Abstract
Multidentate ligands involving tethered pyridyl groups coordinated to transition metal ions have been frequently used to mimic the 3-histidine (3H), 2-histidine-1-carboxylate (2H1C) brace motifs or other combinations of histidine and carboxylate endogenous ligating residues found in bioinorganic metalloenzymes. It is of interest to immobilize these ligand chelates onto heterogeneous supports. This, however, requires the use of bromine-substituted (chloromethyl)pyridines, whose current synthetic routes involve the use of extremely pyrophoric chemicals, such as n-butyllithium that require cryogenic reaction conditions, and toxic chemicals, such as thionyl chloride, that are challenging to handle and require extensive hazard controls. Herein, we report alternative methodologies towards the syntheses of 2-bromo-6-hydroxymethylpyridine and 2-bromo-6-chloromethylpyridine from inexpensive commercially available 2,6-dibromopyridine using isopropylmagnesium chloride lithium chloride complex (Turbo Grignard) and cyanuric chloride which are easier to handle and require milder reaction conditions than the conventional reagents. Gas chromatography-mass spectrometry (GC-MS) methods were developed and simple 1H- and 13C- nuclear magnetic resonance (NMR) and Fourier-transform infrared (FT-IR) spectroscopies were also used to monitor the conversion of both reaction steps and showed that products could be obtained and isolated through simple workups without the presence of unreacted starting material or undesired overchlorinated 2-chloro-6-chloromethylpyridine side product.
1. Introduction
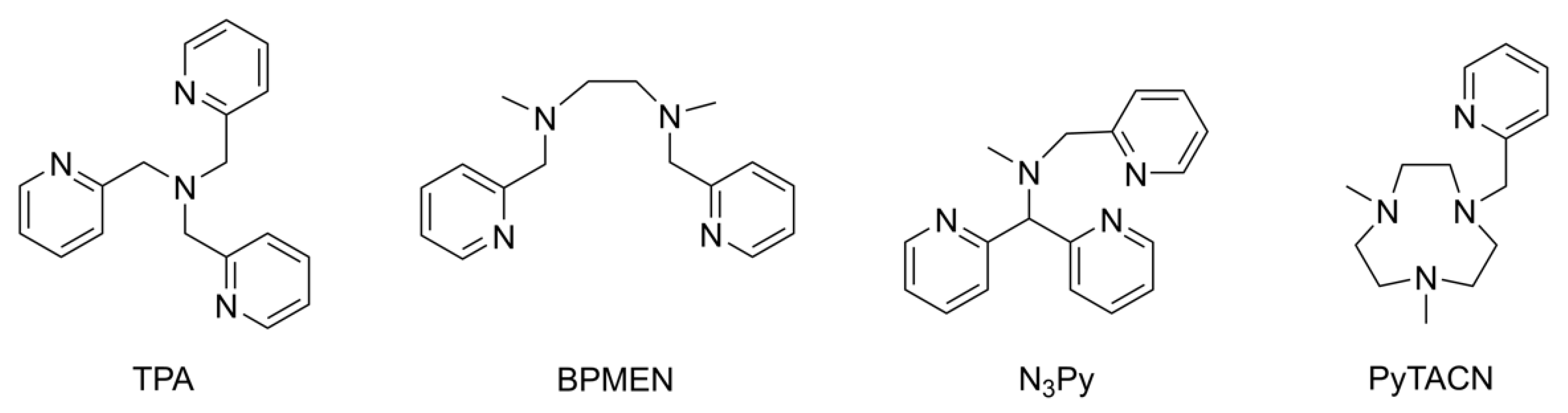
Pyridine based ligands play an important role in supporting transition metal ions for biomimetic chemistry [1,2,3,4,5,6,7,8,9,10]. Ligands containing two bis[2-(2-pyridyl)ethyl]amine (pye) moieties have been used on copper(I) to mimic copper containing oxygenases or hemocyanin by promoting dioxygen oxidation chemistry or reversible dioxygen binding, respectively, depending on the type of central linker [11,12,13]. Many non-haem iron containing metalloenzymes bind iron through moieties known as the 2-His-1-carboxylate, 3-His facial triads or other combinations of histidine and carboxylate endogenous ligating residues [14,15,16,17]. These structural motifs have been extensively modeled using pyridyl based ligands (Figure 1) that have been shown to support high valent metal-oxo species capable of activating C–H bonds of substrates similarly to their enzymatic counterparts [18,19,20,21,22,23]. The appeal of these heterocyclic N-donor groups is that they function as strong σ donors to support high oxidation states and provide flexibility in their functional derivatization for the tuning of primary coordination spheres [24].
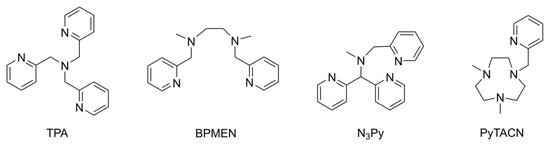
Figure 1.
Select pyridine-based ligands that have been used to synthesize biomimetic metal complexes.
It is of interest to link these biomimetic ligand scaffolds to solid supports towards the development of heterogeneous catalysts. These heterogeneous reactions would theoretically increase the ease of product isolation and catalyst recovery. Zhdankin and coworkers showed attaching N-(2-iodyl-phenyl)-acylamide, a derivative of 2-iodoxybenzoic acid (IBX), onto a polymer support afforded an efficient oxidizing system that could be recycled with only a moderate decline in activity over multiple recovery steps [25]. Polystyrene-based supports for palladium catalysts for Suzuki cross-couplings, rhodium catalysts for ketone synthesis, and ruthenium catalysts for olefin metathesis have also been developed and used to form heterogeneous catalysts that have shown comparable activities to their homogenous analogues and could be reused, some up to five to ten times, with no loss in activity [26]. This recyclability of catalyst increases the efficiency and atom economy of these catalytic transformations.
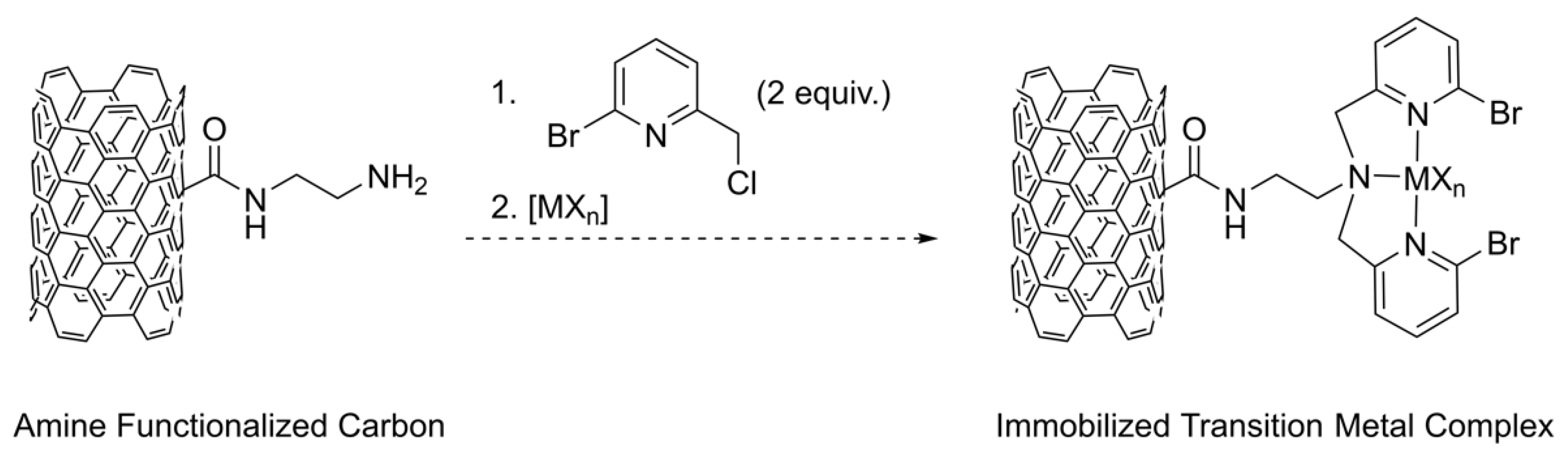
Other solid supports such as silica and activated carbons exist due to the ability to functionalize the surfaces of these materials with amine or oxygen containing groups such as lactones or carboxylic acids. Indeed, Radi and coworkers linked a bis(2-pyridylmethyl)amine chelate to functionalized silica for applications in heavy metal extractions [27]. Furthermore, one of us recently reported the grafting of polyethyleneimine (PEI) onto activated carbons for the removal of Cr(VI) ions from water at pH values ranging from 2 to 5 [28]. Extension of this activated carbon work includes linking the biomimetic pyridine ligand scaffolds onto activated carbon surfaces to study catalytic activity and recyclability of these activated carbon supported heterogeneous systems. However, linking of these pyridine-based ligands first requires the addition of chloromethyl groups onto the heteroaromatic ring to give electrophilic compounds capable of participating in substitution reactions with amine functionalized activated carbon nucleophiles. In addition, bromo substituents on the heteroaromatic ring are also highly desirable as they allow for subsequent functionalization post tethering to the solid support. This led us to search the literature for ways to synthesize the previously reported 2-bromo-6-chloromethylpyridine precursor compound [29,30,31] and we found some concern with the conventional routes and characterization of the compound, which necessitated initial modifications to these methods prior to us attempting our immobilization plans as depicted in Scheme 1.
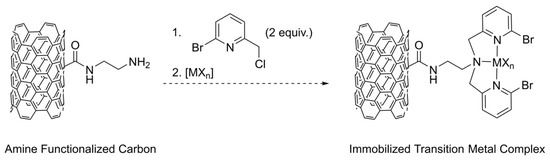
Scheme 1.
Proposed plan for the immobilization of transition metal ions onto amine functionalized carbons using the 2-bromo-6-chloromethylpyridine linker precursor described in this work.
