; pp. 186–198. [Google Scholar] [CrossRef]
- Moldoveanu, S.C. Chapter 7 Pyrolysis of Hydrocarbons. In Techniques and Instrumentation in Analytical Chemistry; Elsevier: Amsterdam, The Netherlands, 2010; Volume 28, pp. 131–229. [Google Scholar] [CrossRef]
- Donnet, J.-B. (Ed.) Carbon Black: Science and Technology, 2nd ed.; Routledge: New York, NY, USA, 2017. [Google Scholar] [CrossRef]
- Chruściel, J.J.; Leśniak, E. Modification of epoxy resins with functional silanes, polysiloxanes, silsesquioxanes, silica and silicates. Prog. Polym. Sci. 2015, 41, 67–121. [Google Scholar] [CrossRef]
- Shin, Y.; Shuiliang, Y.; Satoshi, K.; Chieko, M.; Yuichi, F. Effects of O3 and NO2 on Catalytic Oxidation of Diesel PM. Chem. Lett. 2008, 37, 998–999. [Google Scholar] [CrossRef]
- Ju, Y.; Lefkowitz, J.K.; Reuter, C.B.; Won, S.H.; Yang, X.; Yang, S.; Sun, W.; Jiang, Z.; Chen, Q. Plasma Assisted Low Temperature Combustion. Plasma Chem. Plasma Process. 2016, 36, 85–105. [Google Scholar] [CrossRef]
- Silane Coupling Agent, (n.d.) 12. Available online: http://amchro.at/uct/Silane_Coupling_Agents_2014_4101-32-03.pdf (accessed on 24 April 2022).
- Industrial Silanes for Adhesives, Sealants, Coatings, and Composites, (n.d.) 24. Available online: https://technical.gelest.com/brochures/industrial-silanes-for-adhesives-sealants-coatings-and-composites/industrial-silanes/_2014_4101-32-03.pdf (accessed on 24 April 2022).
- Limitless Silanes—Bonding Organic and Inorganic Materials, (n.d.) 16. Available online: https://www.dow.com/content/dam/dcc/documents/en-us/mark-prod-info/26/26-2350-silanes-bonding-organic-inorganic-materials.pdf (accessed on 24 April 2022).
- Velasco-Santos, C.; Martinez-Hernandez, A.L.; Castano, V.M. Silanization of Carbon Nanotubes: Surface Modification and Polymer Nanocomposites. In Carbon Nanotubes—Polymer Nanocomposites; Yellampalli, S., Ed.; InTech: London, UK, 2011. [Google Scholar] [CrossRef]
- Lee, S.M. Handbook of Composite Reinforcements; VCH: New York, NY, USA, 1993; ISBN 978-1-56081-632-4. [Google Scholar]
- Peerzada, M.; Abbasi, S.; Lau, K.T.; Hameed, N. Additive Manufacturing of Epoxy Resins: Materials, Methods, and Latest Trends. Ind. Eng. Chem. Res. 2020, 59, 6375–6390. [Google Scholar] [CrossRef]
- May, C.A. (Ed.) Epoxy Resins: Chemistry and Technology, 2nd ed.; Routledge: New York, NY, USA, 2017. [Google Scholar] [CrossRef]
- Akindoyo, J.O.; Beg, M.D.H.; Ghazali, S.; Islam, M.R.; Jeyaratnam, N.; Yuvaraj, A.R. Polyurethane types, synthesis and applications—A review. RSC Adv. 2016, 6, 114453–114482. [Google Scholar] [CrossRef]
- Gardziella, A.; Pilato, L.A.; Knop, A. Phenolic Resins; Springer: Berlin/Heidelberg, Germany, 2000. [Google Scholar] [CrossRef]
- Petko, F.; Świeży, A.; Ortyl, J. Photoinitiating systems and kinetics of frontal photopolymerization processes—The prospects for efficient preparation of composites and thick 3D structures. Polym. Chem. 2021, 12, 4593–4612. [Google Scholar] [CrossRef]
- Knaack, P.; Klikovits, N.; Tran, A.D.; Bomze, D.; Liska, R. Radical induced cationic frontal polymerization in thin layers. J. Polym. Sci. Part Polym. Chem. 2019, 57, 1155–1159. [Google Scholar] [CrossRef]
- Klikovits, N.; Liska, R.; D’Anna, A.; Sangermano, M. Successful UV-Induced RICFP of Epoxy-Composites. Macromol. Chem. Phys. 2017, 218, 1700313. [Google Scholar] [CrossRef]
- Puchleitner, R.; Riess, G.; Kern, W. X-ray induced cationic curing of epoxy-bonded composites. Eur. Polym. J. 2017, 91, 31–45. [Google Scholar] [CrossRef]
- Hoppe, H.; Sariciftci, N.S. Organic solar cells: An overview. J. Mater. Res. 2004, 19, 1924–1945. [Google Scholar] [CrossRef]
- Carbon from Decarbonation as a Soil Management Resource—Thomas Prohaska, Montanuniversität Leoben. 2021. Available online: https://www.youtube.com/watch?v=O4miZRodBEg (accessed on 20 July 2022).
- Vasić, M.V.; Goel, G.; Vasić, M.; Radojević, Z. Recycling of waste coal dust for the energy-efficient fabrication of bricks: A laboratory to industrial-scale study. Environ. Technol. Innov. 2021, 21, 101350. [Google Scholar] [CrossRef]
- Gökçe, M.; Akçaözoğlu, S.; Sinani, B. Investigation of Production of Brick with Waste Coal Powder Additive. In Proceedings of the UBT International Conference, Pristina, Kosovo, 27 October 2018; Available online: https://knowledgecenter.ubt-uni.net/conference/2018/all-events/56 (accessed on 20 April 2022).
- Ryms, M.; Januszewicz, K.; Kazimierski, P.; Łuczak, J.; Klugmann-Radziemska, E.; Lewandowski, W.M. Post-Pyrolytic Carbon as a Phase Change Materials (PCMs) Carrier for Application in Building Materials. Materials 2020, 13, 1268. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Bastardo, N.; Schlögl, R.; Ruland, H. Methane Pyrolysis for CO2-Free H2 Production: A Green Process to Overcome Renewable Energies Unsteadiness. Chem. Ing. Tech. 2020, 92, 1596–1609. [Google Scholar] [CrossRef]
- Khoja, A.H.; Azad, A.K.; Saleem, F.; Khan, B.A.; Naqvi, S.R.; Mehran, M.T.; Amin, N.A.S. Hydrogen Production from Methane Cracking in Dielectric Barrier Discharge Catalytic Plasma Reactor using a Nanocatalyst. Energies 2020, 13, 5921. [Google Scholar] [CrossRef]
- Ferreira, F.V.; De Simone Cividanes, L.; Sales Brito, F.; Rossi Canuto de Menezes, B.; Franceschi, W.; Alves Nunes Simonetti, E.; Thim, G.P. Functionalizing Graphene and Carbon Nanotubes; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Gautier, M.; Rohani, V.; Fulcheri, L. Direct decarbonization of methane by thermal plasma for the production of hydrogen and high value-added carbon black. Int. J. Hydrogen Energy 2017, 42, 28140–28156. [Google Scholar] [CrossRef]
- Iqbaldin, M.; Khudzir, I.; Azlan, M.; Zaidi, A.; Surani, B.; Zubri, Z. Properties of Coconut Shell Activated Carbon. J. Trop. For. Sci. 2013, 25, 497–503. Available online: https://www.semanticscholar.org/paper/PROPERTIES-OF-COCONUT-SHELL-ACTIVATED-CARBON-Iqbaldin-Khudzir/b3ebd53586400304ef50f53fe330343cc45253fd (accessed on 20 April 2022).
