Abstract
Natural photosynthesis inspired the scientific community to design and synthesize molecular assemblies that possess advanced light-harvesting and electron-transfer features. In this review, we present the preparation and the photophysical investigation of novel porphyrin–fullerene hybrids acting as artificial photosynthetic systems. Porphyrinoids stand as chlorophyll analogues and have emerged as suitable photosensitizers in supramolecular electron donor–acceptor hybrids. Fullerenes (C60) are versatile electron acceptors with small reorganization energy and low reduction potentials. The novel derivatives presented herein mimic the fundamental features of the photosynthetic reaction center, namely, light harvesting, charge separation, and charge transport. To this end, a comprehensive analysis on these key processes that occur in various porphyrin–fullerene entities is illustrated in this work.
1. Introduction
The primary process in natural photosynthesis is the collection and subsequently the conversion of sunlight into chemical energy [1]. To achieve this conversion, sequential photo-induced energy transfers are required, which are followed by an electron transfer process in the reaction center [2]. The photosynthetic reaction center of photosystem II consists of a protein scaffold that contains porphyrinoids in a self-assembled structure [3]. This porphyrinoid-based scaffold is the light harvesting antenna system which absorbs sunlight and efficiently transfers it to the reaction center. Natural systems have motivated researchers to develop artificial photosynthetic systems that mimic the basic processes of photosynthesis in order to achieve solar energy conversion into valuable fuel products and electricity [4,5].
In an ideal artificial photosynthetic scheme, a light harvesting dye absorbs solar irradiation and, upon its excitation, transfers the energy to another dye—the so-called photosensitizer. Subsequently, an electron transfer process occurs between the photosensitizer and an electron acceptor, forming a charge-separated state [1]. Throughout the last few decades, the research community has developed simple artificial photosynthetic schemes in order to comprehend all the important processes that govern natural photosynthesis [2]. In such systems, the electron and/or energy transfer processes between all components that comprise the artificial scheme were studied extensively. Notably, the majority of such artificial photosynthetic systems in the literature are based on porphyrinoids and fullerene derivatives [6,7].
Porphyrinoids, are extremely versatile materials from a synthetic point of view [8] and thus have been widely utilized in various chemistry fields, such as opto-electronics [9], catalysis [10,11], electrochemistry [12,13], etc. Due to their unique characteristics, namely, strong absorption features, great photo- and thermal stability, and most importantly, tunable electro-hemical and photophysical properties [14], are the ideal photosensitizers in artificial schemes [15]. Thus, tailoring the physical and the chemical characteristics of porphyrin derivatives stimulated their broad use in applications such as artificial photosynthesis and solar energy conversion [5,16]. The most frequently studied systems consist of porphyrin derivatives coupled with an energy donor and/or an electron acceptor moiety. Chromophores such as boron–dipyrromethenes (BODIPYs) or ferrocenes have been widely used as energy donors, while the most commonly tested electron acceptors are fullerenes, carbon nanotubes, and naphthalene diimides [17].
Undoubtedly, the most broadly investigated electron acceptors in artificial schemes are fullerenes and particularly C60, since these carbon-based materials retain distinctive electronic and photophysical features. More specifically, their rigid structure permits the stabilization of charged moieties through the delocalization of electrons or holes. Furthermore, fullerenes and their carbon-based derivatives exhibit rich electrochemical behavior, namely, six equally spaced reversible reductions can be detected in their cyclic voltammograms [18]. Last but not least, fullerenes can diminish the charge recombination processes, due to their small reorganization energy, promoting an increased lifetime of the ion pair.
There are two main methods to connect chromophores with electron acceptors: i) the covalent approach in which the moieties are linked together through a rigid bond and ii) the noncovalent connection of entities via supramolecular interactions (dipoles, hydrogen bonds, π–π interactions, and metal–ligand axial coordination) [6]. In general, the covalently linked donor–acceptor assemblies present increased lifetime of the charge-separated state compared to the supramolecular systems. To date, the most efficient artificial photosynthetic scheme, in terms of intramolecular charge transfer, is a covalently linked ferrocene–porphyrin–fullerene hybrid described by Imahori et al. [19]. More specifically, the reported triad displayed a lifetime of the charge-separated state of 0.38 seconds, a value comparable to that observed for the bacterial photosynthetic reaction center [19].
In this perspective review article, we illustrate examples of porphyrinoid–fullerene conjugates reported in the literature from our and other research groups, noting all significant findings concerning the field of artificial photosynthesis. In the first part (2.1), we present covalent and noncovalent connected porphyrinoid–fullerene derivatives that were synthesized using porphyrin, corrole, and phthalocyanine chromophores. In part 2.2, we illustrate how the distance between zinc metallated porphyrinoids and C60 affects the electron transfer process and the charge recombination. In the last part of this article (2.3), we demonstrate that the increasing number of metal–ligand coordination bonds between the porphyrins and the C60 derivatives can positively influence all the above mentioned processes.
2. Porphyrinoids Connected with Fullerenes
2.1. Covalent and Noncovalent Connected Porphyrinoid–Fullerene Assemblies
Numerous covalent and noncovalent connected systems based on porphyrinoids and carbon allotropes (fullerene C60, graphene, and nanotube) have been prepared and tested as donor–acceptor (D–A) hybrids that mimic photosynthesis [20]. In the majority of these works, the photophysical events were comprehensively examined, revealing the presence of subsequent energy and electron transfer processes [15,21]. Our research group has published plenty of covalent and noncovalent connected porphyrinoid–fullerene assemblies over the last few years. Specifically, porphyrins, phthalocyanines, and corroles have been attached onto appropriate functionalized fullerene moieties, hence exploring different synthetic approaches.
In such multichromophore assemblies, the nature of the spacer that connects the electron donor moiety (e.g., porphyrin) with the electron acceptor (e.g., C60) is of great importance. To be more specific, the linkages between the entities can definitely influence both the dynamics and the kinetics of photo-induced electron transfer process (PET) [22,23]. To identify the effect of the different connection to the photophysical properties; conjugated, nonconjugated, flexible, and rigid bridges were utilized in plenty artificial photosynthetic systems containing porphyrinoids and fullerenes [24,25].