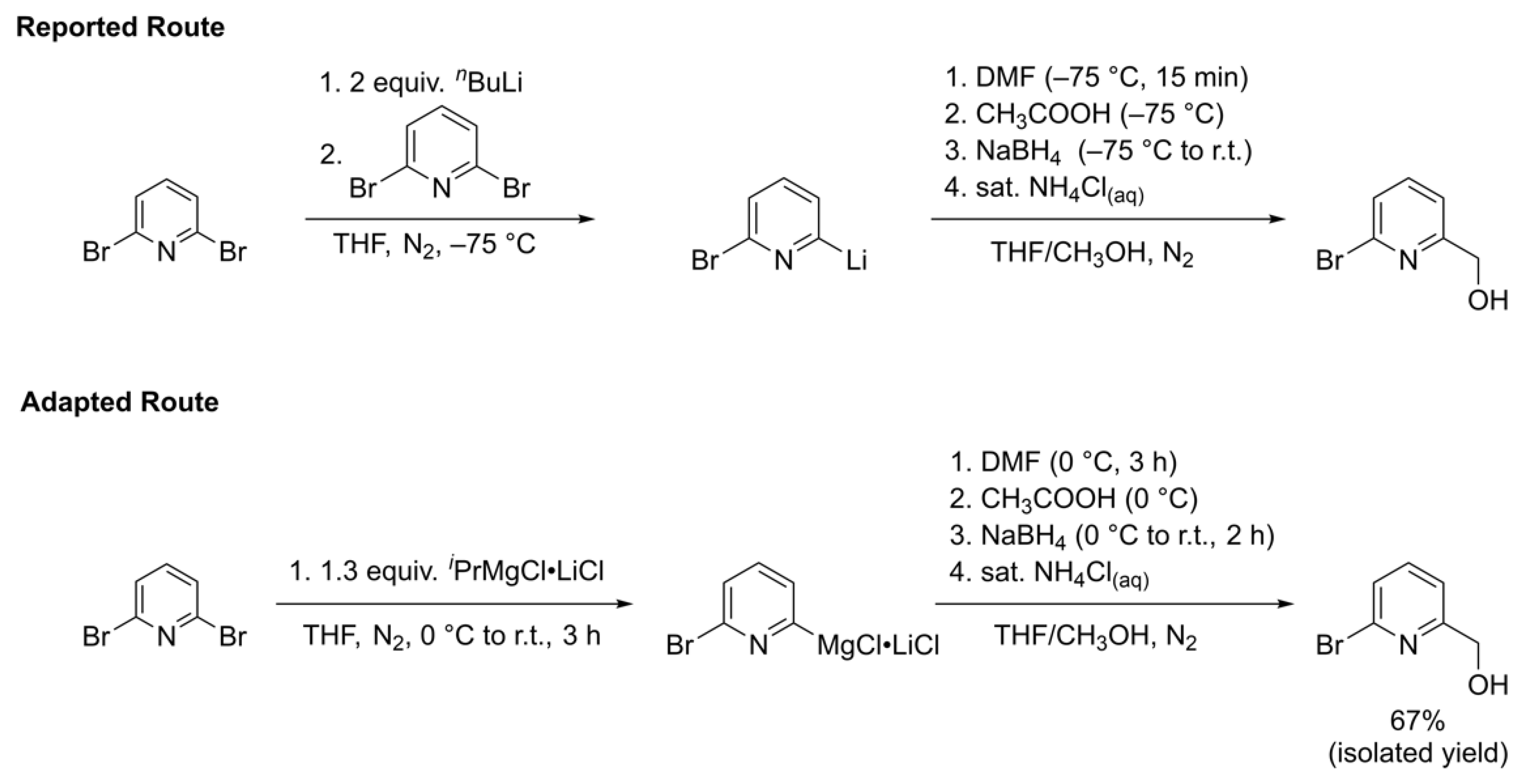
The first step in the synthesis of the 2-bromo-6-chloromethylpyridine precursor involves an initial metal halogen exchange on 2,6-dibromopyridine. As shown at the top of Scheme 2, the procedure published by Cai and coworkers indicates use of n-butyllithium as the metal halogen exchange reagent to achieve the mono-lithiated 2-bromo-6-lithiopyridine nucleophile [32]. Use of extremely pyrophoric materials such as n-butyllithium in synthetic procedures provide safety concerns, which inhibit their scalability. The researchers also warn about problems with selectivity when using n-butyllithium to prepare 2-bromo-6-hydroxymethylpyridine and mentioned in their hands that by adding just one equivalent of n-butyllithium, they observed a mixture of products involving acid-base side reactions rather than the desired metal halogen exchange product. These issues were amplified when scaling up the reaction since the slow addition of n-butyllithium is required to maintain the necessary −78 °C reaction temperature. Reversing the order of addition did not prove to be beneficial; rather, it led to the over-conversion to the di-lithiated pyridine reagent. The researchers were able to circumvent these issues by initially forming the di-lithiated pyridine using two equivalents of n-butyllithium and then subsequently performing halogen-lithium exchange between the 2,6-dilithiated pyridine intermediate and 2,6-dibromopyridine. However, the initial metal-halogen exchange still called for cryogenic temperatures and the handling of n-butyllithium as do the other reported syntheses of 2-bromo-6-hydroxymethylpyridine [33,34,35,36,37,38].
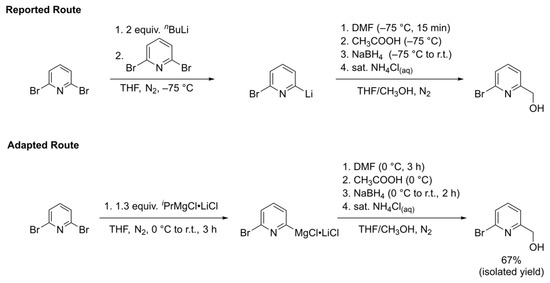
Scheme 2.
Reported route from reference [32] (top) and adapted route from this work (bottom) for the synthesis of 2-bromo-6-hydroxymethylpyridine.
We sought an alternative synthetic route to avoid the use of n-butyllithium, as shown in the bottom of Scheme 2, that involves the use of isopropylmagnesium chloride lithium chloride complex (Turbo Grignard). Knochel and coworkers reported that the addition of 1.05 equivalents of Turbo Grignard to 2,6-dibromopyridine successfully formed the 2-bromo-6-magnesiopyridine intermediate, which upon addition of benzaldehyde gave their desired (6-bromopyridin-2-yl)(phenyl)methanol product in 89% yield [39]. We hypothesized that this methodology could be expanded to use N,N-dimethylformamide (DMF) as the electrophile followed by borohydride reduction to give the desired 2-bromo-6-hydroxymethylpyridine alcohol under milder reaction conditions and without the need for n-butyllithium that could easily be scaled up with less safety concerns.
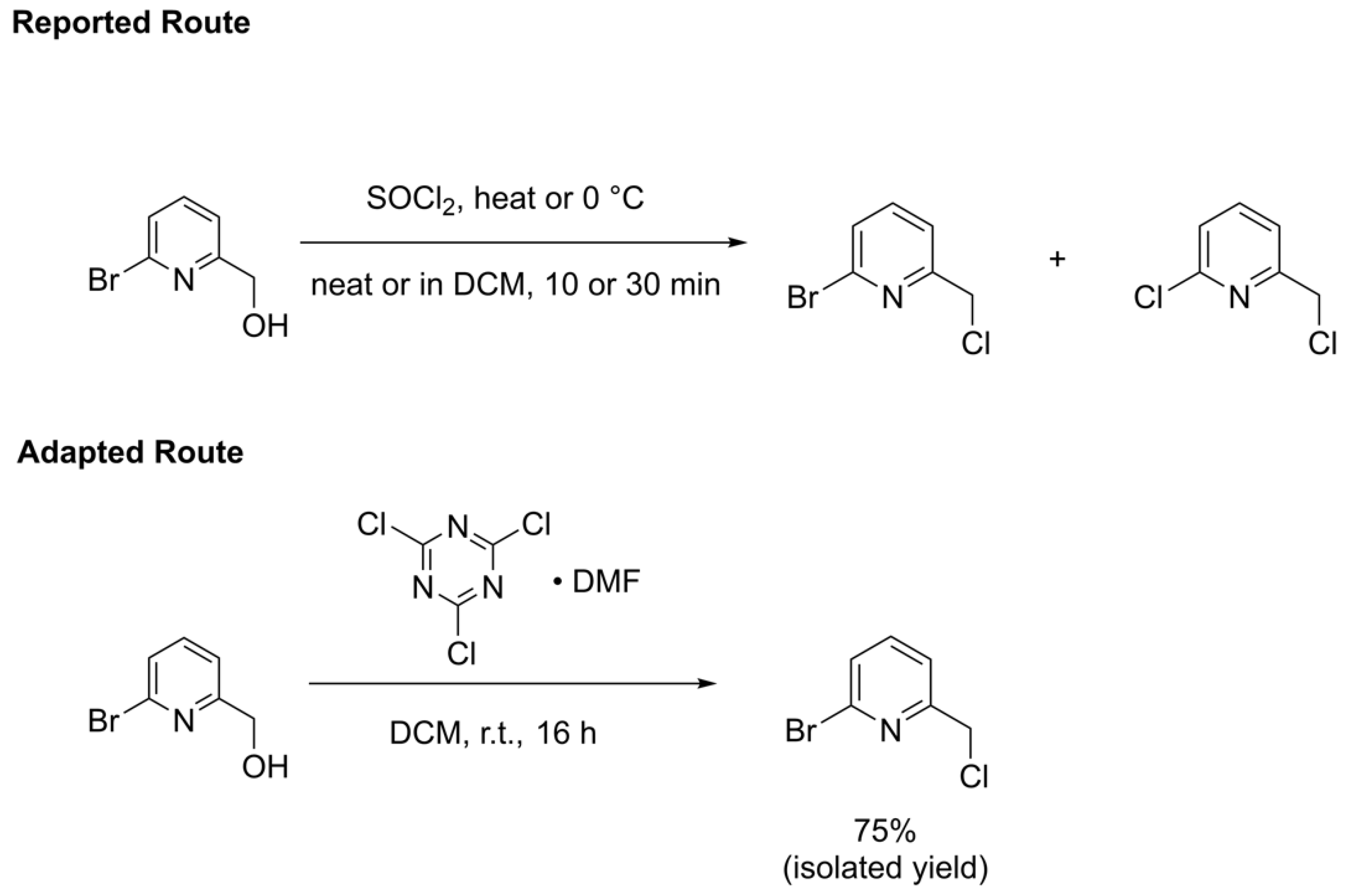
The final step to synthesizing the 2-bromo-6-chloromethylpyridine precursor involves a chlorine transfer to the 2-bromo-6-hydroxymethylpyridine alcohol. Conventional routes [29,30,31] use thionyl chloride (SOCl2) to transfer this chlorine, either neat or in halogenated solvent, at varying temperatures between slight heating to 0 °C (Scheme 3 top). SOCl2 is a robust chlorinating agent and indeed one paper [29] reporting the synthesis of 2-bromo-6-chloromethylpyridine using this reagent warns that allowing the reaction to run too long or heating at too high of a temperature could lead to the formation of 2-chloro-6-(chloromethyl)pyridine from the over-conversion of product, where the bromo substituent on the pyridine ring is also replaced with a chloro substituent. In addition, the reaction using thionyl chloride produces toxic sulfur dioxide gas as a byproduct, which also complicates use of this procedure on large production scales. The synthesis of the 2-bromo-6-chloromethylpyridine compound could, therefore, be improved through the use of a milder chlorine transfer reagent that would reduce the likelihood of over-chlorination. A promising reagent is the cyanuric chloride•DMF adduct reported by De Luca and coworkers [40] that lead cleanly to many alkyl chlorides. We sought to explore whether this reagent could be used as an alternative to SOCl2 for the synthesis of 2-bromo-6-chloromethylpyridine (Scheme 3 bottom). The byproduct of chlorination through the use of cyanuric chloride is solid cyanuric acid, which further helps simplify the scalability of this alternative synthetic route.
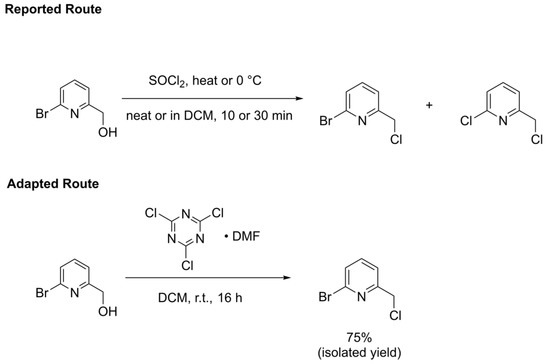
Scheme 3.
Reported route from references [29,30,31] (top) and adapted route from this work (bottom) for the synthesis of 2-bromo-6-chloromethylpyridine.
Herein, we report that these alternative synthetic routes towards the synthesis of 2-bromo-6-hydroxymethylpyridine and 2-bromo-6-chloromethylpyridine using Turbo Grignard and cyanuric chloride, respectively, were successful in cleanly converting to products in good yields. Furthermore, this manuscript also provides a thorough characterization of these compounds by 1H- and 13C-nuclear magnetic resonance (NMR) spectroscopies, Fourier-transform infrared (FT-IR) spectroscopy, and gas chromatography mass spectrometry (GC-MS), which can be used as experimental handles to monitor conversion to these products during synthesis or conversion from these compounds during future modification steps. These alternative synthetic routes can be used by researchers to prepare the 2-bromo-6-hydroxymethylpyridine and 2-bromo-6-chloromethylpyridine precursor compounds in a safer and more practical manner compared to the conventional syntheses currently published in the literature. In addition, the detailed characterization provided herein can be used by researchers for monitoring reactions involving these compounds.