- Keppetipola, N.M.; Dissanayake, M.; Dissanayake, P.; Karunarathne, B.; Dourges, M.A.; Talaga, D.; Servant, L.; Olivier, C.; Toupance, T.; Uchida, S.; et al. Graphite-type activated carbon from coconut shell: A natural source for eco-friendly non-volatile storage devices. RSC Adv. 2021, 11, 2854–2865. [Google Scholar] [CrossRef]
- Ghosh, A.; Rao, K.V.; Voggu, R.; George, S.J. Non-covalent functionalization, solubilization of graphene and single-walled carbon nanotubes with aromatic donor and acceptor molecules. Chem. Phys. Lett. 2010, 488, 198–201. [Google Scholar] [CrossRef]
- Su, P.; Zhang, J.; Tang, J.; Zhang, C. Preparation of nitric acid modified powder activated carbon to remove trace amount of Ni(II) in aqueous solution. Water Sci. Technol. 2019, 80, 86–97. [Google Scholar] [CrossRef]
- Vennerberg, D.; Rueger, Z.; Kessler, M.R. Effect of silane structure on the properties of silanized multiwalled carbon nanotube-epoxy nanocomposites. Polymer 2014, 55, 1854–1865. [Google Scholar] [CrossRef]
- Heidarinejad, Z.; Dehghani, M.H.; Heidari, M.; Javedan, G.; Ali, I.; Sillanpää, M. Methods for preparation and activation of activated carbon: A review. Environ. Chem. Lett. 2020, 18, 393–415. [Google Scholar] [CrossRef]
- Budi, E.; Nasbey, H.; Yuniarti, B.D.P.; Nurmayatri, Y.; Fahdiana, J.; Budi, A.S. Pore structure of the activated coconut shell charcoal carbon. AIP Conf. Proc. 2014, 1617, 130–133. [Google Scholar] [CrossRef]
- Gawande, P.; Kaware, D.J. Characterization and Activation of Coconut Shell Activated Carbon Research Paper. 2017. Available online: https://www.semanticscholar.org/paper/Characterization-and-activation-of-coconut-shell-Gawande-Kaware/3a6ba403afbc8afbcae75296cf35e9c0b246cb7a (accessed on 20 April 2022).
- Gratuito, M.K.B.; Panyathanmaporn, T.; Chumnanklang, R.-A.; Sirinuntawittaya, N.; Dutta, A. Production of activated carbon from coconut shell: Optimization using response surface methodology. Bioresour. Technol. 2008, 99, 4887–4895. [Google Scholar] [CrossRef] [PubMed]
- Strelko, V.; Malik, D.J.; Streat, M. Characterisation of the surface of oxidised carbon adsorbents. Carbon 2002, 40, 95–104. [Google Scholar] [CrossRef]
- Westbroek, P.; Temmerman, E. Mechanism of hydrogen peroxide oxidation reaction at a glassy carbon electrode in alkaline solution. J. Electroanal. Chem. 2000, 482, 40–47. [Google Scholar] [CrossRef]
- Kato, K.; Uchida, E.; Kang, E.-T.; Uyama, Y.; Ikada, Y. Polymer surface with graft chains. Prog. Polym. Sci. 2003, 28, 209–259. [Google Scholar] [CrossRef]
- Bhattacharyaa, A.; Misrab, B.N. Grafting: A versatile means to modify polymers: Techniques, factors and applications. Prog. Polym. Sci. 1978. Available online: https://www.semanticscholar.org/paper/Grafting-%3A-a-versatile-means-to-modify-polymers-%2C-Bhattacharyaa-Misrab/1ca9b18466b5c27ced2653986a749f068f1c43b6 (accessed on 8 June 2022).
- Minko, S. Grafting on Solid Surfaces: “Grafting to” and “Grafting from” Methods. In Polymer Surfaces and Interfaces: Characterization, Modification and Applications; Stamm, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 215–234. [Google Scholar] [CrossRef]
- Mueller, M.; Bandl, C.; Kern, W. Surface-Immobilized Photoinitiators for Light Induced Polymerization and Coupling Reactions. Polymers 2022, 14, 608. [Google Scholar] [CrossRef]
- Khan, R.; Nishina, Y. Covalent functionalization of carbon materials with redox-active organic molecules for energy storage. Nanoscale 2021, 13, 36–50. [Google Scholar] [CrossRef]
- Rehman, A.; Park, M.; Park, S.-J. Current Progress on the Surface Chemical Modification of Carbonaceous Materials. Coatings 2019, 9, 103. [Google Scholar] [CrossRef]
- Sangermano, M.; Razza, N. Light induced grafting-from strategies as powerfull tool for surface modification. Express Polym. Lett. 2019, 13, 135–145. [Google Scholar] [CrossRef]
- Yagci, Y.; Jockusch, S.; Turro, N.J. Photoinitiated Polymerization: Advances, Challenges, and Opportunities. Macromolecules 2010, 43, 6245–6260. [Google Scholar] [CrossRef]
- Sariciftci, N.S.; Smilowitz, L.; Heeger, A.J.; Wudl, F. Photoinduced Electron Transfer from a Conducting Polymer to Buckminsterfullerene. Science 1992, 258, 1474–1476. [Google Scholar] [CrossRef]
- Colavita, P.E.; Sun, B.; Tse, K.-Y.; Hamers, R.J. Photochemical Grafting of n-Alkenes onto Carbon Surfaces: The Role of Photoelectron Ejection. J. Am. Chem. Soc. 2007, 129, 13554–13565. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wang, L.; Liu, L.; Yang, W. Developments and new applications of UV-induced surface graft polymerizations. Prog. Polym. Sci. 2009, 34, 156–193. [Google Scholar] [CrossRef]
- Georgakilas, V.; Tiwari, J.N.; Kemp, K.C.; Perman, J.A.; Bourlinos, A.B.; Kim, K.S.; Zboril, R. Noncovalent Functionalization of Graphene and Graphene Oxide for Energy Materials, Biosensing, Catalytic, and Biomedical Applications. Chem. Rev. 2016, 116, 5464–5519. [Google Scholar] [CrossRef] [PubMed]
- Speranza, G. The Role of Functionalization in the Applications of Carbon Materials: An Overview. C 2019, 5, 84. [Google Scholar] [CrossRef]
- Zhou, Y.; Fang, Y.; Ramasamy, R. Non-Covalent Functionalization of Carbon Nanotubes for Electrochemical Biosensor Development. Sensors 2019, 19, 392. [Google Scholar] [CrossRef]
- Panchakarla, L.S.; Govindaraj, A. Covalent and non-covalent functionalization and solubilization of double-walled carbon nanotubes in nonpolar and aqueous media. J. Chem. Sci. 2008, 120, 607–611. [Google Scholar] [CrossRef]