To that end, we synthesized a series of covalently linked porphyrin–C60 dyads in order to investigate the impact of a rigid bridge between the porphyrin macrocycle and the C60 moiety [26]. More specifically, we studied whether the insertion of a spacer would affect the electron transfer process and consequently the photophysical properties of the dyads. The rigid bridge that we introduced was a triazole ring derived from an azide–alkyne Huisgen cycloaddition [27] between azide–porphyrin and 4-ethynylbenzaldehyde. In general, Huisgen cycloaddition has been proven a very efficient way to link porphyrins with other chromophores under mild conditions, offering high reaction yields [28]. In our case also, the porphyrin–C60 dyads were produced in high yields, performing a Huisgen cycloaddition followed by a Prato reaction with fullerene C60. Transient absorption spectroscopy studies demonstrated that upon photo-excitation of the porphyrin, a charge-separated state (ZnP)+–(C60)− is formed in all dyads. The dyad in which the triazole spacer was inserted between the porphyrin and the fullerene (C60–tr –ZnPCOOH, Figure 1) presented the most long-lived lifetime of the charge-separated state, namely, 1260 ps in benzonitrile. These findings suggested that the presence of the triazole bridge enhances the electronic communication in such dyads and more importantly improves the photovoltaic parameters of the solar device, since those dyads were utilized as sensitizers in photovoltaic devices.
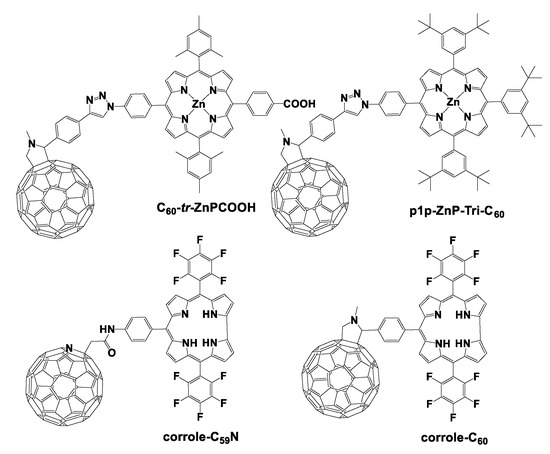
Figure 1.
The covalent linked C60–tr –ZnPCOOH, p1p–ZnP–Tri–C60, corrole–C59N, and corrole–C60.
Aromatic triazoles have been widely utilized in the literature as linkers in order to covalently connect chromophores together [29]. Furthermore, the triazole bridge has been proved as an effective π-conjugated bridge that assists intramolecular electron transfer processes in multichromophore systems comprising porphyrin entities [28]. In 2011, de Miguel et al. published a series of triazole-linked porphyrin–fullerene dyads examining the influence of the connectivity at the triazole spacer to the electron transfer dynamics [30]. From all reported porphyrin–fullerene dyads, p1p–ZnP–Tri–C60 (Figure 1) demonstrated the best performance in terms of lifetime of the charge-separated state. To be more specific, the lifetime of the charge-separated state of p1p–ZnP–Tri–C60 was 646 ns in benzonitrile, indicating that the para–para connection between the donor and acceptor moieties is the optimum.
In another example, our research group replaced the porphyrin chromophore with a corrole derivative and prepared the first corrole–azafullerene dyad (corrole–C59N), studying the electrochemical and photophysical features of this entity (Figure 1) [31]. In order to synthesize this fullerene–corrole dyad, an amide coupling reaction was performed between an amino-substituted corrole and an azafullerene–acid. The photophysical investigation of corrole–C59N revealed that the corrole based fluorescence is strongly quenched by the presence of the azafullerene entity, indicating an efficient electronic communication between the two components in the excited state. The femtosecond transient absorption spectroscopy studies shed light into this assumption by identifying the formation of the (corrole)+–(C59N)− charge-separated state. The lifetime of this charge-separated state corrole–C59N was 1021 ps in o-xylene, which was relatively lower than that of the C60–tr –ZnPCOOH derivative (1260 ps). Nonetheless, the corrole–C59N dyad could be a potential candidate as an artificial photosynthetic system and could be utilized in solar conversion applications.
D’Souza et al. reported the first covalently linked corrole–fullerene dyad which was studied as a mimic model of the photosynthetic reaction center consisting of the fullerene C60 as an electron acceptor and the free base corrole as an electron donor [32]. In this work, the corrole–C60 (Figure 1) dyad yielded higher lifetimes of the charge separation state compared to similar porphyrin analogues. More specifically, the best observed lifetime of the radical ion pair was 6300 ns using cyclohexane as solvent, while in THF, a significantly shorter lifetime was calculated (38 ps). Consequently, the charge recombination rates are highly dependent on the solvent polarity in such corrole–fullerene dyads in contrast to the porphyrin–fullerene analogues.
We have also examined the noncovalent approach via connecting zinc–porphyrins and zinc–phthalocyanines with pyridyl-modified C60 moieties. As illustrated in Figure 2, we synthesized a zinc–porphyrin–C60 derivative (C60–pyr·ZnP) that was formed by metal–ligand coordination between the zinc center of the porphyrin ring and the pyridyl group of the functionalized fullerene [33]. We comprehensively evaluated the photophysical characteristics of this dyad which exhibited charge separation lifetimes of 25 and 37 ps in anisole and chlorobenzene, respectively. Moreover, C60–pyr·ZnP displayed a radical ion pair state that decayed with lifetimes of 1189 ps in anisole and 991 ps in chlorobenzene. In the same manner, only recently, we prepared an analogous phthalocyanine–C60 conjugate [34] (Figure 2). In this study, the C60–pyr·ZnPc hybrid presented a binding constant of 1.3 × 105 M−1 in chlorobenzene and 3.6 × 104 M−1 in anisole. It is worth noting that this binding constant was greater compared to that of the C60–pyr·ZnP dyad. In addition, the lifetime of the charge recombination process of the ZnPc•+–C60•– ion pair state was calculated at approximately 400 ps in chlorobenzene and anisole. Interestingly, these values are in the same order of magnitude with a similar covalently linked ZnPc–C60 assembly (τ ≈ 3 ns) that has been reported in literature [35].