2. Materials and Methods
2.1. General Methods
Air- and moisture-sensitive reactions and manipulations were performed under an inert nitrogen atmosphere using a standard Schlenk (Chemglass) manifold with syringe or cannula transfer techniques or in an AtmosBag (Sigma-Aldrich) purged three times. For moisture sensitive reactions, glassware was oven-dried for at least 3 h at 150 °C in a gravity convection oven (VWR). Volatiles were removed using either a rotary evaporator or the Schlenk line equipped with a Welch 1400B vacuum pump. 1H- and 13C-NMR spectroscopies were performed on a Bruker Avance-III 300 NMR spectrometer at room temperature and chemical shifts were recorded in units of parts per million (ppm) referenced from residual proteo solvent for 1H-NMR spectroscopy and from characteristic solvent peaks for 13C-NMR spectroscopy. Spectra were collected and analyzed using TopSpin (Bruker) software. FT-IR spectroscopy was performed on a Nicolet iS10 FTIR spectrometer (Thermo Scientific) equipped with a Smart iTR Attenuated Total Reflectance (ATR) sampling accessory and solids or oils were measured neat. Spectra were collected and analyzed using OMNIC (Thermo Scientific) software. GC-MS was performed on a Thermo Scientific Trace 1300 Gas Chromatograph with an AI 1310 autosampler paired to a Thermo Scientific ISQ single quadrupole mass spectrometer. Data was analyzed using Chromelon 7.2 chromatography software (Thermo Fisher). CHN Elemental Analysis was conducted by Robertson Microlit Laboratories on a Perkin-Elmer Model 2400 CHN Analyzer.
2.2. Materials
AcroSealed bottles of extra dry unstabilized tetrahydrofuran (THF), extra dry methanol (CH3OH) over molecular sieves, and extra dry N,N-dimethylformamide (DMF) over molecular sieves were purchased from ACROS Organics and used as received for moisture sensitive reactions. For non-moisture sensitive reactions or reaction workups, DMF, methanol, and n-hexane purchased from VWR Chemicals, and dichloromethane (DCM), chloroform (CHCl3), and ethyl acetate purchased from ACROS Organics were used without further modifications. Solid 2,6-dibromopyridine was purchased from TCI America. Turbo Grignard (1.3 M in THF) was purchased from Sigma-Aldrich and titrated against menthol (Sigma-Aldrich) and 1,10-phenanthroline (Sigma-Aldrich) according to published procedure [41]. Glacial acetic acid (CH3COOH) purchased from Fisher Scientific was used as received but sealed with electrical tape between uses to limit moisture intake. Sodium borohydride (NaBH4) was purchased from BeanTown Chemical and stored in a desiccator and sealed with electrical tape between uses to limit moisture intake and maintain packaging under inert atmosphere. Thionyl chloride (SOCl2) was purchased from Alfa Aesar and handled within a chemical fume hood. Cyanuric chloride was purchased from Sigma-Aldrich and stored in the refrigerator. Ammonium chloride (NH4Cl), sodium bicarbonate (NaHCO3), and sodium chloride (NaCl) purchased from Sigma Aldrich and sodium carbonate (Na2CO3) purchased from VWR Chemicals in deionized water were used for aqueous workups. Magnesium sulfate (MgSO4) purchased from VWR Chemicals was used as received to dry organic extracts. Silica (70–230 mesh, pore size 60 Å; Sigma-Aldrich) was used as received for column chromatography. Deuterated CDCl3 was purchased from Cambridge Isotopes, Inc. (Tewksbury, MA, USA) and used to prepare samples for NMR spectroscopy. Pyridine was purchased from Sigma Aldrich and dried over potassium hydroxide purchased from Flinn Scientific Inc. (Batavia, IL, USA) to prepare GC-MS samples.
2.3. Synthetic Procedures
2.3.1. 2-bromo-6-hydroxymethylpyridine
Prior to synthesizing the 2-bromo-6-hydroxymethylpyridine precursor, the bottle of Turbo Grignard was titrated against menthol with 1,10-phenanthroline as an indicator in the THF reaction solvent following a previously developed procedure [41]. This titration found the effective concentration of the first bottle of Turbo Grignard to be 1.0 M rather than 1.3 M as advertised. Performing this titration again on a second bottle of Turbo Grignard revealed an effective concentration of 0.85 M. The reagent masses used for the synthesis were adjusted to account for these effective concentrations.
To a 250 mL 3-necked round bottom flask equipped with a Teflon stir bar was added 50 mL of titrated Turbo Grignard (1.3 mol equiv.) using an airtight syringe. The reaction flask was cooled to 0 °C in an ice/water bath and a solution of 2,6-dibromopyridine (1 mol equiv.) in dry THF (30 mL) was cannula transferred into the flask. The flask was allowed to warm to room temperature and reacted for 3 h, after which a dark green-brown solution formed. The reaction flask was cooled back down to 0 °C in the ice/water bath and 2.2 mol equiv. of extra dry DMF was syringed into the flask. The reaction was maintained at 0 °C over 3 h and a more vivid dark green solution formed. CH3COOH (glacial, 1.4 mol equiv.) was syringed into the reaction flask dropwise forming a bright red solid, which was vigorously stirred to suspend the solid. To this suspension, a solution of NaBH4 (1.4 mol equiv.) in extra dry methanol (50 mL), prepared within an AtmosBag, was slowly cannula transferred into the reaction flask forming a bright yellow suspension. Caution: large amounts of gas releases upon addition of NaBH4 to the reaction flask and care should be taken while performing the transfer. The reaction flask was removed from the ice/water bath and allowed to react at room temperature for 2 h. The reaction was quenched with 100 mL of saturated aqueous NH4Cl, and the layers were separated. The aqueous layer was extracted with 3 × 50 mL CHCl3, dried over MgSO4, filtered, and concentrated on the rotary evaporator. The product was dried overnight under reduced pressure at 70 °C to yield pure 2-bromo-6-hydroxymethylpyridine as a brown oil in 67% yield. Analytically pure compound could be obtained through column chromatography using a methanol:DCM (1:9) solvent mixture (Rf = 0.58) or an ethyl acetate:hexane (1:1) solvent mixture (Rf = 0.42) as eluent. 1H-NMR (300 MHz, CDCl3, ppm): δ 7.56 (t, J = 7.68 Hz, 1H), 7.39 ppm (dd, J = 7.85 Hz and 0.67 Hz, 1 H), 7.32 (dd, J = 7.57 Hz and 0.56 Hz, 1H), 4.75 (s, 2H), 3.62 (br s, 1 H); 13C-NMR (75.5 MHz, CDCl3, ppm): δ 161.7, 141.2, 139.2, 126.6, 119.4, 64.2; FT-IR (cm−1): 3311, 2954, 2868, 2780, 1582, 1556, 1433, 1406, 1325, 1260, 1221, 1153, 1119, 1082, 1059, 985, 833, 780, 740, 704, 675; GC-MS (EI, m/z): 76, 77, 78, 79, 106, 107, 156, 157, 158, 159, 160, 171, 173, 186, 187, 188, 189. Anal. Calcd for: C, 38.33; H, 3.22; N, 7.45. Found: C, 38.37; H, 3.33; N, 7.45.
2.3.2. 2-bromo-6-chloromethylpyridine
To an Erlenmeyer flask equipped with a wide stir bar was added 9.23 g cyanuric chloride (50 mmol, 1.02 equiv.) and 15 mL DMF and allowed to react for 1 h to form the cyanuric chloride•DMF adduct. To this adduct was added a solution of 2-bromo-6-hydroxymethylpyridine (9.23 g, 49 mmol, 1 equiv.) in DCM (75 mL) and the reaction was allowed to react at room temperature overnight. The reaction was quenched with 300 mL of DI water and the layers were separated. The aqueous layer was extracted with 3 × 50 mL DCM and the organic extracts were dried over MgSO4, filtered, and concentrated down to a brown oil on the rotary evaporator. This brown oil was charged onto a column of silica and eluted with 600 mL of DCM. The eluent was concentrated down to a pale yellow oil on the rotary evaporator, triturated with DCM, and dried under reduced pressure at room temperature to yield 7.6 g of 2-bromo-6-hydroxymethylpyridine as an off white solid (75% yield). Analytically pure compound could be obtained via sublimation under reduced pressure at 70 °C. 1H-NMR (300 MHz, CDCl3, ppm): δ 7.60 (t, J = 7.72 Hz, 1H), 7.46 ppm (t, J = 8.32 Hz, 2 H), 4.64 (s, 2H); 13C-NMR (75.5 MHz, CDCl3, ppm): δ 157.9, 141.4, 139.4, 127.5, 121.6, 45.7; FT-IR (cm−1): 3080, 3069, 3048, 3016, 2964, 1604, 1577, 1553, 1474, 1438, 1407, 1349, 1328, 1290, 1259, 1237, 1167, 1115, 1081, 1018, 987, 916, 907, 854, 802, 744, 698, 669; GC-MS (EI, m/z): 79, 90, 91, 125, 126, 127, 128, 170, 171, 172, 205, 206, 207, 208, 209. Anal. Calcd for: C, 34.90; H, 2.44; N, 6.78. Found: C, 35.04; H, 2.34; N, 6.79.
2.4. GC-MS
To a 1.5 mL GC vial was added 30 mg of analyte and 1.0 mL of dried pyridine. Samples were injected (1.00 µL, split 100:1) into the Trace 1300 using a 10 µL Thermo Scientific syringe prewashed with methanol, hexane and sample using the Thermo Scientific AI 1310. The split splitless injector (SSL) was kept at a constant temperature of 280 °C, and was equipped with a glass liner packed with quartz wool to prevent any non-volatile species from contaminating the column. Data was collected using Chromelon 7.2 software for three minutes post injection, with the RTX 5 Sil MS column (Restek) heated to a constant 250 °C and ultrapure hydrogen carrier gas at a constant 1.0 mL min−1. The ISQ mass spectrometer was set to full scan mode with a mass range of 20 m/z to 250 m/z and a dwell time of 0.2 s scan−1. The transfer line was heated to 270 °C, and the mass spectrometer source was heated to 250 °C. The mass spectrometer was tuned prior to analysis using PFTBA.