- Verma, P.; Anoop, S.; Sasidhara Rao, V.; Sharma, A.K.; Uma Rani, R. Multiwalled carbon nanotube-poly vinyl alcohol nanocomposite multifunctional coatings on aerospace alloys. Mater. Today Proc. 2018, 5, 21205–21216. [Google Scholar] [CrossRef]
- Yang, W.; Wang, Y.; Li, J.; Yang, X. Polymer wrapping technique: An effective route to prepare Pt nanoflower/carbon nanotube hybrids and application in oxygenreduction. Energy Environ. Sci 2010, 3, 144–149. [Google Scholar] [CrossRef]
- Vaisman, L.; Wagner, H.D.; Marom, G. The role of surfactants in dispersion of carbon nanotubes. Adv. Colloid Interface Sci. 2006, 128–130, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Fujigaya, T.; Nakashima, N. Non-covalent polymer wrapping of carbon nanotubes and the role of wrapped polymers as functional dispersants. Sci. Technol. Adv. Mater. 2015, 16, 024802. [Google Scholar] [CrossRef] [PubMed]
- Hummers, W.S.; Offeman, R.E. Preparation of Graphitic Oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Feicht, P.; Biskupek, J.; Gorelik, T.E.; Renner, J.; Halbig, C.E.; Maranska, M.; Puchtler, F.; Kaiser, U.; Eigler, S. Brodie’s or Hummers’ Method: Oxidation Conditions Determine the Structure of Graphene Oxide. Chem. Eur. J. 2019, 25, 8955–8959. [Google Scholar] [CrossRef]
- Sakib, N.; Rahman, M.M.; Ali, M.H. Optimization of the Oxidation Temperature of Graphene Oxide. Int. J. Eng. Res. Technol. 2019, 8. [Google Scholar] [CrossRef]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved Synthesis of Graphene Oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef]
- Kovtyukhova, N.I.; Ollivier, P.J.; Martin, B.R.; Mallouk, T.E.; Chizhik, S.A.; Buzaneva, E.V.; Gorchinskiy, A.D. Layer-by-Layer Assembly of Ultrathin Composite Films from Micron-Sized Graphite Oxide Sheets and Polycations. Chem. Mater. 1999, 11, 771–778. [Google Scholar] [CrossRef]
- Shao, G.; Lu, Y.; Wu, F.; Yang, C.; Zeng, F.; Wu, Q. Graphene oxide: The mechanisms of oxidation and exfoliation. J. Mater. Sci. 2012, 47, 4400–4409. [Google Scholar] [CrossRef]
- Zaaba, N.I.; Foo, K.L.; Hashim, U.; Tan, S.J.; Liu, W.-W.; Voon, C.H. Synthesis of Graphene Oxide using Modified Hummers Method: Solvent Influence. Procedia Eng. 2017, 184, 469–477. [Google Scholar] [CrossRef]
- Chen, J.; Yao, B.; Li, C.; Shi, G. An improved Hummers method for eco-friendly synthesis of graphene oxide. Carbon 2013, 64, 225–229. [Google Scholar] [CrossRef]
- Chang, C.-I.; Chang, K.-H.; Shen, H.-H.; Hu, C.-C. A unique two-step Hummers method for fabricating low-defect graphene oxide nanoribbons through exfoliating multiwalled carbon nanotubes. J. Taiwan Inst. Chem. Eng. 2014, 45, 2762–2769. [Google Scholar] [CrossRef]
- Olorunkosebi, A.A.; Eleruja, M.A.; Adedeji, A.V.; Olofinjana, B.; Fasakin, O.; Omotoso, E.; Oyedotun, K.O.; Ajayi, E.O.B.; Manyala, N. Optimization of graphene oxide through various Hummers’ methods and comparative reduction using green approach. Diam. Relat. Mater. 2021, 117, 108456. [Google Scholar] [CrossRef]
- Rosillo-Lopez, M.; Salzmann, C.G. A simple and mild chemical oxidation route to high-purity nano-graphene oxide. Carbon 2016, 106, 56–63. [Google Scholar] [CrossRef]
- Gupta, V.; Sharma, N.; Singh, U.; Arif, M.; Singh, A. Higher oxidation level in graphene oxide. Optik 2017, 143, 115–124. [Google Scholar] [CrossRef]
- Rungrodnimitchai, S.; Hiranphinyophat, S. The Functionalization of Activated Carbon by Oxidation. Key Eng. Mater. 2020, 846, 251–256. [Google Scholar] [CrossRef]
- Saka, C. Overview on the Surface Functionalization Mechanism and Determination of Surface Functional Groups of Plasma Treated Carbon Nanotubes. Crit. Rev. Anal. Chem. 2018, 48, 1–14. Available online: https://www.tandfonline.com/doi/full/10.1080/10408347.2017.1356699 (accessed on 20 April 2022). [CrossRef]
- Senneca, O.; Scala, F.; Chirone, R.; Salatino, P. Relevance of structure, fragmentation and reactivity of coal to combustion and oxy-combustion. Fuel 2017, 201, 65–80. [Google Scholar] [CrossRef]
- Salatino, P.; Senneca, O.; Masi, S. Gasification of a coal char by oxygen and carbon dioxide. Carbon 1998, 36, 443–452. [Google Scholar] [CrossRef]
- Besenhard, J.O.; Schulte, A.; Schur, K.; Jannakoudakis, P.D. Preparation of Voltammetric and Potentiometric Carbon Fibre Microelectrodes. In Microelectrodes: Theory and Applications; Montenegro, M.I., Queirós, M.A., Daschbach, J.L., Eds.; Springer: Dordrecht, The Netherlands, 1991; pp. 189–204. [Google Scholar] [CrossRef]
- Hérold, A. Synthesis of graphite intercalation compounds. In Chemical Physics of Intercalation; Legrand, A.P., Flandrois, S., Eds.; Springer: Boston, MA, USA, 1987; pp. 3–45. [Google Scholar] [CrossRef]
- Du, Z.; Sarofim, A.F.; Longwell, J.P.; Mims, C.A. Kinetic measurement and modeling of carbon oxidation. Energy Fuels 1991, 5, 214–221. [Google Scholar] [CrossRef]
- Lear, A.E.; Brown, T.C.; Haynes, B.S. Formation of metastable oxide complexes during the oxidation of carbons at low temperatures. Symp. Int. Combust. 1991, 23, 1191–1197. [Google Scholar] [CrossRef]
- Haynes, B.S. A turnover model for carbon reactivity I. development. Combust. Flame 2001, 126, 1421–1432. [Google Scholar] [CrossRef]
- Yan, J.-A.; Chou, M.Y. Oxidation functional groups on graphene: Structural and electronic properties. Phys. Rev. B 2010, 82, 125403. [Google Scholar] [CrossRef]
- Sánchez, A.; Mondragón, F. Role of the Epoxy Group in the Heterogeneous CO2 Evolution in Carbon Oxidation Reactions. J. Phys. Chem. C 2007, 111, 612–617. [Google Scholar] [CrossRef]