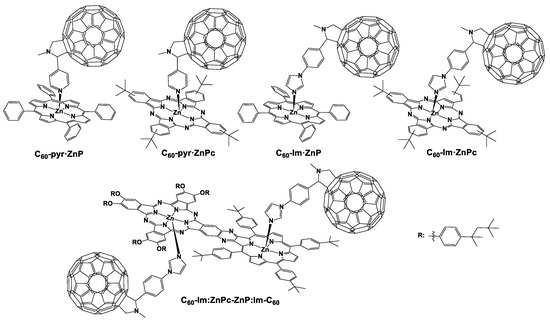
Figure 2.
The chemical structures of the noncovalent linked porphyrinoid–fullerene compounds. C60–pyr·ZnP, C60–pyr·ZnPc, C60–Im·ZnP, C60–Im·ZnPc, and C60–Im:ZnPc–ZnP:Im–C60.
Ito, D’Souza, and coworkers in 2002 synthesized a series of porphyrin–fullerene conjugates in which modified fullerenes, bearing either pyridine or imidazole, were incorporated as electron acceptors [36]. The photophysical investigation of the porphyrin–fullerene dyad connected through an imidazole linker (C60–Im·ZnP, Figure 2) revealed that the binding constant of C60–Im·ZnP was greater compared to the pyridine analogue. Concerning the transient absorption measurements, upon employment of a noncoordinating solvent such as o-dichlorobenzene, an electron transfer process takes place from the singlet excited state of the porphyrin to the fullerene entity. However, when benzonitrile was used as a coordinating solvent, intermolecular electron transfer from the triplet excited state of the porphyrin to the fullerene was clearly detected. The same research groups, one year later, prepared and fully characterized a phthalocyanine–fullerene dyad connected through an imidazole bridge (C60–Im·ZnPc, Figure 2) [37]. Transient absorption experiments showed the successful formation of the radical ion pair [(C60)−–Im·(ZnPc)+] between the electron donating phthalocyanine and the electron accepting fullerene. The charge separation rate of C60–Im·ZnPc was calculated in toluene and o -dichlorobenzene and found 1.4 × 1010 s−1 and 8.9 × 109 s−1, respectively. In benzonitrile, C60–Im·ZnPc undergoes an intermolecular electron transfer from the excited triplet state of the zinc–phthalocyanine to the fullerene similar to the C60–Im·ZnP.
Taking another step forward, we prepared a supramolecular complex consisting of porphyrin, phthalocyanine, and fullerene in order to investigate this compound as an artificial photosynthetic center mimic [38]. Hence, we synthesized a novel zinc phthalocyanine–zinc porphyrin dyad in which the two chromophores were linked through a pyrazine ring. As shown in Figure 2, the zinc centers of both chromophores were connected with an imidazole substituted fullerene forming C60–Im:ZnPc–ZnP:Im–C60. Following this approach, we were able to prepare a supramolecular D–A entity and analyze the electronic interactions occurring in this system. The investigation of the photophysical studies exposed that there is a strong electronic interaction between the two zinc–metallated porphyrinoids. More specifically, the transient absorption studies indicated that upon photoexcitation of the zinc–porphyrin derivative (ZnP) in the supramolecular complex C60–Im:ZnPc–ZnP:Im–C60, a very efficient energy transfer from the poprhyrin to the phthalocyanine takes place. Subsequently, the excited zinc–phthalocyanine is formed (1ZnPc*), which performs an electron transfer to the fullerene moiety. Thus, we observe the formation of a long-lived charge-separated state between the zinc–phthalocyanine and the fullerene (C60Im)−–(ZnPc)+–ZnP:ImC60 with a lifetime of 200 ns, signifying the importance of such complexes in solar harvesting applications. It is worth mentioning that in a recent publication, we synthesized the nonmetallated porphyrin analogue of this supramolecular compound [39]. As expected, in this supramolecular complex, the respective sequential energy and electron transfer events take place as well. However, due to the absence of the second fullerene moiety axially coordinated to the porphyrin moiety in this complex, the photophysical investigation is much more controllable.
2.2. The Influence of the Distance Between Porphyrinoids and C60
There are plenty of covalently linked porphyrinoid–fullerene hybrids reported in the literature in which the thorough investigation of their photophysical properties and electronic features was performed [15,16,17,21,22]. What is more, Segura et al. in 2005 reported that increasing the distance between the electron donor and electron acceptor components by introducing a conjugated bridge leads to increased lifetime of the charge separation state [40]. Our group, in an effort to determine whether the length of a rigid bridge affects the electronic communication in a noncovalent porphyhrin–fullerene dyad, synthesized C60 moieties using oligo(p-phenylenevinylene) (oPPVs) bridges of different sizes [33]. Hence, we synthesized fullerene entities connected with oPPVs of different lengths bearing pyridyl groups. The pyridyl groups of the formed C60–oPPV–pyr moieties are well-known nitrogen ligands which enable the formation of metal–ligand coordination bonds with zinc–porphyrin chromophores. In Figure 3, the chemical structures of the three different porphyrin–fullerene dyads are presented. We synthesized a porphyrin–fullerene dyad in which the C60 entity is directly coordinated onto the zinc of the porphyrin ring through the pyridyl group abbreviated as C60–pyr·ZnP and two new dyads with one or three extra phenylenevinylene groups, C60–PPV1–pyr·ZnP and C60–PPV3–pyr·ZnP, respectively.