3. Results and Discussion
3.1. Metal Halogen Exchange using Turbo Grignard
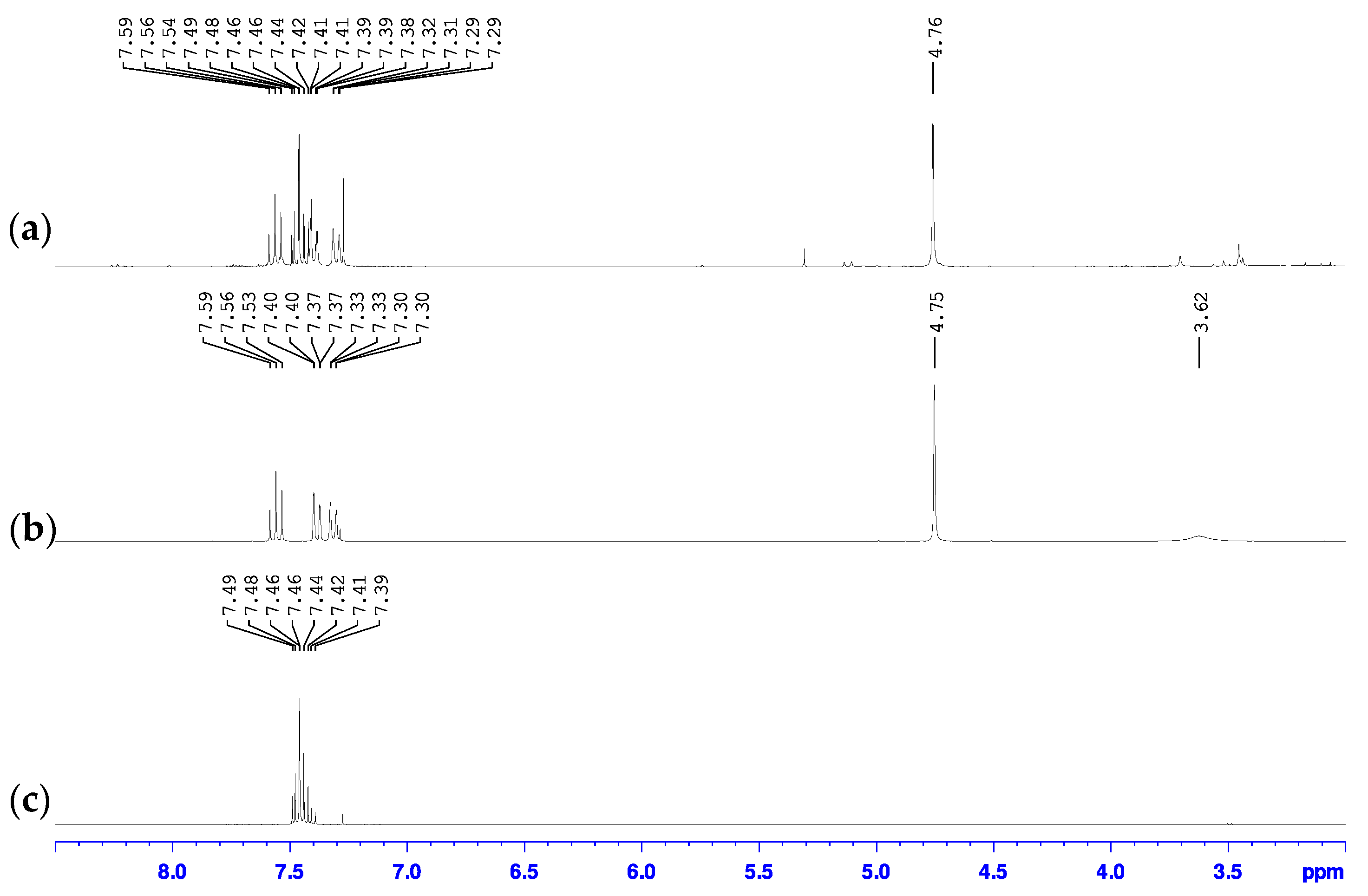
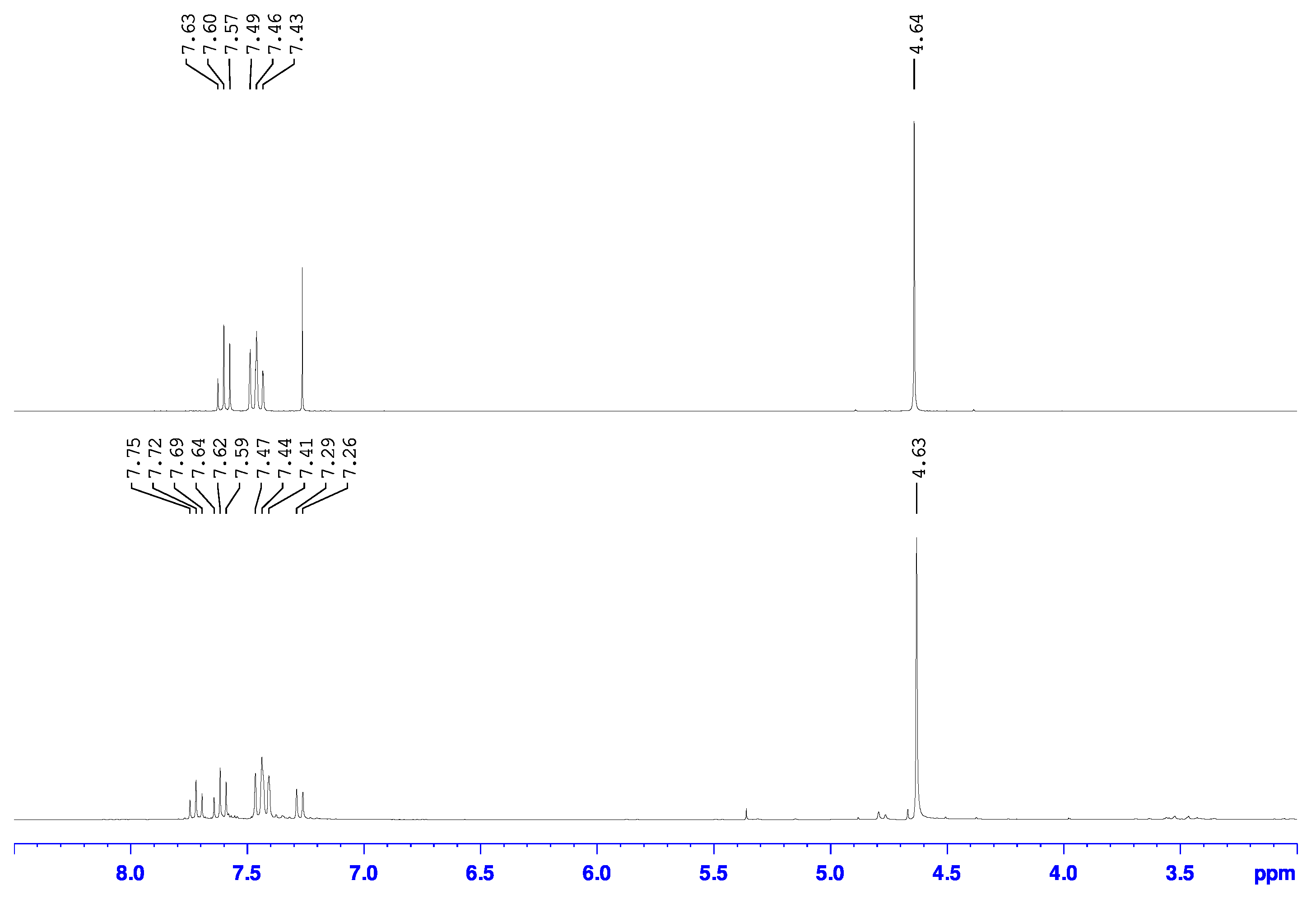
Our findings show that the use of Turbo Grignard to perform the initial metal halogen exchange step in the synthesis of 2-bromo-6-hydroxymethylpyridine is an appealing alternative to the use of n-butyllithium, having formed the desired product in good yields under milder reaction conditions (Scheme 2 bottom). Briefly, the 2-bromo-6-magnesiopyridine intermediate was formed following the procedure detailed by Knochel [39]. Without the need of isolating this intermediate, the reaction was cooled back down to 0 °C and dry N,N-dimethylformamide (DMF) was syringed into the reaction mixture forming a dark green solution which upon addition of glacial acetic acid created a vivid red suspension. Cannula transfer of a methanolic NaBH4 solution into this suspension released large amount of gas and formed a bright yellow suspension. Aqueous workup of this suspension isolated a dark brown oil with additional solid present in some cases. Analysis of these mixtures by 1H- and 13C- NMR spectroscopy and GC-MS indicated that the oil was the desired 2-bromo-6-hydroxymethylpyridine product while the solid was unconverted 2,6-dibromopyridine starting material. In the 1H-NMR spectrum of the mixture with the solid (Figure 2a), chemical shifts were observed at 4.76, 7.30, 7.40, and 7.56 ppm which matched those previously reported for 2-bromo-6-hydroxymethylpyridine [37,38,42], but a multiplet between 7.4 and 7.5 ppm that matched the spectrum for starting 2,6-dibromopyridine [43,44] (Figure 2c) was also observed. The 1H-NMR spectrum of the oil only showed chemical shifts associated with the 2-bromo-6-hydroxymethylpyridine product and the multiplet was not observed (Figure 2b). Furthermore, the 1H-NMR spectrum of the oil had an additional broad singlet at 3.62 ppm that was not observed in the crude mixture with the solid. This additional resonance was attributed to the hydrogen of the hydroxy group. Similarly, these same findings were observed in the 13C-NMR spectra of these samples (Figure S1) showing further support that the oil was the pure 2-bromo-6-hydroxymethylpyridine and the solid was unreacted 2,6-dibromopyridine starting material.
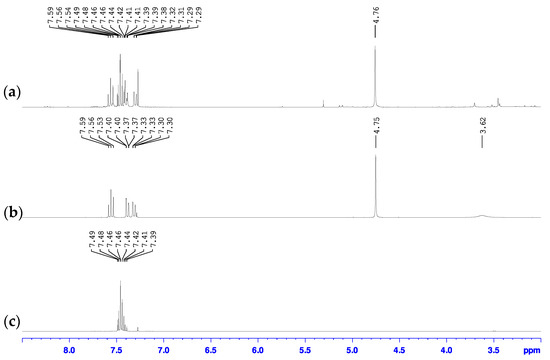
Figure 2.
Overlay of the 1H-NMR spectra of (a) oil mixture with solid, (b) pure oil, and (c) pure 2,6-dibromopyridine starting material in CDCl3.