- Gerber, I.; Oubenali, M.; Bacsa, R.; Durand, J.; Gonçalves, A.; Pereira, M.F.R.; Jolibois, F.; Perrin, L.; Poteau, R.; Serp, P. Theoretical and Experimental Studies on the Carbon-Nanotube Surface Oxidation by Nitric Acid: Interplay between Functionalization and Vacancy Enlargement. Chem. Eur. J. 2011, 17, 11467–11477. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Webley, P.A.; Zhao, D. Comprehensive study of pore evolution, mesostructural stability, and simultaneous surface functionalization of ordered mesoporous carbon (FDU-15) by wet oxidation as a promising adsorbent. Langmuir 2010, 26, 10277–10286. [Google Scholar] [CrossRef]
- Houshmand, A.; Daud, W.M.A.W.; Shafeeyan, M.S. Tailoring the Surface Chemistry of Activated Carbon by Nitric Acid: Study Using Response Surface Method. Bull. Chem. Soc. Jpn. 2011, 84, 1251–1260. [Google Scholar] [CrossRef]
- Moreno-Castilla, C.; López-Ramón, M.V.; Carrasco-Marín, F. Changes in surface chemistry of activated carbons by wet oxidation. Carbon 2000, 38, 1995–2001. [Google Scholar] [CrossRef]
- Moreno-Castilla, C.; Ferro-Garcia, M.A.; Joly, J.P.; Bautista-Toledo, I.; Carrasco-Marin, F.; Rivera-Utrilla, J. Activated Carbon Surface Modifications by Nitric Acid, Hydrogen Peroxide, and Ammonium Peroxydisulfate Treatments. Langmuir 1995, 11, 4386–4392. [Google Scholar] [CrossRef]
- Moreno-Castilla, C.; Carrasco-Marín, F.; Mueden, A. The creation of acid carbon surfaces by treatment with (NH4)2S2O8. Carbon 1997, 35, 1619–1626. [Google Scholar] [CrossRef]
- Kanai, Y.; Khalap, V.R.; Collins, P.G.; Grossman, J.C. Atomistic Oxidation Mechanism of a Carbon Nanotube in Nitric Acid. Phys. Rev. Lett. 2010, 104, 066401. [Google Scholar] [CrossRef]
- Ternero-Hidalgo, J.J.; Rosas, J.M.; Palomo, J.; Valero-Romero, M.J.; Rodríguez-Mirasol, J.; Cordero, T. Functionalization of activated carbons by HNO3 treatment: Influence of phosphorus surface groups. Carbon 2016, 101, 409–419. [Google Scholar] [CrossRef]
- Sengupta, D.; Mazumder, S.; Cole, J.V.; Lowry, S. Controlling Non-Catalytic Decomposition of High Concentration Hydrogen Peroxide. 2004. Available online: https://oa.mg/work/10.21236/ada426795 (accessed on 20 April 2022). [CrossRef]
- Ahmed, S.; Back, M.H.; Roscoe, J.M. A kinetic model for the low temperature oxidation of carbon: I. Combust. Flame 1987, 70, 1–16. [Google Scholar] [CrossRef]
- Feng, S.-H.; Li, G.-H. Chapter 4—Hydrothermal and Solvothermal Syntheses. In Modern Inorganic Synthetic Chemistry, 2nd ed.; Xu, R., Xu, Y., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 73–104. [Google Scholar] [CrossRef]
- Suvaci, E.; Özel, E. Hydrothermal Synthesis. In Encyclopedia of Materials: Technical Ceramics and Glasses; Pomeroy, M., Ed.; Elsevier: Oxford, UK, 2021; pp. 59–68. [Google Scholar] [CrossRef]
- Silva, A.M.T.; Machado, B.F.; Figueiredo, J.L.; Faria, J.L. Controlling the surface chemistry of carbon xerogels using HNO3-hydrothermal oxidation. Carbon 2009, 47, 1670–1679. [Google Scholar] [CrossRef]
- Marques, R.R.N.; Machado, B.F.; Faria, J.L.; Silva, A.M.T. Controlled generation of oxygen functionalities on the surface of Single-Walled Carbon Nanotubes by HNO3 hydrothermal oxidation. Carbon 2010, 48, 1515–1523. [Google Scholar] [CrossRef]
- Morales-Torres, S.; Silva, T.L.S.; Pastrana-Martínez, L.M.; Brandão, A.T.S.C.; Figueiredo, J.L.; Silva, A.M.T. Modification of the surface chemistry of single- and multi-walled carbon nanotubes by HNO3 and H2SO4 hydrothermal oxidation for application in direct contact membrane distillation. Phys. Chem. Chem. Phys. 2014, 16, 12237–12250. [Google Scholar] [CrossRef]
- Wang, Q.; Shi, W.; Zhu, B.; Su, D.S. An effective and green H2O2/H2O/O3 oxidation method for carbon nanotube to reinforce epoxy resin. J. Mater. Sci. Technol. 2020, 40, 24–30. [Google Scholar] [CrossRef]
- Wang, Q.; Zhu, B.; Su, D.S. Hydrothermal method oxidized carbon nanotube: Properties and performances in epoxy composite. Diam. Relat. Mater. 2021, 114, 108321. [Google Scholar] [CrossRef]
- Ang, T.N.; Young, B.R.; Burrell, R.; Taylor, M.; Aroua, M.K.; Baroutian, S. Oxidative hydrothermal surface modification of activated carbon for sevoflurane removal. Chemosphere 2021, 264, 128535. [Google Scholar] [CrossRef]
- Schönherr, J.; Buchheim, J.; Scholz, P.; Stelter, M. Oxidation of carbon nanotubes with ozone and hydroxyl radicals. Carbon 2017, 111, 631–640. [Google Scholar] [CrossRef]
- Glaze, W.H.; Kang, J.-W.; Chapin, D.H. The Chemistry of Water Treatment Processes Involving Ozone, Hydrogen Peroxide and Ultraviolet Radiation. Ozone Sci. Eng. 1987, 9, 335–352. [Google Scholar] [CrossRef]
- Mawhinney, D.B.; John, T.Y., Jr. FTIR study of the oxidation of amorphous carbon by ozone at 300 K—Direct COOH formation. Carbon 2001, 39, 1167–1173. [Google Scholar] [CrossRef]
- Sutherland, I.; Sheng, E.; Bradley, R.H.; Freakley, P.K. Effects of ozone oxidation on carbon black surfaces. J. Mater. Sci. 1996, 31, 5651–5655. [Google Scholar] [CrossRef]
- Cataldo, F. Ozone Reaction with Carbon Nanostructures 2: The Reaction of Ozone with Milled Graphite and Different Carbon Black Grades. J. Nanosci. Nanotechnol. 2007, 7, 1446–1454. [Google Scholar] [CrossRef] [PubMed]
- Razumovskii, S.D.; Gorshenev, V.N.; Kovarskii, A.L.; Kuznetsov, A.M.; Shchegolikhin, A.N. Carbon Nanostructure Reactivity: Reactions of Graphite Powders with Ozone. Fuller. Nanotub. Carbon Nanostruct. 2007, 15, 53–63. [Google Scholar] [CrossRef]
- Fijołek, L.; Świetlik, J.; Frankowski, M. The influence of active carbon contaminants on the ozonation mechanism interpretation. Sci. Rep. 2021, 11, 9934. [Google Scholar] [CrossRef] [PubMed]
- Horwath, J.; Schweickart, D. Corona resistance of low density polyethylene. In Proceedings of the 2000 Annual Report Conference on Electrical Insulation and Dielectric Phenomena (Cat. No.00CH37132), Victoria, BC, Canada, 15–18 October 2000; Volume 2, pp. 613–616. [Google Scholar] [CrossRef]