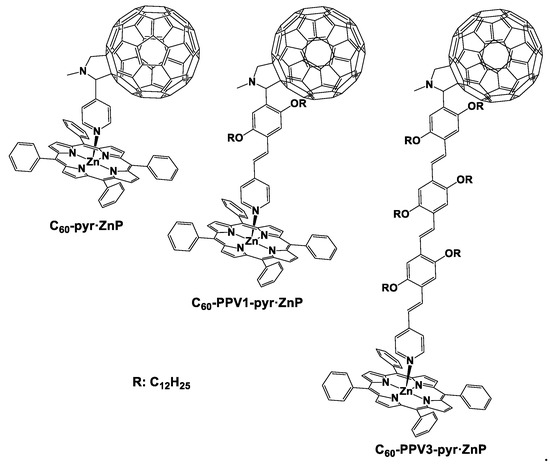
Figure 3.
The noncovalent porphyrin–fullerene dyads C60–pyr·ZnP, C60–PPV1–pyr·ZnP, and C60–PPV3–pyr·ZnP.
In the ground state, there is not any noticeable electronic interaction between porphyrin and C60. Nevertheless, the fluorescence experiments conducted in all three dyads implied that in the excited state, there is a very efficient energy or electron transfer between the zinc–porphyrin and the fullerene. We performed transient absorption measurements that confirmed the above hypothesis—namely, the formation of the reduced C60 (radical anion) and the oxidized zinc–porphyrin (radical cation) were observed. Interestingly, in the case of C60–PPV3–pyr·ZnP, there was a faster charge recombination process despite the greater distance between the porphyrin and the fullerene. This is in contrast with the tendency reported for the covalently linked porphyrin–fullerene entities, since in those cases, increasing the distance leads to higher charge separation lifetimes [40]. In those cases, the different distance between the electron donor and electron acceptor components does not affect the reorganization energy of the system [33,40]. By contrast, in our case, C60–PPV1–pyr·ZnP and C60–PPV3–pyr·ZnP demonstrated reorganization energies of 0.74 and 0.91 eV, respectively. In addition, the theoretical calculations that were performed showed that the pyridinic nitrogen disrupts the electronic pathway from the zinc–porphyrin to the fullerene. All the above observations highlight the importance of controlling the reorganization energy in noncovalent supramolecular assemblies and thus modulate the desired properties of such schemes in the field of energy conversion based on artificial photosynthetic mimics.
In a similar manner, we also examined to what extent the distance between the donor and the acceptor entities affects their electronic communication in phthalocyanine–C60 assemblies. Once more, we achieved that by inserting varied lengths of oligo(p-phenylenevinylene) bridges between the zinc–phtalocyanine and the fullerene (C60–pyr·ZnPc, C60–PPV1-pyr·ZnPc, and C60–PPV3–pyr·ZnPc, Figure 4). Accordingly, in this case, a dependence was observed between the increasing distance of the donor–acceptor moieties in the three studied assemblies (from 8.8 to 29.1 Å), and charge-separated state lifetime was increased from 0.4 to 86.6 ns, and reorganization energy was also amplified from 0.43 to 0.63 eV.
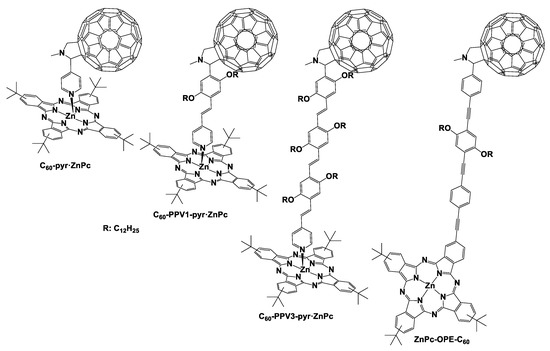
Figure 4.
The noncovalent porphyrin–fullerene dyads C60–pyr·ZnPc, C60–PPV1–pyr·ZnPc, C60–PPV3–pyr·ZnPc, and ZnPc–OPE–C60.
Plenty of research groups have dealt with the preparation and photophysical investigation of similar phthalocyanine–fullerene entities over the years [22,23]. As illustrated in Figure 4, Göransson et al. synthesized a phthalocyanine–fullerene dyad linked through an oligophenylene–ethynylene bridge [41]. This kind of connection was used as a bridging unit in plenty of research works, promoting the electron transfer over long distances [42,43,44]. In addition, the ethylene groups between donor and acceptor moieties assist the high rates of electron transfer processes [45,46]. The photophysical investigation of the phthalocyanine–fullerene dyad (ZnPc–OPE–C60) proved that an efficient photo-induced electron transfer occurs between the electron donor zinc–phthalocyanine and the electron acceptor C60 forming the radical ion pair [(ZnPc)+–OPE–(C60)−] of the dyad. The rate of the electron transfer was 1.1 × 109 s−1, whereas the recombination process was very fast at approximately 5 × 1010 s−1.
2.3. Increasing the Number of Porphyrin Derivatives in Porphyrin–C60 Entities
Consequently, we explored the impact of increasing the number of zinc–porphyrins that were linked to fullerene derivatives in supramolecular porphyrin–fullerene conjugates. Accordingly, we synthesized fullerene entities functionalized with two or four pyridyl groups offering binding sites for zinc–porphyrin derivatives. More specifically, fullerenes bearing one or two conjugated bridges which enable the coordination of one or two flexible porphyrin dimers (ZnP2) in order to create the D–A porphyrin–fullerene hybrids C60–dipyr·ZnP2 and C60–tpyr·[(ZnP)2]2, respectively (Figure 5) [47,48]. As illustrated, in both cases, the zinc centers of the ZnP2 are coordinated with the pyridyl groups of the functionalized fullerenes. In these reports, by utilizing increased number of porphyrin chromophores onto a fullerene, we additionally simulated the structure of the photosynthetic reaction center compared to our previously reported C60–PPV–pyr·ZnP derivatives [33]. The supramolecular hybrids also presented enhanced solubility in solvents with varied polarity such as THF, DCM, Toluene, and o-DCB. This property is significant for potential application in solar harvesting devices.