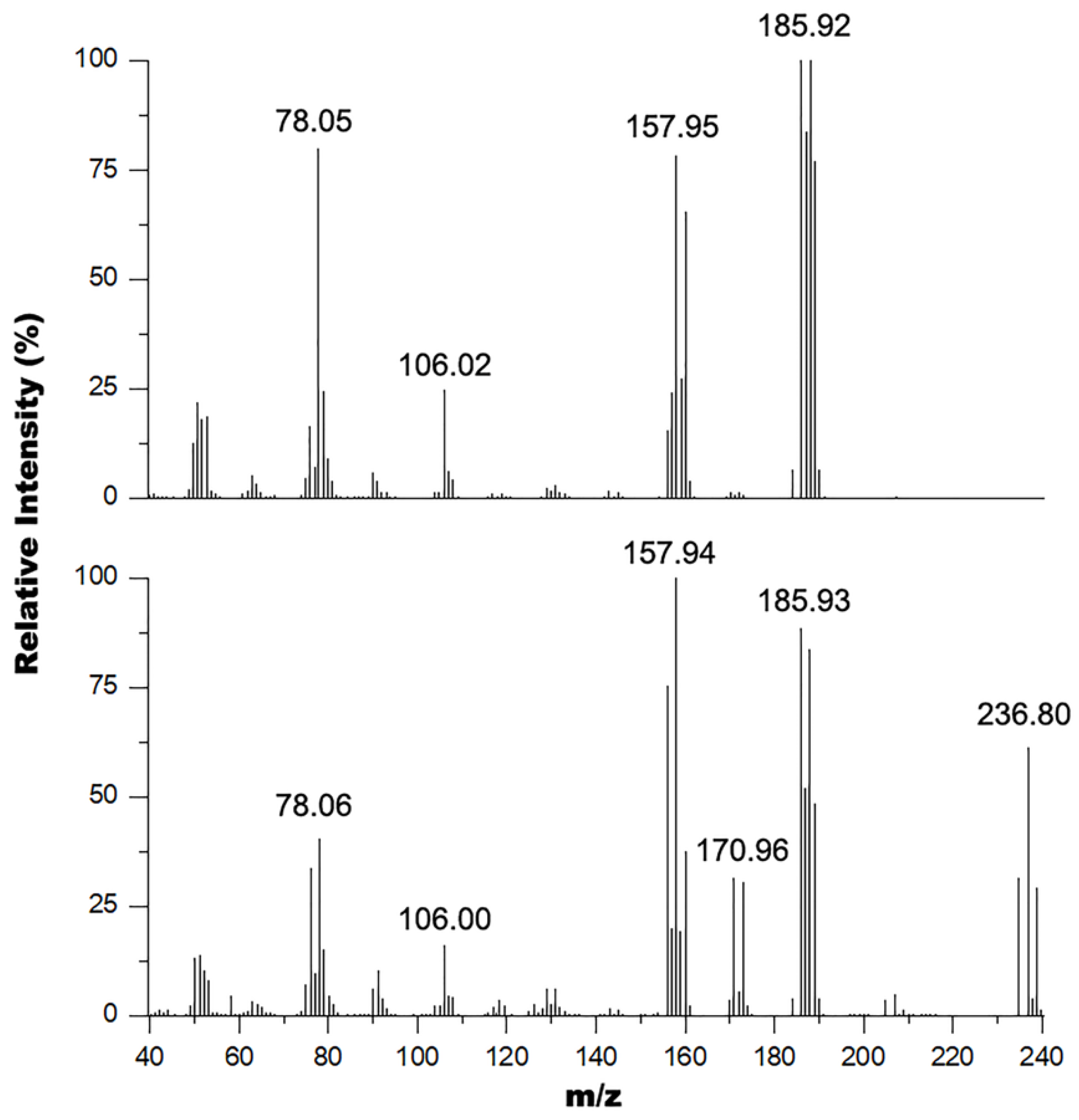
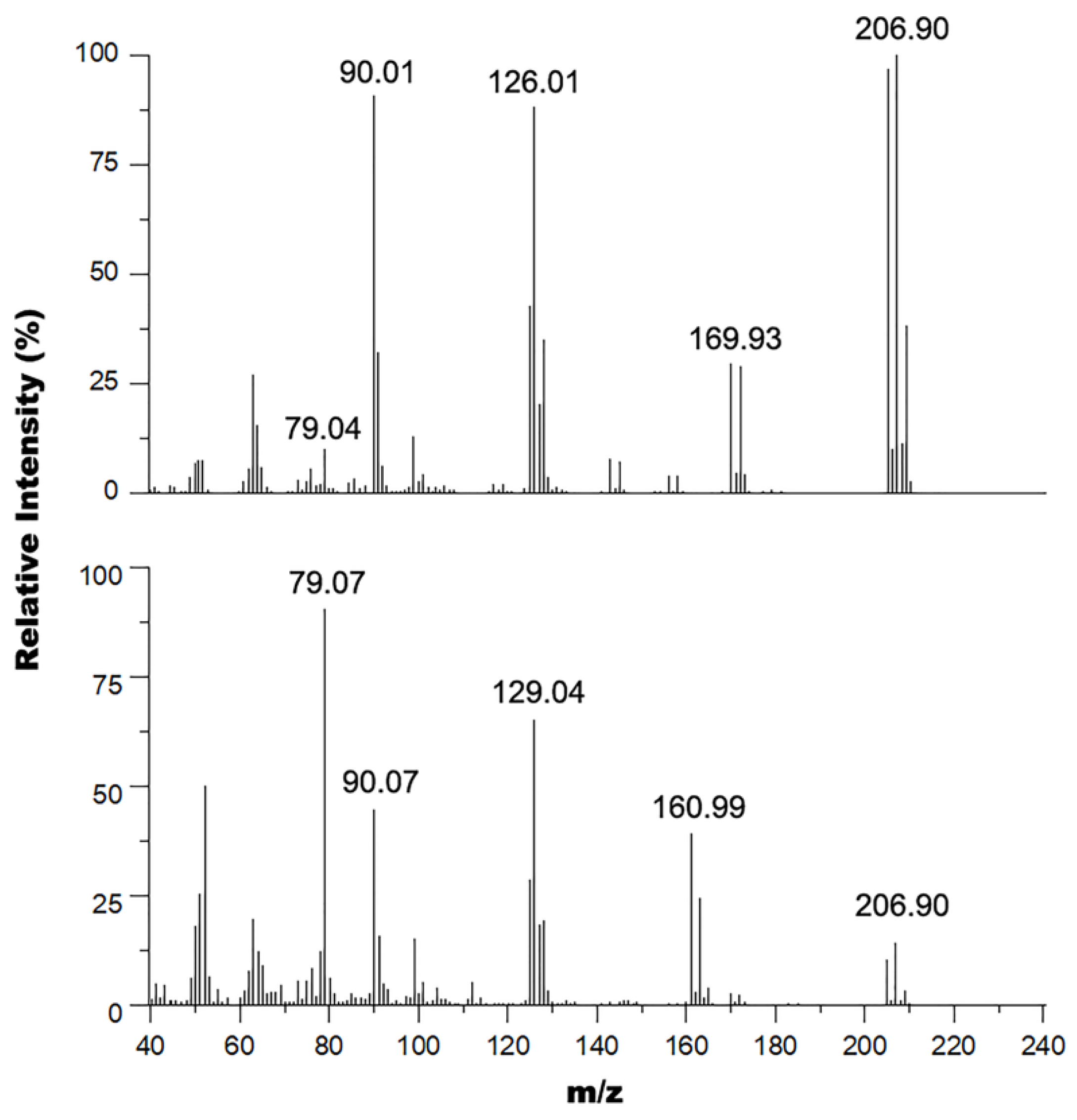
These results were corroborated by GC-MS experiments performed on these samples where the mass spectrum of the oil and solid mixture (Figure 3 bottom) shows signals at 235, 237, and 239 m/z in a 1:2:1 isotope pattern indicating the presence of two bromine atoms in the structure consistent with the 2,6-dibromopyrdine starting material (exact mass 234.86 amu). This spectrum also shows peaks at 186 and 188 m/z in a 1:1 isotope pattern indicating the presence of a single bromine atom in the structure consistent with the 2-bromo-6-hydroxymethylpyridine product (exact mass 186.96 amu). The other major peaks observed in this spectrum were 171, 158, 106 and 78 m/z corresponding to the 2-bromo-6-methylpyridine (exact mass 172.03 amu), 2-bromopyrdine (exact mass 156.95 amu), 2-hydroxymethylpyridine (exact mass 109.05 amu), and pyridine (exact mass 79.04 amu) fragments of the parent ions respectively within the mass spectrometer. Comparatively, the mass spectrum of the oil alone (Figure 3 top) does not contain peaks around the 237 m/z region, and only contains peaks corresponding to the 2-bromo-6-hydroxymethylpyridine product and its predominant fragments. The spectrum for this product is similar to the EI mass spectrum obtained by Bolm and coworkers [42] with the exception that they did not observe the 106 m/z fragment.
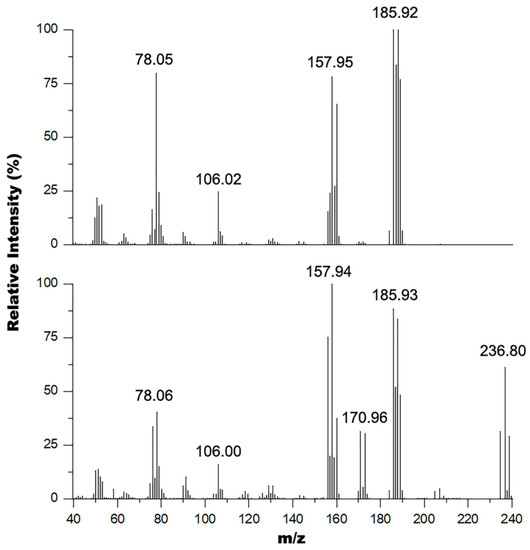
Figure 3.
Overlay of the mass spectra of the pure oil (top) and oil mixture with solid (bottom).
After adjusting the stoichiometry of reagents to account for the appropriate effective concentration of Turbo Grignard found through titration, we were able to successfully synthesize only the 2-bromo-6-hydroxymethylpyridine oil product with no solid 2,6-dibromopyridine present. In our hands, there were no issues with scaling up this adapted synthetic route to a similar scale as that used by Cai et al. [32]. The absence of starting material in these samples was confirmed by 1H- and 13C-NMR spectroscopies and GC-MS. Elemental analysis on analytically pure oil that was isolated found C, H, and N percentage values within 0.11%, confirming no impurities or byproducts were present. This oil could be used in a subsequent chlorine transfer step to form the final 2-bromo-6-chloromethylpyridine product without further purification.
3.2. Chlorine Transfer Using Cyanuric Chloride versus Thionyl Chloride
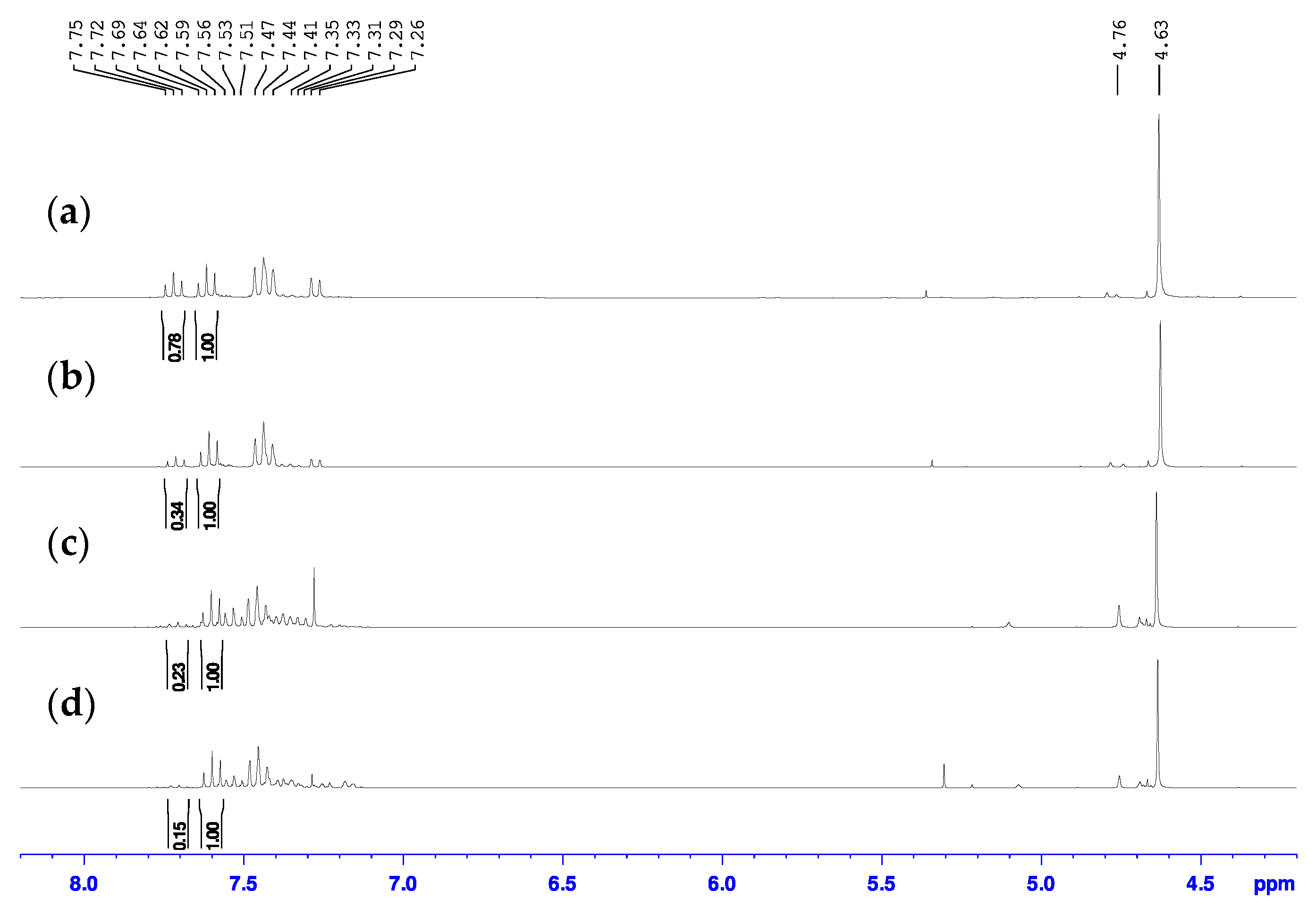
Initial attempts to complete this chlorine transfer reaction were performed following the literature procedures using thionyl chloride [29,30,31]. In our hands, however, the use of thionyl chloride did not lead to clean results. The first reaction attempt towards 2-bromo-6-chloromethylpyridine product involved the addition of neat SOCl2 following the procedure reported by Chuang et al. [29]. While the researchers specify in their work that they performed the reaction for 10 min, they did not indicate the reaction temperature, so we first attempted the reaction at 40 °C and analyzed the reaction mixture by NMR spectroscopy. In order for the 2-bromo-6-chloromethylpyridinium chloride in the reaction mixture to dissolve in CDCl3 for 1H-NMR spectroscopic analysis to compare with the literature data, we first had to rinse the reaction mixture dissolved in chloroform with aqueous sodium bicarbonate, followed by extraction of the organic layer. We were surprised to clearly see two species in the 1H-NMR spectrum (Figure 4a) since the authors claimed that the overconverted product was indistinguishable from the desired 2-bromo-6-chloromethylpyridine product by NMR spectroscopy and could only be identified by running subsequent reaction products through liquid chromatography (LC)-MS [29]. One of the species was the desired product which had triplets at 7.46 and 7.62 ppm in the aromatic region, which matched reported data [29,30,31]. The other species had a doublet at 7.28 ppm, an unresolved overlapping peak at 7.48 ppm, and a triplet at 7.72 ppm in the aromatic region. We believe this other species to in fact be the 2-chloro-6-chloromethylpyridine since the peaks match very closely to the known 1H-NMR spectrum of that compound as reported by Pesti and coworkers [45]. Again, a similar observation was made when analyzing the 13C-NMR spectra of these samples (Figure S2) and determined that the desired 2-bromo-6-chloromethylpyridine product had chemical shifts of 45.7, 121.6, 127.5, 139.4, 141.4, and 157.8 ppm. To the best of our knowledge this is the first report in the literature of the 13C-NMR spectrum of this compound.