- Park, S.-J.; Jin, J.-S. Effect of Corona Discharge Treatment on the Dyeability of Low-Density Polyethylene Film. J. Colloid Interface Sci. 2001, 236, 155–160. [Google Scholar] [CrossRef]
- Haq, A.; Boyd, A.; Acheson, J.; McLaughlin, J.; Meenan, B.J. Corona Discharge-Induced Functional Surfaces of Polycarbonate and Cyclic Olefins Substrates. Surf. Coat. Technol. 2019, 362, 185–190. [Google Scholar] [CrossRef]
- Pego, M.F.F.; Bianchi, M.L.; Carvalho, J.A.; Veiga, T.R.L.A. Surface modification of activated carbon by corona treatment. An. Acad. Bras. Ciênc. 2019, 91, e20170947. [Google Scholar] [CrossRef]
- Lu, H.; Yao, X.; Li, J.; Yao, S.; Wu, Z.; Zhang, H.; Lin, H.; Nozaki, T. Mechanism on the plasma-catalytic oxidation of graphitic carbon over Au/γ-Al2O3 by in situ plasma DRIFTS-mass spectrometer. J. Hazard. Mater. 2020, 396, 122730. [Google Scholar] [CrossRef]
- Kong, L.; Wang, X.; Zheng, W.; Tian, S.; Qi, Y.; Xue, Y.; Wang, B. Effects of plasma treatment on properties of carbon fiber and its reinforced resin composites. Mater. Res. Express 2020, 7, 065304. [Google Scholar] [CrossRef]
- Ortiz-Ortega, E.; Hosseini, S.; Martinez-Chapa, S.O.; Madou, M.J. Aging of plasma-activated carbon surfaces: Challenges and opportunities. Appl. Surf. Sci. 2021, 565, 150362. [Google Scholar] [CrossRef]
- Chen, C.; Liang, B.; Ogino, A.; Wang, X.; Nagatsu, M. Oxygen Functionalization of Multiwall Carbon Nanotubes by Microwave-Excited Surface-Wave Plasma Treatment. J. Phys. Chem. C 2009, 113, 7659–7665. [Google Scholar] [CrossRef]
- Bormashenko, E.; Chaniel, G.; Grynyov, R. Towards understanding hydrophobic recovery of plasma treated polymers: Storing in high polarity liquids suppresses hydrophobic recovery. Appl. Surf. Sci. 2013, 273, 549–553. [Google Scholar] [CrossRef]
- Enciso, B.; Abenojar, J.; Martínez, M.A. Influence of plasma treatment on the adhesion between a polymeric matrix and natural fibres. Cellulose 2017, 24, 1791–1801. [Google Scholar] [CrossRef]
- Guckenberger, D.J.; Berthier, E.; Young, E.W.K.; Beebe, D.J. Induced hydrophobic recovery of oxygen plasma-treated surfaces. Lab Chip 2012, 12, 2317. [Google Scholar] [CrossRef]
- Fritz, J.L.; Owen, M.J. Hydrophobic Recovery of Plasma-Treated Polydimethylsiloxane. J. Adhes. 1995, 54, 33–45. [Google Scholar] [CrossRef]
- Jongwannasiri, C.; Watanabe, S. Improvement of Hydrophilic Stability of Diamond-Like Carbon Films by O2/CF4 Plasma Post-Treatment. Adv. Mater. Res. 2015, 1125, 38–44. [Google Scholar] [CrossRef]
- Li, O.L.; Qin, L.; Takeuchi, N.; Kim, K.; Ishizaki, T. Effect of hydrophilic/hydrophobic properties of carbon materials on plasma-sulfonation process and their catalytic activities in cellulose conversion. Catal. Today 2019, 337, 155–161. [Google Scholar] [CrossRef]
- Liston, E.M.; Martinu, L.; Wertheimer, M.R. Plasma surface modification of polymers for improved adhesion: A critical review. J. Adhes. Sci. Technol. 1993, 7, 1091–1127. [Google Scholar] [CrossRef]
- Temmel, S.; Kern, W.; Luxbacher, T. Surface Modification of Polyethylene by Photosulfonation. In Proceedings of the 5th International Symposium on Polymer Surface Modification, Toronto, ON, Canada, 5 June 2007. [Google Scholar] [CrossRef]
- Yao, S. Plasma Reactors for Diesel Particulate Matter Removal. Recent Pat. Chem. Eng. 2009, 2, 67–75. [Google Scholar] [CrossRef]
- Erden, S.; Ho, K.K.C.; Lamoriniere, S.; Lee, A.F.; Yildiz, H.; Bismarck, A. Continuous Atmospheric Plasma Oxidation of Carbon Fibres: Influence on the Fibre Surface and Bulk Properties and Adhesion to Polyamide 12. Plasma Chem. Plasma Process. 2010, 30, 471–487. [Google Scholar] [CrossRef]
- Donnet, J.B.; Brendle, M.; Dhami, T.L.; Bahl, O.P. Plasma treatment effect on the surface energy of carbon and carbon fibers. Carbon 1986, 24, 757–770. [Google Scholar] [CrossRef]
- Tiwari, S.; Sharma, M.; Panier, S.; Mutel, B.; Mitschang, P.; Bijwe, J. Influence of cold remote nitrogen oxygen plasma treatment on carbon fabric and its composites with specialty polymers. J. Mater. Sci. 2011, 46, 964–974. [Google Scholar] [CrossRef]
- Materne, T.; Buyl, F.D.; Witucki, G. Organosilane Technology in Coating Applications: Review and Perspectives 1645 By, Undefined. 2012. Available online: https://www.semanticscholar.org/paper/Organosilane-Technology-in-Coating-Applications-%3A-Materne-Buyl/c15e4edbb3a210ef580ca0c23b69ab97f4b2b8fb (accessed on 20 April 2022).
- Kausar, A.; Anwar, Z.; Muhammad, B. Recent Developments in Epoxy/Graphite, Epoxy/Graphene, and Epoxy/Graphene Nanoplatelet Composites: A Comparative Review. Polym. Plast. Technol. Eng. 2016, 55, 1192–1210. [Google Scholar] [CrossRef]
- He, Q.; Xu, Y.; Wang, C.; She, S.; Zhou, S.; Wang, R. Silane modification and characterization of activated carbon. Adsorption 2012, 18, 23–29. [Google Scholar] [CrossRef]
- Aujara, K.M.; Chieng, B.W.; Ibrahim, N.A.; Zainuddin, N.; Thevy Ratnam, C. Gamma-Irradiation Induced Functionalization of Graphene Oxide with Organosilanes. Int. J. Mol. Sci. 2019, 20, 1910. [Google Scholar] [CrossRef]
- Lubis, H.; Noer, Z.; Lubis, R.Y.; Siregar, I.; Hasibuan, E.S. The Influence of Coconut Shell Actived Carbon (X = 0; 0.05; 0.10; 0.15) to Lightweight Concrete. Int. J. Sci. Technol. Res. 2021, 5, 8–12. Available online: https://web.archive.org/web/20210813231433/http://www.ijstr.org/final-print/mar2021/The-Influence-Of-Coconut-Shell-Actived-Carbon-X-0-005-010-015-To-Lightweight-Concrete.pdf (accessed on 20 April 2022).