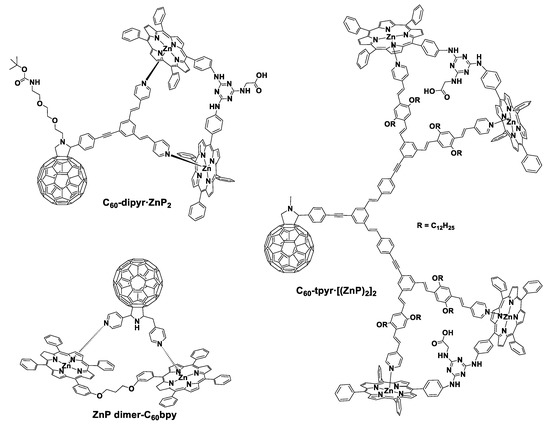
Figure 5.
The supramolecular porphyrin–fullerene conjugates C60–dipyr·ZnP2 and C60–tpyr·[(ZnP)2]2 and ZnP dimer–C60bpy.
In order to verify the successful formation of both porphyrin–fullerene hybrids (C60–dipyr·ZnP2 and C60–tpyr·[(ZnP)2]2), absorption and fluorescence titration experiments were carried out. In either case, the binding constant was greater (5 × 105 M−1 in anisole) compared to that of similar systems with more than one coordination binding site [49]. The femtosecond transient absorption studies for C60–dipyr·ZnP2 and C60–tpyr·[(ZnP)2]2 showed that upon photo-excitation of the porphyrin entity, an efficient electron transfer process occurs from the first excited singlet state of porphyrin dimer to the fullerene. This leads to the rise of the ZnP2 radical cation as well as the C60 radical anion forming the charge-separated states (C60)−–dipyr·(ZnP2)+ and (C60)−–tpyr·[(ZnP2)2]+. The lifetime of the charge-separated states, using toluene as solvent, was 1500 ps for C60–dipyr·ZnP2 and 2400 ps for C60–tpyr·[(ZnP)2]2. It is worth noting that the formation of these states was very rapid, implying that in both cases, the pyridyl groups promote the charge separation process without shortening the lifetime of the charge-separated state. Overall, we established that increasing the number of porphyrinoids in porphyrin–fullerene hybrids is beneficial for the binding constant, the fast charge separation process, and the increased lifetime of the charge-separated state.
In a previous report, D’Souza et al. also assembled a zinc porphyrin dimer with a fullerene derivative that was functionalized with two pyridine groups [50], similar to our work [47]. The fullerene chromophore was suitably modified with two pyridine groups offering binding sites for the zinc–porphyrin dimer, as illustrated in Figure 5, forming the supramolecular complex ZnP dimer–C60bpy. Steady-state and time resolved fluorescence studies showed that efficient intramolecular photo-induced electron transfer process occurs from the zinc–porphyrin dimer to C60. The nanosecond transient absorption studies indicated that the charge recombination process was very fast. More precisely, the rate was 2 × 108 s−1 using o-dichlorobenzene as solvent.
3. Conclusions
In this perspective review article, we presented various covalent and supramolecular artificial photosynthetic schemes that our group have studied over the past decade, analyzing the most important findings of these works. All these D–A systems consist of fullerene derivatives that act as electron acceptors and porphyrinoids, such as corroles, porphyrins, and phthalocyanines, utilized as electron donating units. The thorough photophysical investigation in all systems revealed efficient electronic communication between the donor and acceptor entities. What is more, in all cases, the conducted transient absorption measurements proved the formation of a charge-separated state, which is formed after an electron transfer originating from the singlet excited state of each photosensitizer.
Regarding the covalently linked assemblies, it is clear that their preparation is rather demanding, since multistep reactions are required. In addition to that, these hybrids displayed lifetimes of charge-separated state comparable to those of the noncovalent systems; hence, the metal–ligand axial coordination approach is an advantageous strategy for the construction of such hybrids. Concerning the noncovalent assemblies where electron donor monomers (ZnP and ZnPc) were linked to fullerene derivatives, a longer-lived charge-separated state was calculated for the C60–pyr·ZnP hybrid, whereas the phthalocyanine analogue (C60–pyr·ZnPc) demonstrated a larger binding constant. In an effort to determine whether the length of a rigid bridge affects electronic communication in a noncovalent porphyrin–fullerene dyad, we studied two series of porphyrinoid–C60 derivatives with bridges of different sizes containing both ZnP and ZnPc chromophores. Even though the different distance between the electron donor and acceptor units does not affect the reorganization energy of covalently linked systems, a dependence between the increasing distance and both the ion pair lifetime and reorganization energy was observed in the supramolecular systems.
Interestingly, the supramolecular schemes C60–dipyr·ZnP2 and C60–tpyr·[(ZnP)2]2, in which properly functionalized C60 derivatives are coordinated to metallated porphyrin dimers, presented binding constants that were over one order of magnitude higher than those reported for porphyrin–fullerene hybrids with a single binding site. In addition, the lifetime of their charge-separated state was greater than that of the respective porphyrin–fullerene hybrid with a single binding site. Finally, out of all the described artificial photosynthetic schemes, those which showed the longer-lived charge-separated state are systems incorporating dimeric sensitizer moieties, namely, C60–Im:ZnPc–ZnP:Im–C60 and C60–tpyr·[(ZnP)2]2.
