Highly Active, High Specific Surface Area Fe/C/N ORR Electrocatalyst from Liquid Precursors by Combination of CO2 Laser Pyrolysis and Single NH3 Thermal Post-Treatment

 ,
,
Abstract
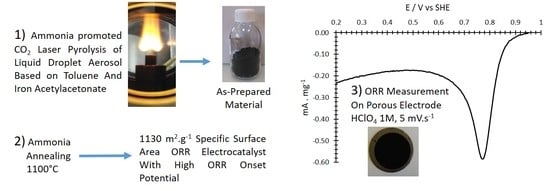
:
1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. Characterization
2.3. Thermal Treatments
2.4. XPS Analysis
2.5. Electrochemical Measurement
3. Results
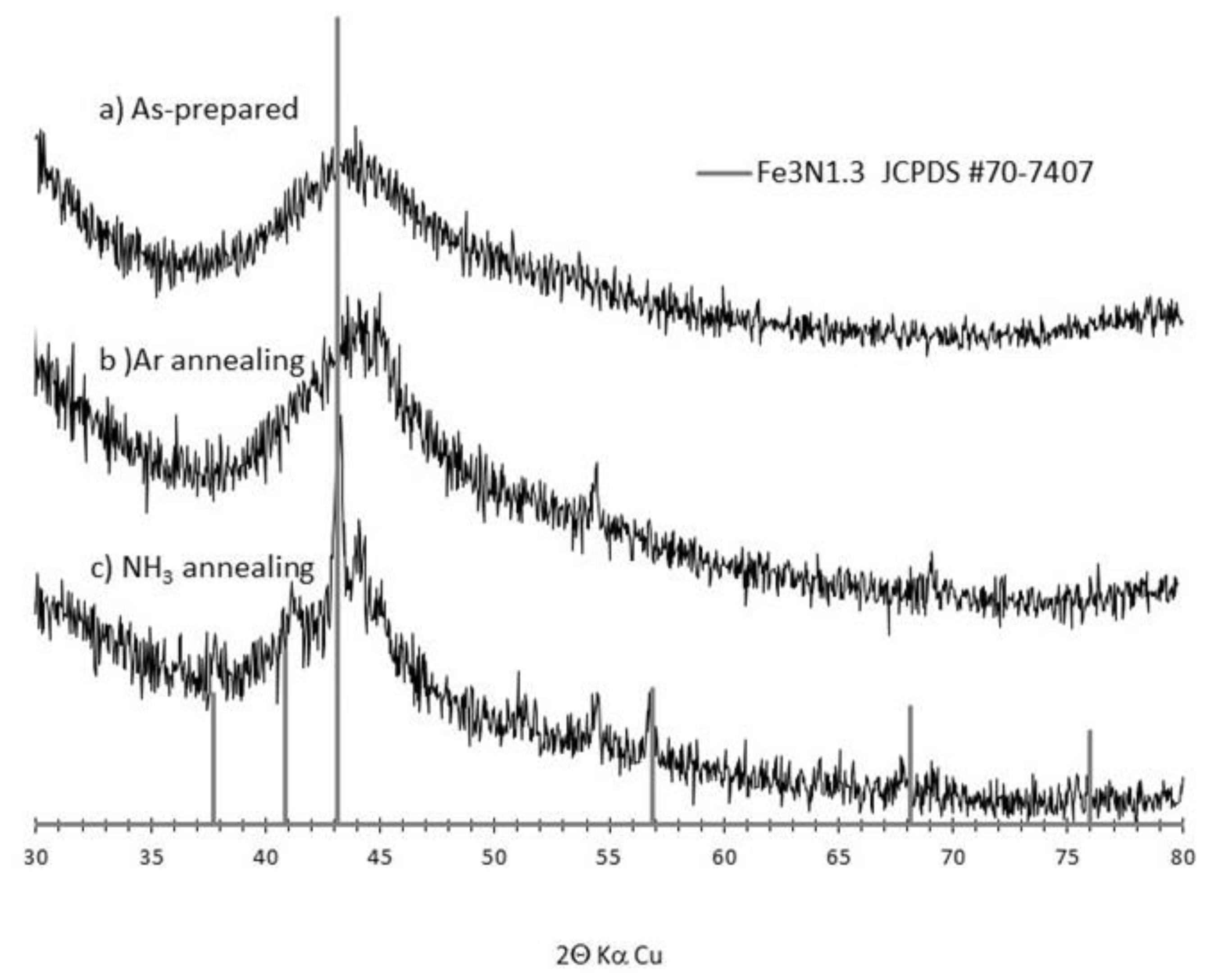
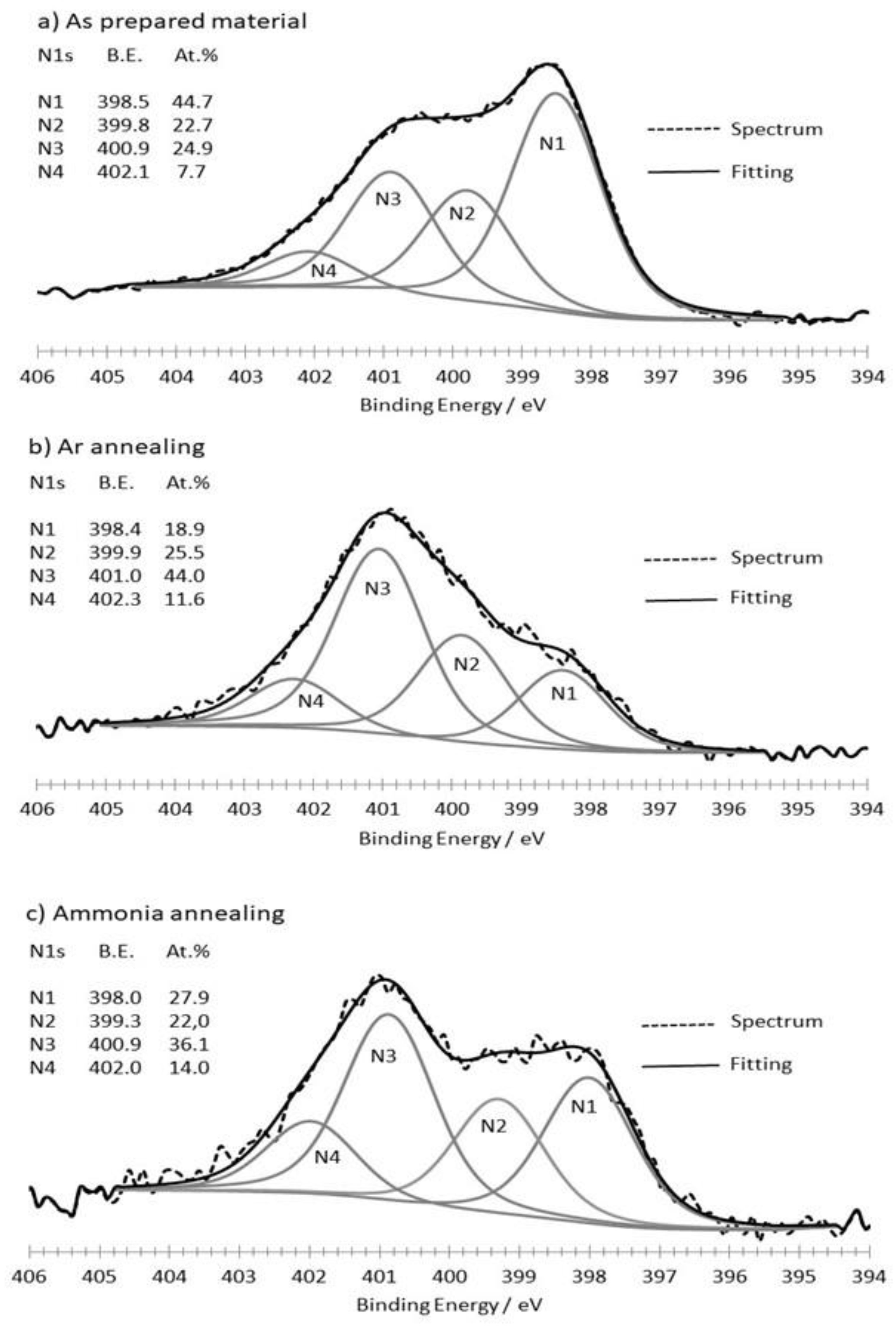
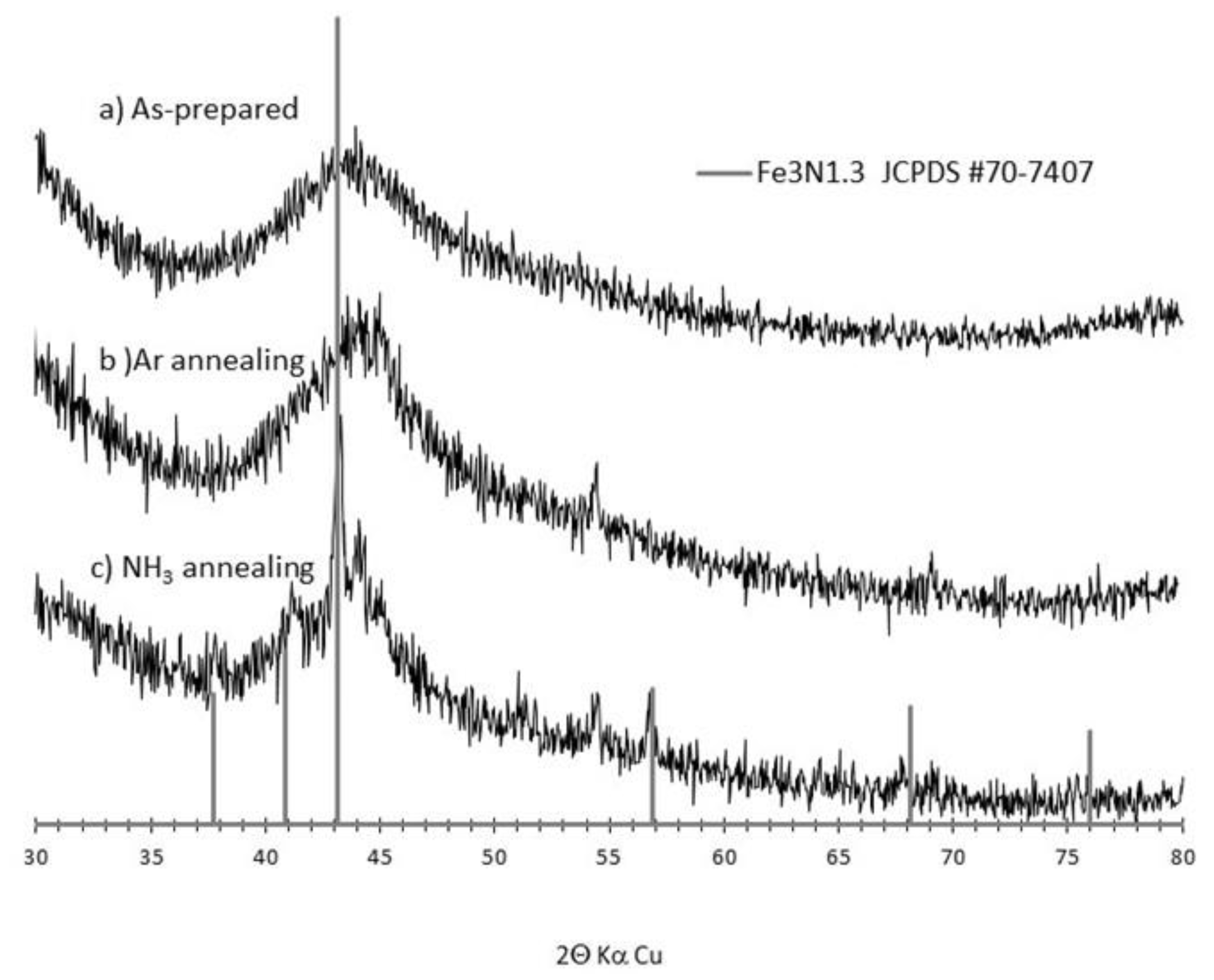