- Jankovský, O.; Lojka, M.; Lauermannová, A.-M.; Antončík, F.; Pavlíková, M.; Záleská, M.; Pavlík, Z.; Pivák, A.; Sedmidubský, D. Towards novel building materials: High-strength nanocomposites based on graphene, graphite oxide and magnesium oxychloride. Appl. Mater. Today 2020, 20, 100766. [Google Scholar] [CrossRef]
- Sheikh, T.M.; Anwar, M.P.; Muthoosamy, K.; Jaganathan, J.; Chan, A.; Mohamed, A.A. The mechanics of carbon-based nanomaterials as cement reinforcement—A critical review. Constr. Build. Mater. 2021, 303, 124441. [Google Scholar] [CrossRef]
- Silvestro, L.; Ruviaro, A.; Lima, G.; de Matos, P.; de Azevedo, A.R.G.; Monteiro, S.N.; Gleize, P. Influence of Ultrasonication of Functionalized Carbon Nanotubes on the Rheology, Hydration, and Compressive Strength of Portland Cement Pastes. Materials 2021, 14, 5248. [Google Scholar] [CrossRef]
- Lavagna, L.; Musso, S.; Ferro, G.; Pavese, M. Cement-based composites containing functionalized carbon fibers. Cem. Concr. Compos. 2018, 88, 165–171. [Google Scholar] [CrossRef]
- Konsta-Gdoutos, M.S.; Danoglidis, P.A.; Falara, M.G.; Nitodas, S.F. Fresh and mechanical properties, and strain sensing of nanomodified cement mortars: The effects of MWCNT aspect ratio, density and functionalization. Cem. Concr. Compos. 2017, 82, 137–151. [Google Scholar] [CrossRef]
- Sadiq, M.M.; Soroushian, P.; Bakker, M.G.; Balachandra, A.M. Ultra-high-performance cementitious composites with enhanced mechanical and durability characteristics. SN Appl. Sci. 2021, 3, 676. [Google Scholar] [CrossRef]
- Silvestro, L.; Jean Paul Gleize, P. Effect of carbon nanotubes on compressive, flexural and tensile strengths of Portland cement-based materials: A systematic literature review. Constr. Build. Mater. 2020, 264, 120237. [Google Scholar] [CrossRef]
- Silvestro, L.; Spat Ruviaro, A.; Ricardo de Matos, P.; Pelisser, F.; Zambelli Mezalira, D.; Jean Paul Gleize, P. Functionalization of multi-walled carbon nanotubes with 3-aminopropyltriethoxysilane for application in cementitious matrix. Constr. Build. Mater. 2021, 311, 125358. [Google Scholar] [CrossRef]
- Wang, Y.-L.; Stanzione, M.; Xia, H.; Buonocore, G.G.; Fortunati, E.; Kaciulis, S.; Lavorgna, M. Effect of mercapto-silanes on the functional properties of highly amorphous vinyl alcohol composites with reduced graphene oxide and cellulose nanocrystals. Compos. Sci. Technol. 2020, 200, 108458. [Google Scholar] [CrossRef]
- He, X.; Zhu, J.; Wang, H.; Zhou, M.; Zhang, S. Surface Functionalization of Activated Carbon with Phosphonium Ionic Liquid for CO2 Adsorption. Coatings 2019, 9, 590. [Google Scholar] [CrossRef]
- Ehlert, G.J.; Lin, Y.; Sodano, H.A. Carboxyl functionalization of carbon fibers through a grafting reaction that preserves fiber tensile strength. Carbon 2011, 49, 4246–4255. [Google Scholar] [CrossRef]
- Liu, J.; Tang, J.; Gooding, J.J. Strategies for chemical modification of graphene and applications of chemically modified graphene. J. Mater. Chem. 2012, 22, 12435. [Google Scholar] [CrossRef]
- Criado, A.; Melchionna, M.; Marchesan, S.; Prato, M. The Covalent Functionalization of Graphene on Substrates. Angew. Chem. Int. Ed. 2015, 54, 10734–10750. [Google Scholar] [CrossRef] [PubMed]
- Korivand, M.; Zamani, M. Surface modification of graphene by coupling with electron deficient radicals. J. Solid State Chem. 2021, 294, 121851. [Google Scholar] [CrossRef]
- Bou-Hamdan, F.R.; Lévesque, F.; O’Brien, A.G.; Seeberger, P.H. Continuous flow photolysis of aryl azides: Preparation of 3 H-azepinones. Beilstein J. Org. Chem. 2011, 7, 1124–1129. [Google Scholar] [CrossRef] [PubMed]
- Zan, W. Chemical functionalization of graphene by carbene cycloaddition: A density functional theory study. Appl. Surf. Sci. 2014, 311, 377–383. [Google Scholar] [CrossRef]
- de Frémont, P.; Marion, N.; Nolan, S.P. Carbenes: Synthesis, properties, and organometallic chemistry. Coord. Chem. Rev. 2009, 253, 862–892. [Google Scholar] [CrossRef]
- Djordjevic, I.; Wicaksono, G.; Šolić, I.; Singh, J.; Kaku, T.; Lim, S.; Ang, E.W.J.; Blancafort, L.; Steele, T. Rapid Activation of Diazirine Biomaterials with the Blue Light Photocatalyst. ACS Appl. Mater. Interfaces 2021, 13, 36839–36848. [Google Scholar] [CrossRef]
- Hesari, M.; Workentin, M.S. Covalent modification of graphene and micro-diamond with redox active substrates via photogenerated carbenes. Carbon 2015, 85, 159–167. [Google Scholar] [CrossRef]
- Sainsbury, T.; Passarelli, M.; Naftaly, M.; Gnaniah, S.; Spencer, S.J.; Pollard, A.J. Covalent Carbene Functionalization of Graphene: Toward Chemical Band-Gap Manipulation. ACS Appl. Mater. Interfaces 2016, 8, 4870–4877. [Google Scholar] [CrossRef]
- Georgakilas, V.; Otyepka, M.; Bourlinos, A.B.; Chandra, V.; Kim, N.; Kemp, K.C.; Hobza, P.; Zboril, R.; Kim, K.S. Functionalization of Graphene: Covalent and Non-Covalent Approaches, Derivatives and Applications. Chem. Rev. 2012, 112, 6156–6214. [Google Scholar] [CrossRef]
- Urdl, K.; Kandelbauer, A.; Kern, W.; Müller, U.; Thebault, M.; Zikulnig-Rusch, E. Self-healing of densely crosslinked thermoset polymers—A critical review. Prog. Org. Coat. 2017, 104, 232–249. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, H.; Gao, B.; Wang, X.; Cai, Q.; Wang, X. Chemical functionalization of graphene via aryne cycloaddition: A theoretical study. J. Mol. Model. 2012, 18, 2861–2868. [Google Scholar] [CrossRef] [PubMed]
- Guenes, S.; Neugebauer, H.; Sariciftci, N.S. Conjugated Polymer-Based Organic Solar Cells. ChemInform 2007, 38, 1324–1338. [Google Scholar] [CrossRef]
- Sariciftci, N.S.; Braun, D.; Zhang, C.; Srdanov, V.I.; Heeger, A.J.; Stucky, G.; Wudl, F. Semiconducting polymer-buckminsterfullerene heterojunctions: Diodes, photodiodes, and photovoltaic cells. Appl. Phys. Lett. 1993, 62, 585–587. [Google Scholar] [CrossRef]
- Bracher, P.J.; Schuster, D.I. Electron Transfer in Functionalized Fullerenes. In Fullerenes: From Synthesis to Optoelectronic Properties; Guldi, D.M., Martin, N., Eds.; Springer: Dordrecht, The Netherlands, 2002; pp. 163–212. [Google Scholar] [CrossRef]
- Mackiewicz, N.; Bark, T.; Cao, B.; Delaire, J.A.; Riehl, D.; Ling, W.L.; Foillard, S.; Doris, E. Fullerene-functionalized carbon nanotubes as improved optical limiting devices. Carbon 2011, 49, 3998–4003. [Google Scholar] [CrossRef]
- Maggini, M.; Menna, E. Fullerenes: From Synthesis to Optoelectronic Properties, 1st ed.; Guldi, D.M., Martin, N., Eds.; Springer: Amsterdam, The Netherlands, 2002; Available online: http://gen.lib.rus.ec/book/index.php?md5=701830c2ac13fc4de453dc1c57a5f1f8 (accessed on 3 August 2022).