Supramolecular hybrids comprising porphyrinoids and fullerene moieties display plenty of imperative as well as desirable features. Specifically, all of them are molecular systems with reliable stability, thus offering the opportunity of straightforward characterization through widely used spectroscopic methods. In addition, for the synthesis of these complexes, well-known and simple experimental procedures are followed, utilizing low-cost reagents and ease purification methods as well. Lastly, the electron transfer processes that take place in such systems can be identified using transient absorption measurements. However, there are a few drawbacks when this class of hybrids is employed that have not been solved yet by the research community. To be more specific, it is very difficult to scale up the synthesis of such compounds, limiting their application in solar energy devices. Moreover, in all cases, the back electron transfer process from the reduced fullerene moiety to the oxidized porphyrinoid entity occurs, limiting the lifetime of the charge-separated state. Overall, such porphyrinoid–fullerene assemblies can provide beneficial directions towards the successful preparation of efficient D–A hybrids as potential candidates for solar energy conversion schemes.
Funding
This research was funded by the General Secretariat for Research and Technology (GSRT) and Hellenic Foundation for Research and Innovation (HFRI) (project code: 508). This research has also been co-financed by the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship, and Innovation, under the call RESEARCH—CREATE—INNOVATE (project code: T1EDK-01504). In addition, this research has been co-financed by the European Union and Greek national funds through the Regional Operational Program “Crete 2014-2020”, project code OPS: 5029187. Moreover, the European Commission’s Seventh Framework Program (FP7/2007-2013) under grant agreement no. 229927 (FP7-REGPOT-2008-1, Project BIO-SOLENUTI) and the Special Research Account of the University of Crete are gratefully acknowledged for the financial support of this research.
Acknowledgments
Dirk Guldi and Axel Kahnt are gratefully acknowledged for performing the photophysical investigation to the majority of the porphyrinoid–fullerene hybrids presented herein.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tachibana, Y.; Vayssieres, L.; Durrant, J.R. Artificial photosynthesis for solar water-splitting. Nat. Photonics 2012, 6, 511. [Google Scholar] [CrossRef]
- Zhang, B.; Sun, L. Artificial photosynthesis: opportunities and challenges of molecular catalysts. Chem. Soc. Rev. 2019, 48, 2216–2264. [Google Scholar] [CrossRef] [PubMed]
- Bredas, J.L.; Sargent, E.H.; Scholes, G.D. Photovoltaic concepts inspired by coherence effects in photosynthetic systems. Nature Mater. 2017, 16, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-W.; Zhong, D.-C.; Lu, T.-B. Artificial photosynthesis: Catalytic water oxidation and CO2 reduction by dinuclear non-noble-metal molecular catalysts. Coord. Chem. Rev. 2018, 377, 225–236. [Google Scholar] [CrossRef]
- Fukuzumi, S. Development of bioinspired artificial photosynthetic systems. Phys. Chem. Chem. Phys. 2008, 10, 2283–2297. [Google Scholar] [CrossRef]
- Rudolf, M.; Kirner, S.V.; Guldi, D.M. A multicomponent molecular approach to artificial photosynthesis—The role of fullerenes and endohedral metallofullerenes. Chem. Soc. Rev. 2016, 45, 612–630. [Google Scholar] [CrossRef]
- Bottari, G.; Suanzes, J.A.; Trukhina, O.; Torres, T. Phthalocyanine−carbon nanostructure materials assembled through supramolecular interactions. J. Phys. Chem. Lett. 2011, 2, 905–913. [Google Scholar] [CrossRef]
- Hiroto, S.; Miyake, Y.; Shinokubo, H. Synthesis and Functionalization of Porphyrins through Organometallic Methodologies. Chem. Rev. 2017, 117, 2910–3043. [Google Scholar] [CrossRef]
- Chen, Y.; Royal, G.; Flahaut, E.; Cobo, S.; Bouchiat, V.; Marty, L.; Bendiab, N. Light control of charge transfer and excitonic transitions in a carbon nanotube/porphyrin hybrid. Adv. Mater. 2017, 29, 1605745. [Google Scholar] [CrossRef]
- Zhang, W.; Lai, W.; Cao, R. Energy-related small molecule activation reactions: oxygen reduction and hydrogen and oxygen evolution reactions catalyzed by porphyrin- and corrole-based systems. Chem. Rev. 2017, 117, 3717–3797. [Google Scholar] [CrossRef]
- Ladomenou, K.; Natali, M.; Iengo, E.; Charalampidis, G.; Scandola, F.; Coutsolelos, A.G. Photochemical hydrogen generation with porphyrin-based systems. Coord. Chem. Rev. 2015, 304–305, 38–54. [Google Scholar] [CrossRef]
- Pascal, S.; Bucher, L.; Desbois, N.; Bucher, C.; Andraud, C.; Gros, C.P. Synthesis, electrochemistry, and photophysics of Aza-BODIPY porphyrin dyes. Chem. Eur. J. 2016, 22, 4971–4979. [Google Scholar] [CrossRef] [PubMed]
- Hebié, S.; Dimé, A.K.D.; Devillers, C.H.; Lucas, D. Electrochemistry as an attractive and effective tool for the synthesis and immobilization of porphyrins on an electrode surface. Chem. Eur. J. 2015, 21, 8281–8289. [Google Scholar] [CrossRef] [PubMed]