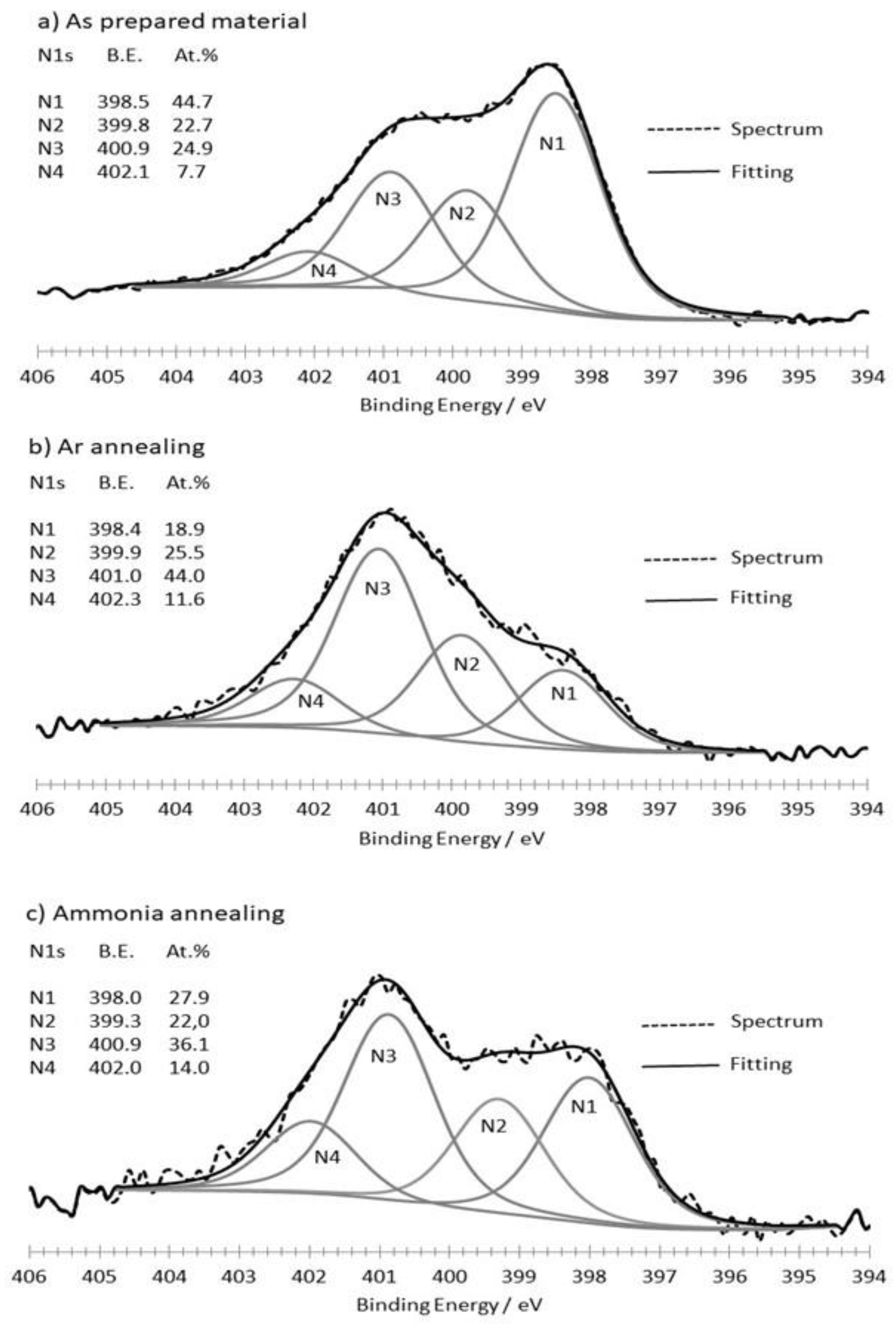
3.1. Synthesis and Characterization of the