- Yamada, M.; Akasaka, T.; Nagase, S. Carbene Additions to Fullerenes. Chem. Rev. 2013, 113, 7209–7264. [Google Scholar] [CrossRef] [PubMed]
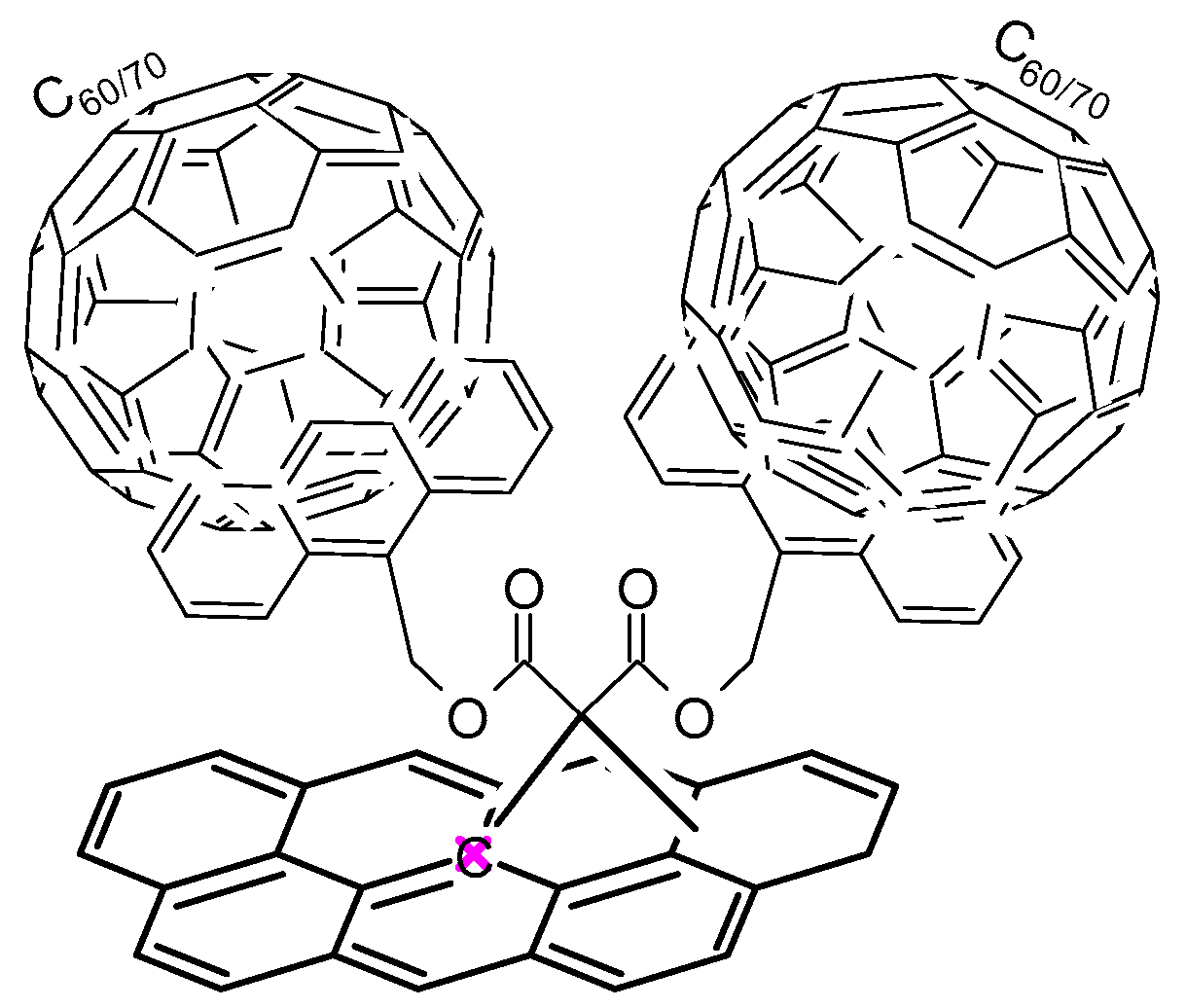
- Fedorczyk, A.; Krogul-Sobczak, A.; Piotrowski, P. Anthracene modified graphene for C60/C70 fullerenes capture and construction of energy storage materials. Chem. Pap. 2022, 76, 2041–2050. [Google Scholar] [CrossRef]
- Olsson, R.T.; Fogelström, L.; Martínez-Sanz, M.; Henriksson, M. Cellulose Nanofillers for Food Packaging. In Multifunctional and Nanoreinforced Polymers for Food Packaging; Elsevier: Amsterdam, The Netherlands, 2011; pp. 86–107. [Google Scholar] [CrossRef]
- Zhang, Y.; Tamijani, A.A.; Taylor, M.E.; Zhi, B.; Haynes, C.L.; Mason, S.E.; Hamers, R.J. Molecular Surface Functionalization of Carbon Materials via Radical-Induced Grafting of Terminal Alkenes. J. Am. Chem. Soc. 2019, 141, 8277–8288. [Google Scholar] [CrossRef]
- Moaseri, E.; Maghrebi, M.; Baniadam, M. Improvements in mechanical properties of carbon fiber-reinforced epoxy composites: A microwave-assisted approach in functionalization of carbon fiber via diamines. Mater. Des. 2014, 55, 644–652. [Google Scholar] [CrossRef]
- Li, H.; Liu, M.; Qi, Y.; Shen, Y. Study on properties of epoxy resin and polyurethane modified with organic silicon. Chem. J. 2018, 1. Available online: https://purkh.com/index.php/tochem/article/view/114 (accessed on 20 April 2022).