- Ladomenou, K.; Nikolaou, V.; Charalambidis, G.; Coutsolelos, A.G. Artificial hemes for DSSC and/or BHJ applications. Dalton Trans. 2016, 45, 1111–1126. [Google Scholar] [CrossRef]
- Bottari, G.; Trukhina, O.; Ince, M.; Torres, T. Towards artificial photosynthesis: Supramolecular, donor–acceptor, porphyrin- and phthalocyanine/carbon nanostructure ensembles. Coord. Chem. Rev. 2012, 256, 2453–2477. [Google Scholar] [CrossRef]
- Guldi, D.M. Fullerene-porphyrin architectures; photosynthetic antenna and reaction center models. Chem. Soc. Rev. 2002, 31, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Frischmann, P.D.; Mahata, K.; Würthner, F. Powering the future of molecular artificial photosynthesis with light-harvesting metallosupramolecular dye assemblies. Chem. Soc. Rev. 2013, 42, 1847–1870. [Google Scholar] [CrossRef]
- Collavini, S.; Delgado, J.L. Fullerenes: the stars of photovoltaics. Sustain. Energy Fuels 2018, 2, 2480–2493. [Google Scholar] [CrossRef]
- Imahori, H.; Guldi, D.M.; Tamaki, K.; Yoshida, Y.; Luo, C.; Sakata, Y.; Fukuzumi, S. Charge separation in a novel artificial photosynthetic reaction center lives 380 ms. J. Am. Chem. Soc. 2001, 123, 6617–6628. [Google Scholar] [CrossRef]
- Zieleniewska, A.; Lodermeyer, F.; Roth, A.; Guldi, D.M. Fullerenes—How 25 years of charge transfer chemistry have shaped our understanding of (interfacial) interactions. Chem. Soc. Rev. 2018, 47, 702–714. [Google Scholar] [CrossRef]
- D’Souza, F.; Ito, O. Photoinduced electron transfer in supramolecular systems of fullerenes functionalized with ligands capable of binding to zinc porphyrins and zinc phthalocyanines. Coord. Chem. Rev. 2005, 249, 1410–1422. [Google Scholar] [CrossRef]
- Konarev, D.V.; Khasanov, S.S.; Lyubovskaya, R.N. Fullerene complexes with coordination assemblies of metalloporphyrins and metal phthalocyanines. Coord. Chem. Rev. 2014, 262, 16–36. [Google Scholar] [CrossRef]
- Kc, C.B.; D’Souza, F. Design and photochemical study of supramolecular donor–acceptor systems assembled via metal–ligand axial coordination. Coord. Chem. Rev. 2016, 322, 104–141. [Google Scholar] [CrossRef]
- Bottari, G.; de la Torre, G.; Guldi, D.M.; Torres, T. Covalent and noncovalent phthalocyanine-carbon nanostructure systems: Synthesis, photoinduced electron transfer, and application to molecular photovoltaics. Chem. Rev. 2010, 110, 6768–6816. [Google Scholar] [CrossRef] [PubMed]
- Ito, O. Photosensitizing electron transfer processes of fullerenes, carbon nanotubes, and carbon nanohorns. Chem. Rec. 2017, 17, 326–362. [Google Scholar] [CrossRef]
- Nikolaou, V.; Plass, F.; Planchat, A.; Charisiadis, A.; Charalambidis, G.; Angaridis, P.A.; Kahnt, A.; Odobel, F.; Coutsolelos, A.G. Effect of the triazole ring in zinc porphyrin-fullerene dyads on the charge transfer processes in NiO-based devices. Phys. Chem. Chem. Phys. 2018, 20, 24477–24489. [Google Scholar] [CrossRef] [PubMed]
- Huisgen, R. 1,3-Dipolar cycloadditions. Past and future. Angew. Chem. Int. Ed. 1963, 2, 565–598. [Google Scholar] [CrossRef]
- Ladomenou, K.; Nikolaou, V.; Charalambidis, G.; Coutsolelos, A.G. "Click"-reaction: An alternative tool for new architectures of porphyrin based derivatives. Coord. Chem. Rev. 2016, 306, 1–42. [Google Scholar] [CrossRef]
- Hein, J.E.; Fokin, V.V. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) and beyond: New reactivity of copper(I) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. [Google Scholar] [CrossRef]
- de Miguel, G.; Wielopolski, M.; Schuster, D.I.; Fazio, M.A.; Lee, O.P.; Haley, C.K.; Ortiz, A.L.; Echegoyen, L.; Clark, T.; Guldi, D.M. Triazole bridges as versatile linkers in electron donor-acceptor conjugates. J. Am. Chem. Soc. 2011, 133, 13036–13054. [Google Scholar] [CrossRef]
- Rotas, G.; Charalambidis, G.; Glatzl, L.; Gryko, D.T.; Kahnt, A.; Coutsolelos, A.G.; Tagmatarchis, N. A corrole-azafullerene dyad: Synthesis, characterization, electronic interactions and photoinduced charge separation. Chem. Commun. 2013, 49, 9128–9130. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, F.; Chitta, R.; Ohkubo, K.; Tasior, M.; Subbaiyan, N.K.; Zandler, M.E.; Rogacki, M.K.; Gryko, D.T.; Fukuzumi, S. Corrole-Fullerene Dyads: Formation of Long-Lived Charge-Separated States in Nonpolar Solvents. J. Am. Chem. Soc. 2008, 130, 14263–14272. [Google Scholar] [CrossRef] [PubMed]
- Stangel, C.; Schubert, C.; Kuhri, S.; Rotas, G.; Margraf, J.T.; Regulska, E.; Clark, T.; Torres, T.; Tagmatarchis, N.; Coutsolelos, A.G.; et al. Tuning the reorganization energy of electron transfer in supramolecular ensembles—metalloporphyrin, oligophenylenevinylenes, and fullerene—and the impact on electron transfer kinetics. Nanoscale 2015, 7, 2597–2608. [Google Scholar] [CrossRef] [PubMed]
- Krug, M.; Stangel, C.; Schubert, C.; Clark, T.; Torres, T.; Coutsolelos, A.G.; Guldi, D.M. Combining zinc phthalocyanines, oligo(p-phenylenevinylenes), and fullerenes to impact reorganization energies and attenuation factors. ChemPhysChem 2019, accepted. [Google Scholar] [CrossRef]