as Formed and Annealed Materials
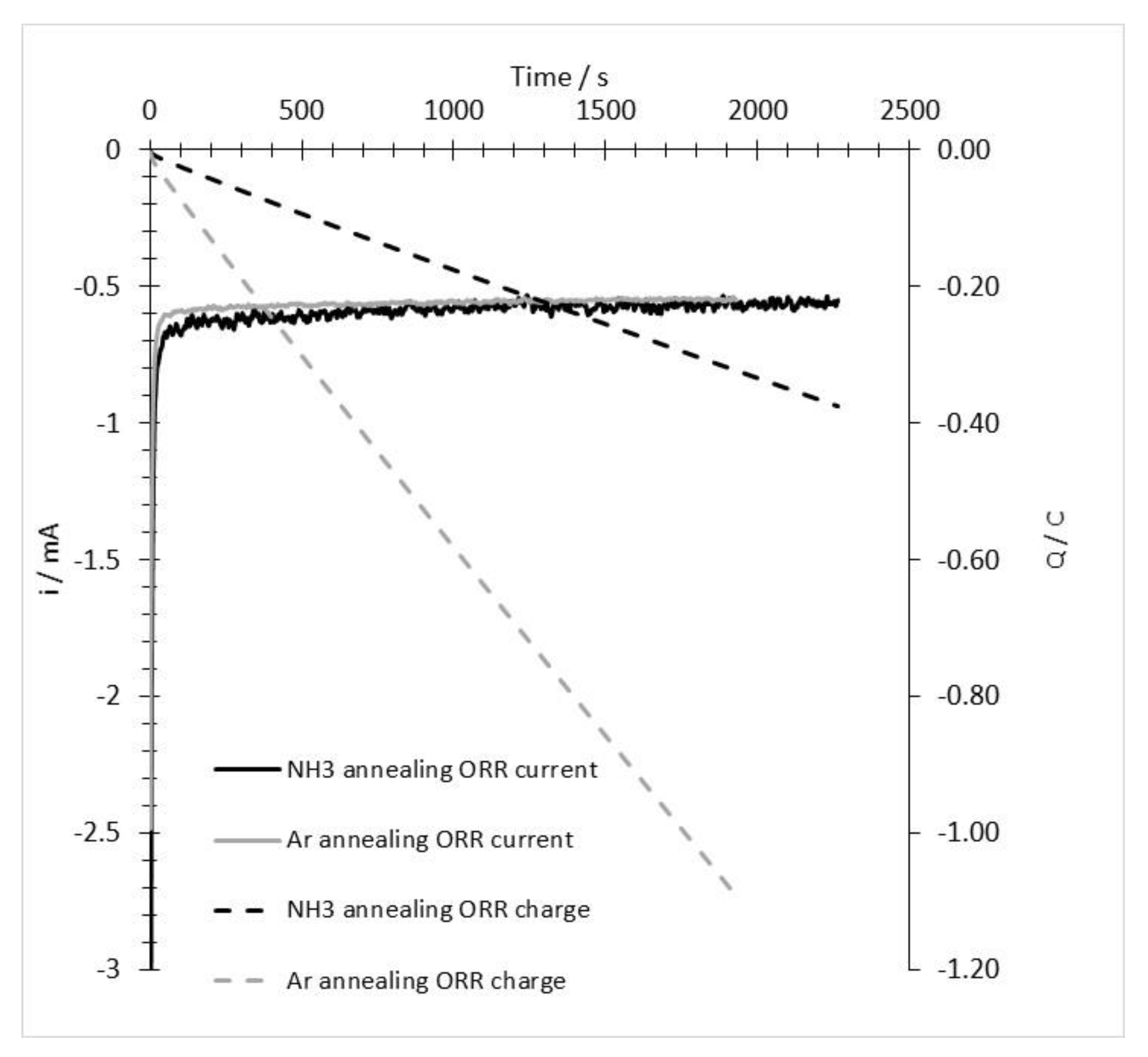
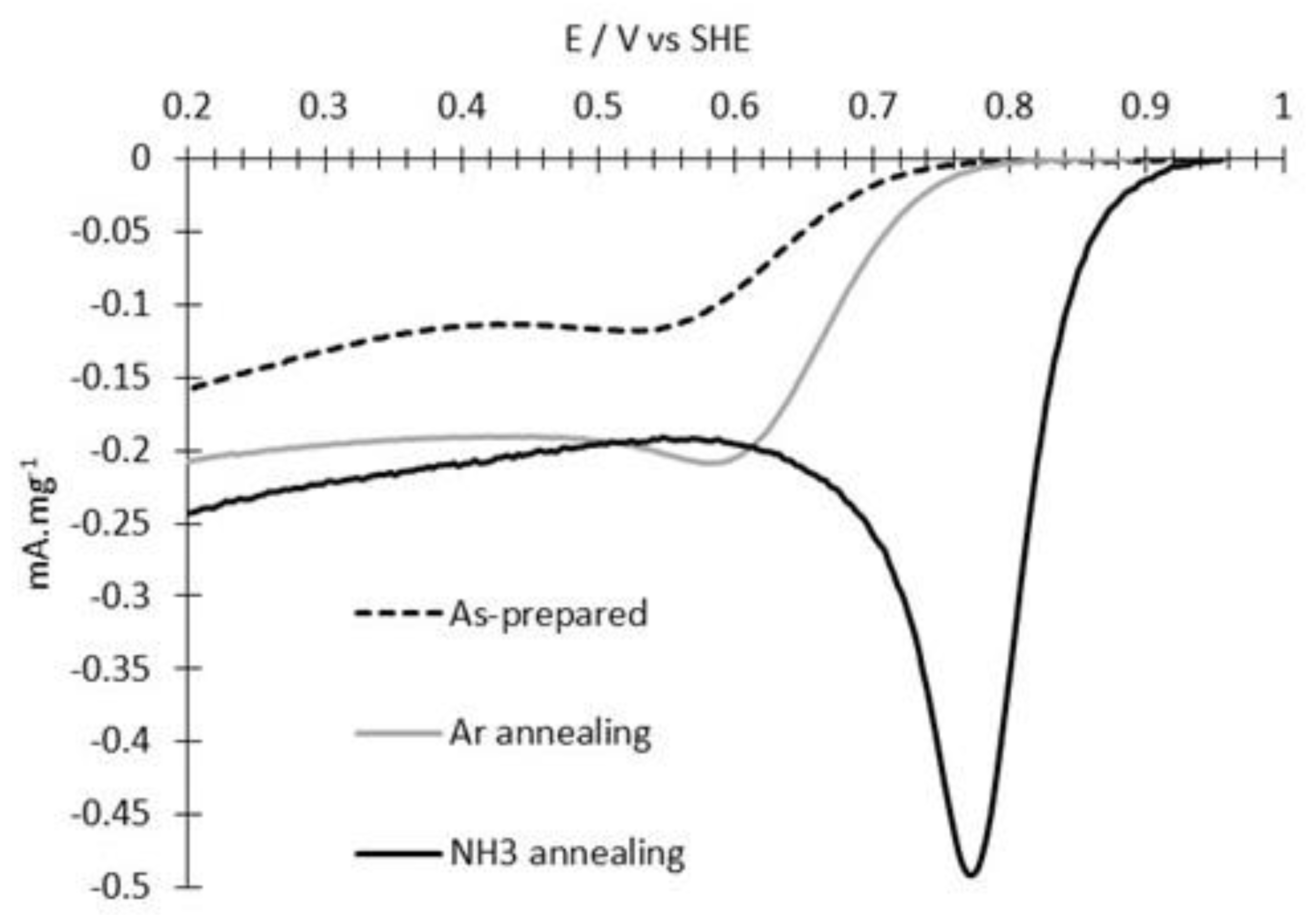