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Zhang, M.; June, S.M.; Long, T.E.; Kong, J. Principles of Step-Growth Polymerization (Polycondensation and Polyaddition). In Reference Module in Materials Science and Materials Engineering; Elsevier: Amsterdam, The Netherlands, 2016; p. 9780128035818015000. [Google Scholar] [CrossRef]
- Plueddemann, E.P. Adhesion through Silane Coupling Agents. In Fundamentals of Adhesion; Lee, L.-H., Ed.; Springer: Boston, MA, USA, 1991; pp. 279–290. [Google Scholar] [CrossRef]
- Silverstein, M.S. Interpenetrating polymer networks: So happy together? Polymer 2020, 207, 122929. [Google Scholar] [CrossRef]
- Kathi, J.; Rhee, K.-Y.; Lee, J.H. Effect of chemical functionalization of multi-walled carbon nanotubes with 3-aminopropyltriethoxysilane on mechanical and morphological properties of epoxy nanocomposites. Compos. Part Appl. Sci. Manuf. 2009, 40, 800–809. [Google Scholar] [CrossRef]
- Yu, Z.-Q.; You, S.-L.; Baier, H. Effect of organosilane coupling agents on microstructure and properties of nanosilica/epoxy composites. Polym. Compos. 2012, 33, 1516–1524. [Google Scholar] [CrossRef]
- Jiang, S.; Li, Q.; Zhao, Y.; Wang, J.; Kang, M. Effect of surface silanization of carbon fiber on mechanical properties of carbon fiber reinforced polyurethane composites. Compos. Sci. Technol. 2015, 110, 87–94. [Google Scholar] [CrossRef]
- Hoepfner, J.C.; Pezzin, S.H. Functionalization of carbon nanotubes with (3-glycidyloxypropyl)-trimethoxysilane: Effect of wrapping on epoxy matrix nanocomposites. J. Appl. Polym. Sci. 2016, 133, 44245. [Google Scholar] [CrossRef]
- Noparvar-Qarebagh, A.; Roghani-Mamaqani, H.; Salami-Kalajahi, M. Organic–inorganic nanohybrids of novolac phenolic resin and carbon nanotube: High carbon yields by using carbon nanotube aerogel and resin incorporation into aerogel network. Micropor. Mesopor. Mater. 2016, 224, 58–67. [Google Scholar] [CrossRef]
- Rafique, I.; Kausar, A.; Muhammad, B. Epoxy Resin Composite Reinforced with Carbon Fiber and Inorganic Filler: Overview on Preparation and Properties. Polym.-Plast. Technol. Eng. 2016, 55, 1653–1672. [Google Scholar] [CrossRef]
- Abdollahi, A.; Roghani-Mamaqani, H.; Salami-Kalajahi, M.; Mousavi, A.; Razavi, B.; Shahi, S. Preparation of organic-inorganic hybrid nanocomposites from chemically modified epoxy and novolac resins and silica-attached carbon nanotubes by sol-gel process: Investigation of thermal degradation and stability. Prog. Org. Coat. 2018, 117, 154–165. [Google Scholar] [CrossRef]
- Karaeva, A.R.; Kazennov, N.V.; Zhukova, E.A.; Mordkovich, V.Z. Carbon nanotubes by continuous growth, pulling and harvesting into big spools. Mater. Today Proc. 2018, 5, 25951–25955. [Google Scholar] [CrossRef]
- Wang, Q.; Su, D.S. Reinforcing epoxy resin with activated carbon: A way of high rate of quality and price. Compos. Commun. 2018, 9, 54–57. [Google Scholar] [CrossRef]
- Mao, L.; Shen, H.; Han, W.; Chen, L.; Li, J.; Tang, Y. Hybrid polyurethane and silane sized carbon fibre/epoxy composites with enhanced impact resistance. Compos. Part Appl. Sci. Manuf. 2019, 118, 49–56. [Google Scholar] [CrossRef]
- Long, J.C.; Zhan, H.; Wu, G.; Zhang, Y.; Wang, J.N. High-strength carbon nanotube/epoxy resin composite film from a controllable cross-linking reaction. Compos. Part Appl. Sci. Manuf. 2021, 146, 106409. [Google Scholar] [CrossRef]
- Fazeli, M.; Liu, X.; Rudd, C. The effect of waterborne polyurethane coating on the mechanical properties of epoxy-based composite containing recycled carbon fibres. Surf. Interfaces 2022, 29, 101684. [Google Scholar] [CrossRef]
- Malucelli, G.; Amerio, E.; Minelli, M.; De Angelis, M.G. Epoxy-siloxane hybrid coatings by a dual-curing process. Adv. Polym. Technol. 2009, 28, 77–85. [Google Scholar] [CrossRef]
- Lu, Y.; Li, H.; Lin, M.; Ng, W.; Liu, H. Mechanical properties of 3-glycidoxypropyltrimethoxysilane functionalized multi-walled carbon nanotubes/epoxy composites cured by electron beam irradiation. J. Compos. Mater. 2013, 47, 1685–1694. [Google Scholar] [CrossRef]
- Borda, J.; Kéki, S.; Ráthy, I.; Bodnár, I.; Zsuga, M. Novel polyurethane elastomer continuous carbon fiber composites: Preparation and characterization. J. Appl. Polym. Sci. 2007, 103, 287–292. [Google Scholar] [CrossRef]
- Sánchez-Adsuar, M.; Linares-Solano, Á.; Cazorla-Amorós, D.; Ibarra-Rueda, L. Influence of the nature and the content of carbon fiber on properties of thermoplastic polyurethane-carbon fiber composites. J. Appl. Polym. Sci. 2003, 90, 2676–2683. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adhesion Effect | Substrates |
|---|---|
| Excellent effect | Silica, alumina, glass, quartz, porcelain clay |
| Good effect | Mica, Talc, clay, water and alumina, grammiterion dust, potassium titanic acid |
| Slight effect | Asbestos, ferric oxides, zinc oxides, carborundum, silicon nitride |
| Poor effect | CaCO3, BaSO4, boron, carbon |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pustahija, L.; Kern, W. Surface Functionalization of (Pyrolytic) Carbon—An Overview. C 2023, 9, 38. https://doi.org/10.3390/c9020038
Pustahija L, Kern W. Surface Functionalization of (Pyrolytic) Carbon—An Overview. C. 2023; 9(2):38. https://doi.org/10.3390/c9020038
Chicago/Turabian StylePustahija, Lucija, and Wolfgang Kern. 2023. "Surface Functionalization of (Pyrolytic) Carbon—An Overview" C 9, no. 2: 38. https://doi.org/10.3390/c9020038
APA StylePustahija, L., & Kern, W. (2023). Surface Functionalization of (Pyrolytic) Carbon—An Overview. C, 9(2), 38. https://doi.org/10.3390/c9020038