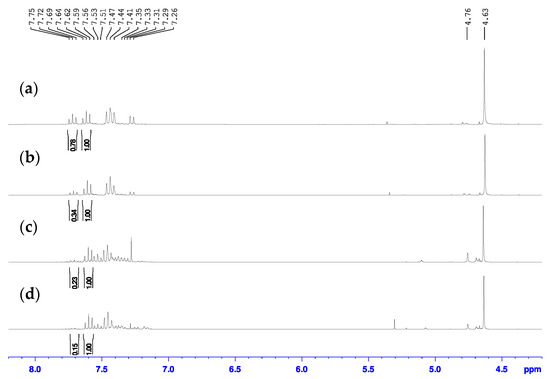
Figure 4.
Overlay of the 1H-NMR spectra for the product mixtures from the reaction of 2-bromo-6-hydroxymethylpyridine with SOCl2 (a) neat at 40 °C, (b) neat at r.t., (c) neat at 0 °C, (d) in DCM at 0 °C in CDCl3.
According to the ratio of the integration of the triplet at 7.72 ppm to the triplet at 7.62 ppm of 0.78 to 1.00 in the 1H-NMR spectrum, the reaction at 40 °C had a 56% conversion to desired 2-bromo-6-chloromethylpyridine and 44% overconversion to 2-chloro-6-chloromethylpyridine. The 2-bromo-6-chloromethylpyridine and 2-chloro-6-chloromethylpyridine compounds had similar retention factors on silica using dichloromethane as eluent and could not easily be separated. Therefore, to reduce the amount of overconverted 2-chloro-6-chloromethylpyridine side product present in these reaction mixtures, we also performed the reaction at room temperature and 0 °C and the 1H-NMR spectra of these attempts are also shown in Figure 4b,c. According to the ratios of the integration of the triplet at 7.72 ppm to that at 7.62 ppm of 0.34 to 1.00 and 0.23 to 1.00, the reaction at room temperature had a 75% conversion to 2-bromo-6-chloromethylpyridine and 25% conversion to 2-chloro-6-chloromethylpyridine while that at 0 °C had approximately an 81% conversion to 2-bromo-6-chloromethylpyridine and 19% conversion to 2-chloro-6-chloromethylpyridine, respectively. The other resonances within the aromatic region of the 0 °C reaction are speculated to be unconverted starting material based on their chemical shifts.
While the overconversion to 2-chloro-6-chloromethylpyridine did reduce upon reduction of the temperature, it was still present. To combat these issues, a final attempt was performed using thionyl chloride following the reported procedure by Tanaka and coworkers where thionyl chloride was added to a DCM solution of starting alcohol rather than neat and stirred at 0 °C [30]. While it was difficult to detect overconverted 2-chloro-6-chloromethylpyridine side product in the 1H-NMR spectrum of the reaction mixture from this attempt (Figure 4d), there still seemed to be a small triplet present at 7.72 ppm that integrated to 0.15 to the 1.00 integration of the triplet at 7.62 ppm, which corresponds to an approximate 87% conversion to 2-bromo-6-chloromethylpyridine and 13% conversion to 2-chloro-6-chloromethylpyridine. This reaction mixture is also speculated to contain small amounts of unconverted starting material. These results are summarized in Table 1.

Table 1.
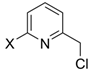
Conversion percentages of 2-bromo-6-hydroxymethylpyridine to 2-bromo-6-chloromethylpyridine (x = Br) and 2-chloro-6-chloromethylpyridine (x = Cl) using SOCl2 under specified reaction conditions.
The use of the milder cyanuric chloride•DMF adduct, reported by De Luca et al. [40], successfully overcame challenges associated with over-chlorination. Briefly, a DCM solution of 2-bromo-6-hydroxymethylpyridine was added to the freshly prepared cyanuric chloride•DMF adduct and reacted overnight (Scheme 3, bottom). Upon aqueous workup and quick purification through a short plug of silica using DCM as eluent, the desired 2-bromo-6-chloromethylpryidine was isolated cleanly in 75% yield as an off-white solid without the presence of any overconverted 2-chloro-6-chloromethylpyridine side product or unreacted starting alcohol, even on a similar scale as that used by Chuang et al. [29]. Elemental analysis on analytically pure sublimed solid found C, H, and N percentage values within 0.14%, confirming no impurities or byproducts were present.
In the 1H-NMR spectrum of the compound synthesized through the use of cyanuric chloride (Figure 5 top), only the triplets at 7.46 and 7.60 ppm in the aromatic region were observed without any detection of the resonances corresponding to the 2-chloro-6-chloromethylpyridine side product. The methylene hydrogens were observed at 4.64 ppm which indicates an upfield shift of 0.11 ppm compared to the starting alcohol and is consistent with the less deshielding of the hydrogens by the less electronegative chloro group attached to that carbon as has been reported [29,30,31]. Again, a similar observation was observed in the 13C-NMR spectrum, which also only had the chemical shifts associated with the species assigned to be 2-bromo-6-chloromethylpyridine.
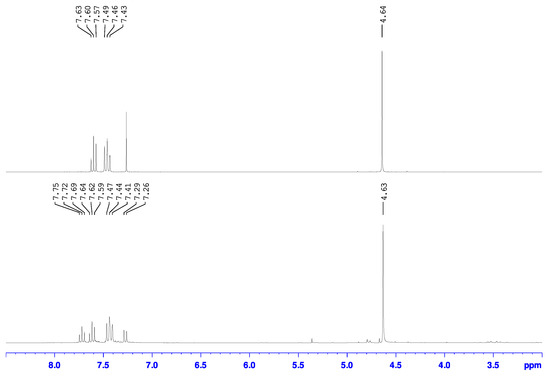
Figure 5.
Overlay of the 1H-NMR spectra for the product mixture from the reaction of 2-bromo-6-hydroxymethylpyridine with cyanuric chloride•DMF in DCM at r.t (top) and SOCl2 at 40 °C (bottom) in CDCl3.
GC-MS spectra of the reaction mixtures when using thionyl chloride (Figure 6, bottom) also confirmed presence of 2-chloro-6-chloromethylpyridine side product as it contained peaks at 161 and 163 m/z in a 1:0.66 isotope pattern indicating two chlorine atoms in the structure consistent with the 2-chloro-6-chloromethylpyridine (exact mass 160.98 amu) side product. The spectrum also shows peaks at 205 and 207 m/z in a 1:1.33 isotope pattern indicating a bromine atom and a chlorine atom in the structure consistent with the 2-bromo-6-chloromethylpyridine product (exact mass 204.930 amu). The other major peaks observed in this spectrum were 129 m/z, corresponding to the 2-chloro-6-methylpyridine (exact mass 127.57 amu) and/or 2-chloromethylpyridine (exact mass 127.02 amu), 90 m/z, corresponding to the 2-methylpyridine (exact mass 93.06 amu), and 79 m/z corresponding to the pyridine fragments of the parent ions.
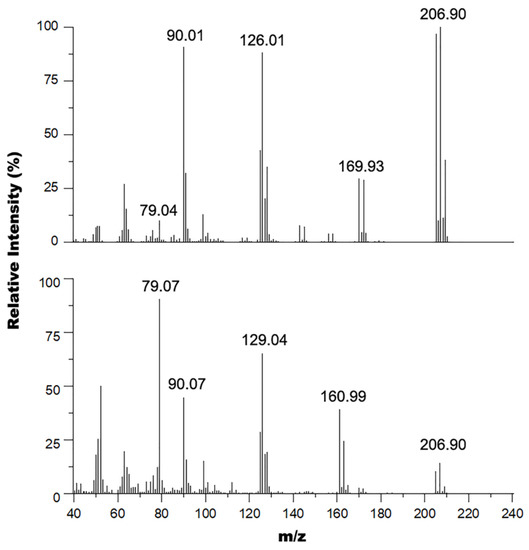
Figure 6.
Overlay of the mass spectra of the product mixture from the reaction of 2-bromo-6-hydroxymethylpyridine with cyanuric chloride•DMF in DCM at r.t (top) and SOCl2 at 40 °C (bottom).
Similarly, formation of the 2-bromo-6-chloromethylpyridine compound without any presence of 2-chloro-6-chloromethylpyridine side product using the cyanuric chloride•DMF adduct as chlorine transfer reagent was also confirmed by GC-MS experiments. The GC-MS spectrum of the reaction mixture when using cyanuric chloride•DMF (Figure 6, top) shows a similar fragmentation pattern to the one discussed above for the mixture containing the 2-chloro-6-chloromethylpyridine (Figure 6, bottom) side product with the key distinction being the lack of a signal for the side product’s parent ion (161 m/z region). In addition, there is a more predominant signal for the fragment at 170 m/z corresponding to 2-bromo-6-methylpyridine, which could only have formed from fragmentation of the 2-bromo-6-chloromethylpyridine desired product within the GC-MS system. Another distinction we observed was a slight difference in fragmentation pattern at the 126 m/z region due to the presence of only the chloromethylpyridine fragment and lack of the 2-chloro-6-methylpyridine fragment from the side product’s parent ion.