- Guldi, D.M.; Gouloumis, A.; Vazquez, P.; Torres, T.; Georgakilas, V.; Prato, M. Nanoscale organization of a phthalocyanine-fullerene system: Remarkable stabilization of charges in photoactive 1-D nanotubules. J. Am. Chem. Soc. 2005, 127, 5811–5813. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, F.; Deviprasad, G.R.; Zandler, M.E.; Hoang, V.T.; Klykov, A.; VanStipdonk, M.; Perera, A.; El-Khouly, M.E.; Fujitsuka, M.; Ito, O. Spectroscopic, electrochemical, and photochemical studies of self-assembled via axial coordination zinc porphyrin-fulleropyrrolidine dyads. J. Phys. Chem. A 2002, 106, 3243–3252. [Google Scholar] [CrossRef]
- El-Khouly, M.E.; Rogers, L.M.; Zandler, M.E.; Suresh, G.; Fujitsuka, M.; Ito, O.; D’Souza, F. Studies on intra-supramolecular and intermolecular electron-transfer processes between zinc naphthalocyanine and imidazole-appended fullerene. ChemPhysChem 2003, 4, 474–481. [Google Scholar] [CrossRef]
- Follana-Berna, J.; Seetharaman, S.; Martin-Gomis, L.; Charalambidis, G.; Trapali, A.; Karr, P.A.; Coutsolelos, A.G.; Fernandez-Lazaro, F.; D’Souza, F.; Sastre-Santos, A. Supramolecular complex of a fused zinc phthalocyanine-zinc porphyrin dyad assembled by two imidazole-C-60 units: Ultrafast photoevents. Phys. Chem. Chem. Phys. 2018, 20, 7798–7807. [Google Scholar] [CrossRef]
- Seetharaman, S.; Follana-Berna, J.; Martin-Gomis, L.; Charalambidis, G.; Trapali, A.; Karr, P.A.; Coutsolelos, A.G.; Fernandez-Lazaro, F.; Sastre-Santos, A.; D’Souza, F. Sequential, Ultrafast Energy Transfer and Electron Transfer in a Fused Zinc Phthalocyanine-free-base Porphyrin-C-60 Supramolecular Triad. ChemPhysChem 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Segura, J.L.; Giacalone, F.; Gomez, R.; Martin, N.; Guldi, D.M.; Luo, C.P.; Swartz, A.; Riedel, I.; Chirvase, D.; Parisi, J.; et al. Design, synthesis and photovoltaic properties of [60]fullerene based molecular materials. Mater. Sci. Eng. C 2005, 25, 835–842. [Google Scholar] [CrossRef]
- Goransson, E.; Boixel, J.; Fortage, J.; Jacquemin, D.; Becker, H.C.; Blart, E.; Hammarstrom, L.; Odobel, F. Long-Range Electron Transfer in Zinc-Phthalocyanine-Oligo(Phenylene-ethynylene)-Based Donor-Bridge-Acceptor Dyads. Inorg. Chem. 2012, 51, 11500–11512. [Google Scholar] [CrossRef] [PubMed]
- Donhauser, Z.J.; Mantooth, B.A.; Kelly, K.F.; Bumm, L.A.; Monnell, J.D.; Stapleton, J.J.; Price, D.W.; Rawlett, A.M.; Allara, D.L.; Tour, J.M.; et al. Conductance switching in single molecules through conformational changes. Science 2001, 292, 2303–2307. [Google Scholar] [CrossRef] [PubMed]
- Weber, H.B.; Reichert, J.; Weigend, F.; Ochs, R.; Beckmann, D.; Mayor, M.; Ahlrichs, R.; von Lohneysen, H. Electronic transport through single conjugated molecules. Chem. Phys. 2002, 281, 113–125. [Google Scholar] [CrossRef]
- Andréasson, J.; Kodis, G.; Ljungdahl, T.; Moore, A.L.; Moore, T.A.; Gust, D.; Mårtensson, J.; Albinsson, B. Photoinduced Hole Transfer from the Triplet State in a Porphyrin-Based Donor−Bridge−Acceptor System. J. Phys. Chem. A 2003, 107, 8825–8833. [Google Scholar] [CrossRef]
- Yang, S.I.; Lammi, R.K.; Prathapan, S.; Miller, M.A.; Seth, J.; Diers, J.R.; Bocian, D.F.; Lindsey, J.S.; Holten, D. Synthesis and excited-state photodynamics of perylene-porphyrin dyads Part 3. Effects of perylene, linker, and connectivity on ultrafast energy transfer. J. Mater. Chem. 2001, 11, 2420–2430. [Google Scholar] [CrossRef]
- Redmore, N.P.; Rubtsov, I.V.; Therien, M.J. Synthesis, electronic structure, and electron transfer dynamics of (aryl)ethynyl-bridged donor-acceptor systems. J. Am. Chem. Soc. 2003, 125, 8769–8778. [Google Scholar] [CrossRef]
- Stangel, C.; Charisiadis, A.; Zervaki, G.E.; Nikolaou, V.; Charalambidis, G.; Kahnt, A.; Rotas, G.; Tagmatarchis, N.; Coutsolelos, A.G. Case Study for Artificial Photosynthesis: Noncovalent Interactions between C-60-Dipyridyl and Zinc Porphyrin Dimer. J. Phys. Chem. C 2017, 121, 4850–4858. [Google Scholar] [CrossRef]
- Stangel, C.; Plass, F.; Charisiadis, A.; Giannoudis, E.; Chararalambidis, G.; Karikis, K.; Rotas, G.; Zervaki, G.E.; Lathiotakis, N.N.; Tagmatarchis, N.; et al. Interfacing tetrapyridyl-C-60 with porphyrin dimers via -conjugated bridges: artificial photosynthetic systems with ultrafast charge separation. Phys. Chem. Chem. Phys. 2018, 20, 21269–21279. [Google Scholar] [CrossRef]
- D’Souza, F.; Gadde, S.; Zandler, M.E.; Itou, M.; Araki, Y.; Ito, O. Supramolecular complex composed of a covalently linked zinc porphyrin dimer and fulleropyrrolidine bearing two axially coordinating pyridine entities. Chem. Commun. 2004. [Google Scholar] [CrossRef]
- Kc, C.B.; Ohkubo, K.; Karr, P.A.; Fukuzumi, S.; D’Souza, F. A ‘two-point’ bound zinc porphyrin-zinc phthalocyanine-fullerene supramolecular triad for sequential energy and electron transfer. Chem. Commun. 2013, 49, 7614–7616. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).