Finally, the replacement of the hydroxy group in 2-bromo-6-hydroxymethylpyridine with the chloro group to give 2-bromo-6-chloromethylpyridine was monitored by FT-IR spectroscopy with the broad ν(OH) stretching vibration being used as an easy probe. As shown in Figure S18, the ν(OH) peak at 3311 cm−1 in the spectrum for 2-bromo-6-hydroxymethylpyridine, which matches closely with the reported frequency values for that vibrational mode [36,42,46], is clearly missing in the spectrum for the 2-bromo-6-chloromethylpyridine product. This confirms that IR spectroscopy provides a good handle for loss of the hydroxy group during the chlorine transfer step and could be used to monitor the conversion of 2-bromo-6-hydroxymethylpyridine to further functionalized products such as 2-bromo-6-chloromethylpyridine.
4. Conclusions
As described, the two pyridine derivatives, 2-bromo-6-hydroxymethylpyridine and 2-bromo-6-chloromethylpyridine were successfully prepared by modified synthetic routes that bypass the use of harsh reagents that require careful handling and difficult reaction conditions. We show that the Turbo Grignard reagent, after titrating to ensure correct reaction stoichiometry, could be used as an alternative to n-butyllithium for metal halogen exchange of 2,6-dibromopyridine that only required reaction conditions with temperatures no colder than 0 °C rather than −78 °C. Similarly, in the following chlorine transfer step towards the formation of 2-bromo-6-chloromethylpyridine from the alcohol precursor, the adduct between cyanuric chloride and DMF was shown to be a much more benign reagent which cleanly lead to product with greater selectivity compared to the conventional thionyl chloride reagent, which was shown by us to easily lead to over-chlorinated 2-chloro-6-chloromethylpyridine side product. The alternative routes described in this manuscript provide more practical ways of synthesizing these important pyridine precursors, which will be of benefit to researchers who desire to incorporate pyridyl groups into their materials.
This manuscript also provides thorough analyses of the reaction steps by 1H- and 13C-NMR spectroscopies, GC-MS, and FT-IR spectroscopy. These various characterization techniques all provide consistent results indicating that without titrating the Turbo Grignard and maintaining the correct stoichiometry of the metal halogen exchange reaction, final reaction mixtures contained a combination of desired 2-bromo-6-hydroxymethylpyridine product and unreacted 2,6-dibromopyridine starting material. Maintaining the correct reaction stoichiometry, however, led to clean and complete conversion to desired alcohol product. Similarly, in the chlorine transfer step, these analyses show that the use of thionyl chloride led to final reaction mixtures that contained both desired 2-bromo-6-chloromethylpyridine product as well as undesired 2-chloro-6-chloromethylpyridine side product. The use of the adduct between cyanuric chloride and DMF, however, led to reaction mixtures that only contained desired 2-bromo-6-chloromethylpyridine product.
With pure 2-bromo-6-hydroxymethylpyridine and 2-bromo-6-chloromethylpyridine in hand, a complete spectroscopic characterization of these compounds was obtained. While the 1H-NMR spectrum [29,30,31] and high resolution EI mass spectrum [31] of 2-bromo-6-chloromethylpyridine have been obtained, this report, to the best of our knowledge, provides the first collection of the 13C-NMR spectrum and EI mass spectrum obtained through gas chromatography. Furthermore, while the 2-bromo-6-hydroxymethylpyridine was run through GC-MS analysis by Huang et al. [47], they only report the m/z peak of the parent ion with largest relative intensity while this manuscript provides a complete analysis and thorough assignment of the entire GC mass spectrum (EI) of this parent compound including regions of the most predominant ion fragments. These more thorough and detailed characterization methods and analyses of the 2-bromo-6-hydroxymethylpyridine and 2-bromo-6-chloromethylpyridine compounds will assist researchers in monitoring reactions involving use of these compounds.
Future experiments involve the linking of the 2-bromo-6-chloromethylpyridine precursor synthesized through these improved adapted routes onto amine functionalized carbons, the characterization of these materials, and affinity studies of these immobilized chelates to bind transition metal ions.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/c7030054/s1, Figure S1: Overlay of the 13C-NMR spectra of (a) oil mixture with solid, (b) pure oil, and (c) pure 2,6-dibromopyridine starting material in CDCl3., Figure S2: Overlay of the 13C-NMR spectra for the product mixture from the reaction of 2-bromo-6-hydroxymethylpyridine with cyanuric chloride•DMF in DCM at r.t (top) and SOCl2 at 40 °C (bottom) in CDCl3., Figure S3: Overlay of the aromatic regions of the 1H-NMR spectra for the product mixtures from the reaction of 2-bromo-6-hydroxymethylpyridine with SOCl2 (a) neat at 40 °C, (b) neat at r.t., (c) neat at 0 °C, (d) in DCM at 0 °C in CDCl3., Figure S4: 1H-NMR Spectrum of the reaction mixture containing 2-bromo-6-hydroxymethylpyridine and 2,6-dibromopyridine in CDCl3., Figure S5: 13C-NMR Spectrum of the reaction mixture containing 2-bromo-6-hydroxymethylpyridine and 2,6-dibromopyridine in CDCl3., Figure S6: 1H-NMR Spectrum of 2-bromo-6-hydroxymethylpyridine in CDCl3., Figure S7: 13C-NMR Spectrum of 2-bromo-6-hydroxymethylpyridine in CDCl3., Figure S8: 1H-NMR Spectrum of 2,6-dibromopyridine in CDCl3., Figure S9: 13C-NMR Spectrum of 2,6-dibromopyridine in CDCl3., Figure S10: 1H-NMR Spectrum of the reaction mixture containing 2-bromo-6-chloromethylpyridine and 2-chloro-6-chloromethylpyridine in CDCl3., Figure S11: 13C-NMR Spectrum of the reaction mixture containing 2-bromo-6-chloromethylpyridine and 2-chloro-6-chloromethylpyridine in CDCl3., Figure S12: 1H-NMR Spectrum of 2-bromo-6-chloromethylpyridine in CDCl3., Figure S13: 13C-NMR Spectrum of 2-bromo-6-chloromethylpyridine in CDCl3., Figure S14: Mass spectrum of the reaction mixture containing 2-bromo-6-hydroxymethylpyridine and 2,6-dibromopyridine., Figure S15: Mass spectrum of 2-bromo-6-hydroxymethylpyridine., Figure S16: Mass spectrum of the reaction mixture containing 2-bromo-6-chloromethylpyridine and 2-chloro-6-chloromethylpyridine., Figure S17: Mass spectrum of 2-bromo-6-chloromethylpyridine., Figure S18: Overlay of the FT-IR spectra of 2-bromo-6-hydroxymethylpyridine (red) and the product of the reaction with cyanuric chloride (blue).
Author Contributions
Conceptualization, J.A.B.; methodology, J.A.B. and T.T.H.; software, J.A.B. and M.R.E.; validation, J.A.B., T.T.H., T.M., A.K., H.S., Y.K. and M.R.E.; formal analysis, J.A.B.; investigation, J.A.B., T.T.H., T.M., A.K., H.S., Y.K. and M.R.E.; resources, J.A.B., and M.R.E.; data curation, J.A.B., and T.T.H.; writing—original draft preparation, J.A.B., and T.T.H.; writing—review and editing, J.A.B., T.T.H. and M.R.E.; visualization, J.A.B.; supervision, J.A.B. and M.R.E.; project administration, J.A.B.; funding acquisition, J.A.B. and M.R.E. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not Applicable.
Acknowledgments
This work was supported by the Becton College Grant-In-Aid program and the Department of Chemistry, Biochemistry and Physics at Fairleigh Dickinson University. The authors of this manuscript would like to thank graduate student Joshua Nelson and supervisor, Eric J. Schelter, of the University of Pennsylvania for lending of reagents for preliminary testing and helpful discussion.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Sun, W.; Sun, Q. Bioinspired Manganese and Iron Complexes for Enantioselective Oxidation Reactions: Ligand Design, Catalytic Activity, and Beyond. Acc. Chem. Res. 2019, 52, 2370–2381. [Google Scholar] [CrossRef]
- Costas, M.; Tipton, A.K.; Chen, K.; Jo, D.-H.; Que, L. Modeling Rieske Dioxygenases: The First Example of Iron-Catalyzed Asymmetric Cis-Dihydroxylation of Olefins. J. Am. Chem. Soc. 2001, 123, 6722–6723. [Google Scholar] [CrossRef]
- White, M.C.; Doyle, A.G.; Jacobsen, E.N. A Synthetically Useful, Self-Assembling MMO Mimic System for Catalytic Alkene Epoxidation with Aqueous H2O2. J. Am. Chem. Soc. 2001, 123, 7194–7195. [Google Scholar] [CrossRef]
- Oddon, F.; Girgenti, E.; Lebrun, C.; Marchi-Delapierre, C.; Pécaut, J.; Ménage, S. Iron Coordination Chemistry of N2Py2 Ligands Substituted by Carboxylic Moieties and Their Impact on Alkene Oxidation Catalysis. Eur. J. Inorg. Chem. 2012, 2012, 85–96. [Google Scholar] [CrossRef]
- Chen, M.S.; White, M.C. A Predictably Selective Aliphatic C–H Oxidation Reaction for Complex Molecule Synthesis. Science 2007, 318, 783–787. [Google Scholar] [CrossRef]
- Gómez, L.; Garcia-Bosch, I.; Company, A.; Benet-Buchholz, J.; Polo, A.; Sala, X.; Ribas, X.; Costas, M. Stereospecific C-H Oxidation with H2O2 Catalyzed by a Chemically Robust Site-Isolated Iron Catalyst. Angew. Chem. Int. Ed. 2009, 48, 5720–5723. [Google Scholar] [CrossRef]
- Bukowski, M.R.; Comba, P.; Lienke, A.; Limberg, C.; de Laorden, C.L.; Mas-Ballesté, R.; Merz, M.; Que, L., Jr. Catalytic Epoxidation and 1,2-Dihydroxylation of Olefins with Bispidine–Iron(II)/H2O2 Systems. Angew. Chem. Int. Ed. 2006, 45, 3446–3449. [Google Scholar] [CrossRef]
- Costas, M.; Mehn, M.P.; Jensen, M.P.; Que, L. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev. 2004, 104, 939–986. [Google Scholar] [CrossRef] [PubMed]
- Tshuva, E.Y.; Lippard, S.J. Synthetic Models for Non-Heme Carboxylate-Bridged Diiron Metalloproteins: Strategies and Tactics. Chem. Rev. 2004, 104, 987–1012. [Google Scholar] [CrossRef]
- Keown, W.; Gary, J.B.; Stack, T.D.P. High-Valent Copper in Biomimetic and Biological Oxidations. JBIC J. Biol. Inorg. Chem. 2017, 22, 289–305. [Google Scholar] [CrossRef]
- Itoh, S.; Abe, T.; Morimoto, Y.; Sugimoto, H. 2-(2-Pyridyl)Ethylamine (Pye) Ligands in Copper(I)-Dioxygen Chemistry. Inorg. Chim. Acta 2018, 481, 38–46. [Google Scholar] [CrossRef]
- Karlin, K.D.; Hayes, J.C.; Gultneh, Y.; Cruse, R.W.; McKown, J.W.; Hutchinson, J.P.; Zubieta, J. Copper-Mediated Hydroxylation of an Arene: Model System for the Action of Copper Monooxygenases. Structures of a Binuclear Copper(I) Complex and Its Oxygenated Product. J. Am. Chem. Soc. 1984, 106, 2121–2128. [Google Scholar] [CrossRef]
- Karlin, K.D.; Haka, M.S.; Cruse, R.W.; Meyer, G.J.; Farooq, A.; Gultneh, Y.; Hayes, J.C.; Zubieta, J. Dioxygen-Copper Reactivity Models for Hemocyanin: Reversible O2 and CO Binding to Structurally Characterized Dicopper(I) Complexes Containing Hydrocarbon-Linked Bis[2-(2-Pyridyl)Ethyl]Amine Units. J. Am. Chem. Soc. 1988, 110, 1196–1207. [Google Scholar] [CrossRef]
- Koehntop, K.D.; Emerson, J.P.; Que, L. The 2-His-1-Carboxylate Facial Triad: A Versatile Platform for Dioxygen Activation by Mononuclear Non-Heme Iron(II) Enzymes. JBIC J. Biol. Inorg. Chem. 2005, 10, 87–93. [Google Scholar] [CrossRef]
- Diebold, A.R.; Neidig, M.L.; Moran, G.R.; Straganz, G.D.; Solomon, E.I. The Three-His Triad in Dke1: Comparisons to the Classical Facial Triad. Biochemistry 2010, 49, 6945–6952. [Google Scholar] [CrossRef] [Green Version]
- Monkcom, E.C.; de Bruin, D.; de Vries, A.J.; Lutz, M.; Ye, S.; Gebbink, R.J.M.K. Structurally Modelling the 2-His-1-Carboxylate Facial Triad with a Bulky N,N,O Phenolate Ligand. Chem. Eur. J. 2021, 27, 5191–5204. [Google Scholar] [CrossRef]
- Monkcom, E.C.; Ghosh, P.; Folkertsma, E.; Negenman, H.A.; Lutz, M.; Gebbink, R.J.M.K. Bioinspired Non-Heme Iron Complexes: The Evolution of Facial N, N, O Ligand Design. Chim. Int. J. Chem. 2020, 74, 450–466. [Google Scholar] [CrossRef]
- Lindhorst, A.C.; Haslinger, S.; Kühn, F.E. Molecular Iron Complexes as Catalysts for Selective C–H Bond Oxygenation Reactions. Chem. Commun. 2015, 51, 17193–17212. [Google Scholar] [CrossRef] [Green Version]
- Rohde, J.-U.; Stubna, A.; Bominaar, E.L.; Münck, E.; Nam, W.; Que, L. Nonheme Oxoiron(IV) Complexes of Tris(2-Pyridylmethyl)Amine with Cis-Monoanionic Ligands. Inorg. Chem. 2006, 45, 6435–6445. [Google Scholar] [CrossRef]
- Andris, E.; Jašík, J.; Gómez, L.; Costas, M.; Roithová, J. Spectroscopic Characterization and Reactivity of Triplet and Quintet Iron(IV) Oxo Complexes in the Gas Phase. Angew. Chem. Int. Ed. 2016, 55, 3637–3641. [Google Scholar] [CrossRef] [Green Version]
- Dantignana, V.; Serrano-Plana, J.; Draksharapu, A.; Magallón, C.; Banerjee, S.; Fan, R.; Gamba, I.; Guo, Y.; Que, L.; Costas, M.; et al. Spectroscopic and Reactivity Comparisons between Nonheme Oxoiron(IV) and Oxoiron(V) Species Bearing the Same Ancillary Ligand. J. Am. Chem. Soc. 2019, 141, 15078–15091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Lutz, M.; Milan, M.; Costas, M.; Otte, M.; Gebbink, R.J.M.K. Non-Heme Iron Catalysts with a Rigid Bis-Isoindoline Backbone and Their Use in Selective Aliphatic C−H Oxidation. Adv. Synth. Catal. 2017, 359, 2590–2595. [Google Scholar] [CrossRef]
- Singh, R.; Ganguly, G.; Malinkin, S.O.; Demeshko, S.; Meyer, F.; Nordlander, E.; Paine, T.K. A Mononuclear Nonheme Iron(IV)-Oxo Complex of a Substituted N4Py Ligand: Effect of Ligand Field on Oxygen Atom Transfer and C–H Bond Cleavage Reactivity. Inorg. Chem. 2019, 58, 1862–1876. [Google Scholar] [CrossRef]
- Park, H.; Lee, D. Ligand Taxonomy for Bioinorganic Modeling of Dioxygen-Activating Non-Heme Iron Enzymes. Chem. Eur. J. 2020, 26, 5916–5926. [Google Scholar] [CrossRef] [PubMed]
- Karimov, R.R.; Kazhkenov, Z.-G.M.; Modjewski, M.J.; Peterson, E.M.; Zhdankin, V.V. Preparation and Reactivity of Polymer-Supported 2-Iodylphenol Ethers, an Efficient Recyclable Oxidizing System. J. Org. Chem. 2007, 72, 8149–8151. [Google Scholar] [CrossRef]
- McNamara, C.A.; Dixon, M.J.; Bradley, M. Recoverable Catalysts and Reagents Using Recyclable Polystyrene-Based Supports. Chem. Rev. 2002, 102, 3275–3300. [Google Scholar] [CrossRef] [PubMed]
- Radi, S.; Tighadouini, S.; Bacquet, M.; Degoutin, S.; Janus, L.; Mabkhot, Y.N. Fabrication and Covalent Modification of Highly Chelated Hybrid Material Based on Silica-Bipyridine Framework for Efficient Adsorption of Heavy Metals: Isotherms, Kinetics and Thermodynamics Studies. RSC Adv. 2016, 6, 82505–82514. [Google Scholar] [CrossRef]
- Smith, V.A.; Rivera, J.F.A.; Bello, R.; Rodríguez-Aguado, E.; Elshaer, M.R.; Wodzinski, R.L.; Bashkova, S. The Role of Surface Chemistry and Polyethylenimine Grafting in the Removal of Cr (VI) by Activated Carbons from Cashew Nut Shells. C 2021, 7, 27. [Google Scholar] [CrossRef]
- Chuang, C.-L.; Dos Santos, O.; Xu, X.; Canary, J.W. Synthesis and Cyclic Voltammetry Studies of Copper Complexes of Bromo- and Alkoxyphenyl-Substituted Derivatives of Tris(2-Pyridylmethyl)Amine: Influence of Cation-Alkoxy Interactions on Copper Redox Potentials. Inorg. Chem. 1997, 36, 1967–1972. [Google Scholar] [CrossRef]
- Tanaka, K.; Ewing, W.R.; Yu, J.-Q. Hemilabile Benzyl Ether Enables γ-C(Sp3)–H Carbonylation and Olefination of Alcohols. J. Am. Chem. Soc. 2019, 141, 15494–15497. [Google Scholar] [CrossRef]
- Kmentova, I.; Sutherland, H.S.; Palmer, B.D.; Blaser, A.; Franzblau, S.G.; Wan, B.; Wang, Y.; Ma, Z.; Denny, W.A.; Thompson, A.M. Synthesis and Structure−Activity Relationships of Aza- and Diazabiphenyl Analogues of the Antitubercular Drug (6S)-2-Nitro-6-{[4-(Trifluoromethoxy)Benzyl]Oxy}-6,7-Dihydro-5H-Imidazo[2,1-b][1,3]Oxazine (PA-824). J. Med. Chem. 2010, 53, 8421–8439. [Google Scholar] [CrossRef]
- Cai, D.; Hughes, D.L.; Verhoeven, T.R. A Study of the Lithiation of 2,6-Dibromopyridine with Butyllithium, and Its Application to Synthesis of L-739,010. Tetrahedron Lett. 1996, 37, 2537–2540. [Google Scholar] [CrossRef]
- Meth-Cohn, O.; Jiang, H. Ligands Containing Alternating 2,6-Linked Pyridine and 2,5-Linked Thiophene Units 1. J. Chem. Soc. Perkin Trans. 1998, 22, 3737–3746. [Google Scholar] [CrossRef]
- Li, X.; Gibb, C.L.D.; Kuebel, M.E.; Gibb, B.C. Two New Ligands for Carbonic Anhydrase Mimicry. Tetrahedron 2001, 57, 1175–1182. [Google Scholar] [CrossRef]
- Landa, A.; Minkkilä, A.; Blay, G.; Jørgensen, K.A. Bis(Oxazoline) Lewis Acid Catalyzed Aldol Reactions of Pyridine N-Oxide Aldehydes—Synthesis of Optically Active 2-(1-Hydroxyalkyl)Pyridine Derivatives: Development, Scope, and Total Synthesis of an Indolizine Alkaloid. Chem. Eur. J. 2006, 12, 3472–3483. [Google Scholar] [CrossRef]
- Tsukube, H.; Uenishi, J.; Higaki, H.; Kikkawa, K.; Tanaka, T.; Wakabayashi, S.; Oae, S. Side Arm Effects on Cation Binding, Extraction, and Transport Functions of Oligopyridine-Functionalized Aza-Crown Ethers. J. Org. Chem. 1993, 58, 4389–4397. [Google Scholar] [CrossRef]
- Ulrich, S.; Buhler, E.; Lehn, J.-M. Reversible Constitutional Switching between Macrocycles and Polymers Induced by Shape Change in a Dynamic Covalent System. New J. Chem. 2009, 33, 271–292. [Google Scholar] [CrossRef]
- Maindron, N.; Poupart, S.; Hamon, M.; Langlois, J.-B.; Plé, N.; Jean, L.; Romieu, A.; Renard, P.-Y. Synthesis and Luminescence Properties of New Red-Shifted Absorption Lanthanide(Iii) Chelates Suitable for Peptide and Protein Labelling. Org. Biomol. Chem. 2011, 9, 2357–2370. [Google Scholar] [CrossRef] [PubMed]
- Krasovskiy, A.; Knochel, P. A LiCl-Mediated Br/Mg Exchange Reaction for the Preparation of Functionalized Aryl- and Heteroarylmagnesium Compounds from Organic Bromides. Angew. Chem. Int. Ed. 2004, 43, 3333–3336. [Google Scholar] [CrossRef]
- De Luca, L.; Giacomelli, G.; Porcheddu, A. An Efficient Route to Alkyl Chlorides from Alcohols Using the Complex TCT/DMF. Org. Lett. 2002, 4, 553–555. [Google Scholar] [CrossRef]
- Lin, H.-S.; Paquette, L.A. A Convenient Method for Determining the Concentration of Grignard Reagents. Synth. Commun. 1994, 24, 2503–2506. [Google Scholar] [CrossRef]
- Bolm, C.; Schlingloff, G.; Harms, K. Catalyzed Enantioselective Alkylation of Aldehydes. Chem. Ber. 1992, 125, 1191–1203. [Google Scholar] [CrossRef]
- Do, H.-Q.; Daugulis, O. A Simple Base-Mediated Halogenation of Acidic Sp2 C−H Bonds under Noncryogenic Conditions. Org. Lett. 2009, 11, 421–423. [Google Scholar] [CrossRef] [Green Version]
- Popov, I.; Do, H.-Q.; Daugulis, O. In Situ Generation and Trapping of Aryllithium and Arylpotassium Species by Halogen, Sulfur, and Carbon Electrophiles. J. Org. Chem. 2009, 74, 8309–8313. [Google Scholar] [CrossRef] [Green Version]
- Pesti, J.A.; Huhn, G.F.; Yin, J.; Xing, Y.; Fortunak, J.M.; Earl, R.A. Efficient Pyridinylmethyl Functionalization: Synthesis of 10,10-Bis[(2-Fluoro-4-Pyridinyl)Methyl]-9(10H)-Anthracenone (DMP 543), an Acetylcholine Release Enhancing Agent. J. Org. Chem. 2000, 65, 7718–7722. [Google Scholar] [CrossRef]
- Shawcross, A.P.; Stanforth, S.P. Preparation of Some Substituted Imidazo[1,2-a]Pyridine Derivatives. J. Heterocycl. Chem. 1993, 30, 563–565. [Google Scholar] [CrossRef]
- Huang, S.; Hong, X.; Cui, H.-Z.; Zhan, B.; Li, Z.-M.; Hou, X.-F. Bimetallic Bis-NHC-Ir(III) Complex Bearing 2-Arylbenzo[d]Oxazolyl Ligand: Synthesis, Catalysis, and Bimetallic Effects. Organometallics 2020, 39, 3514–3523. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).