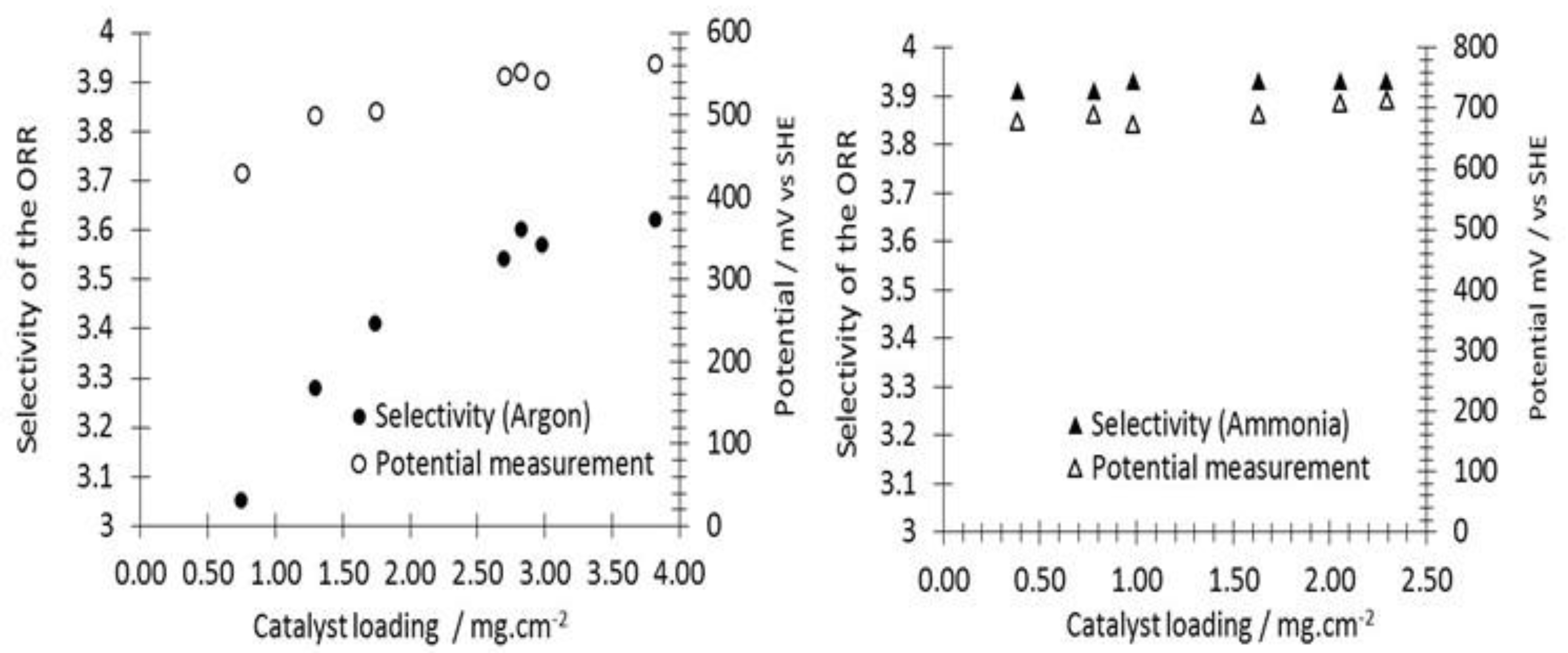
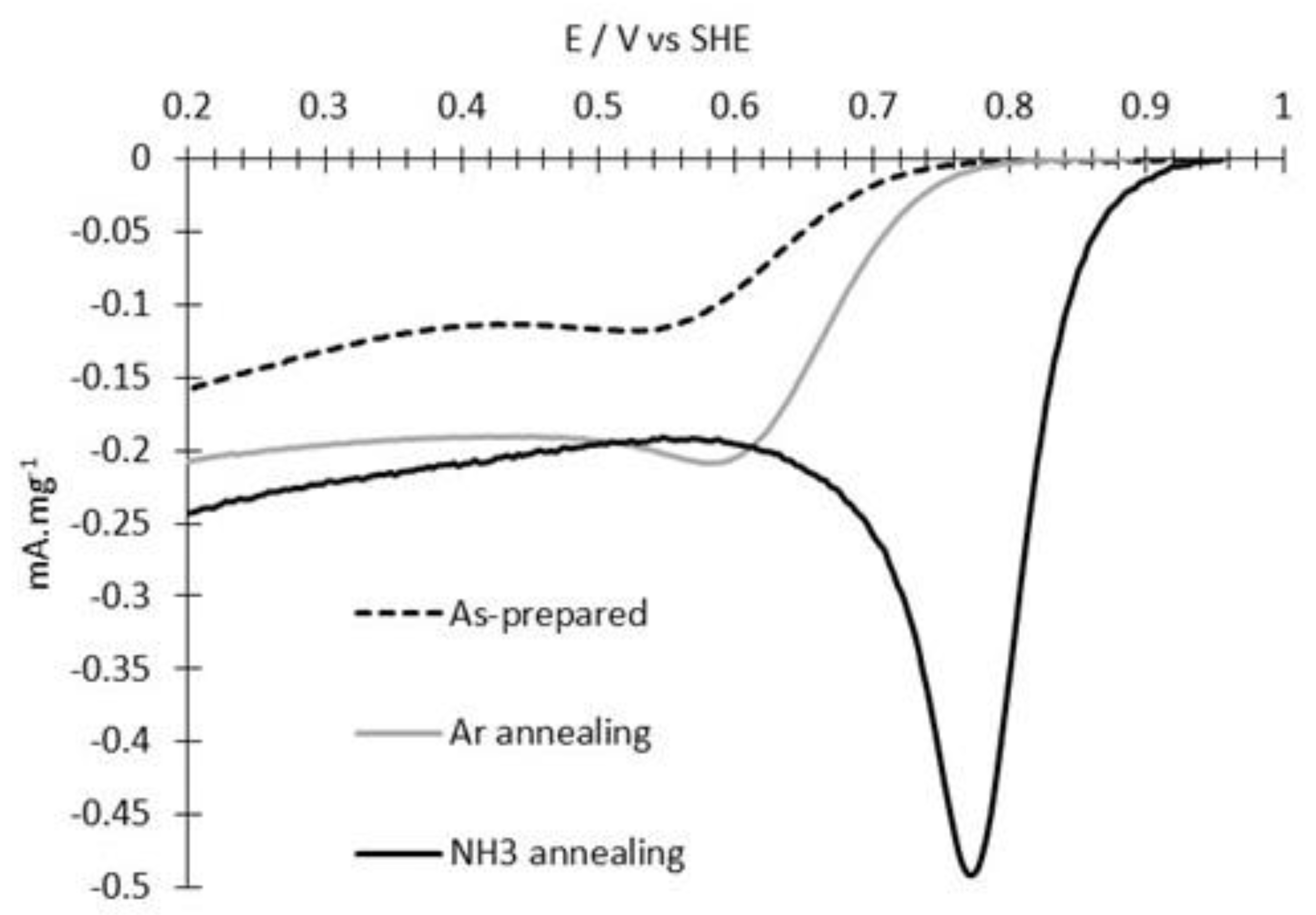
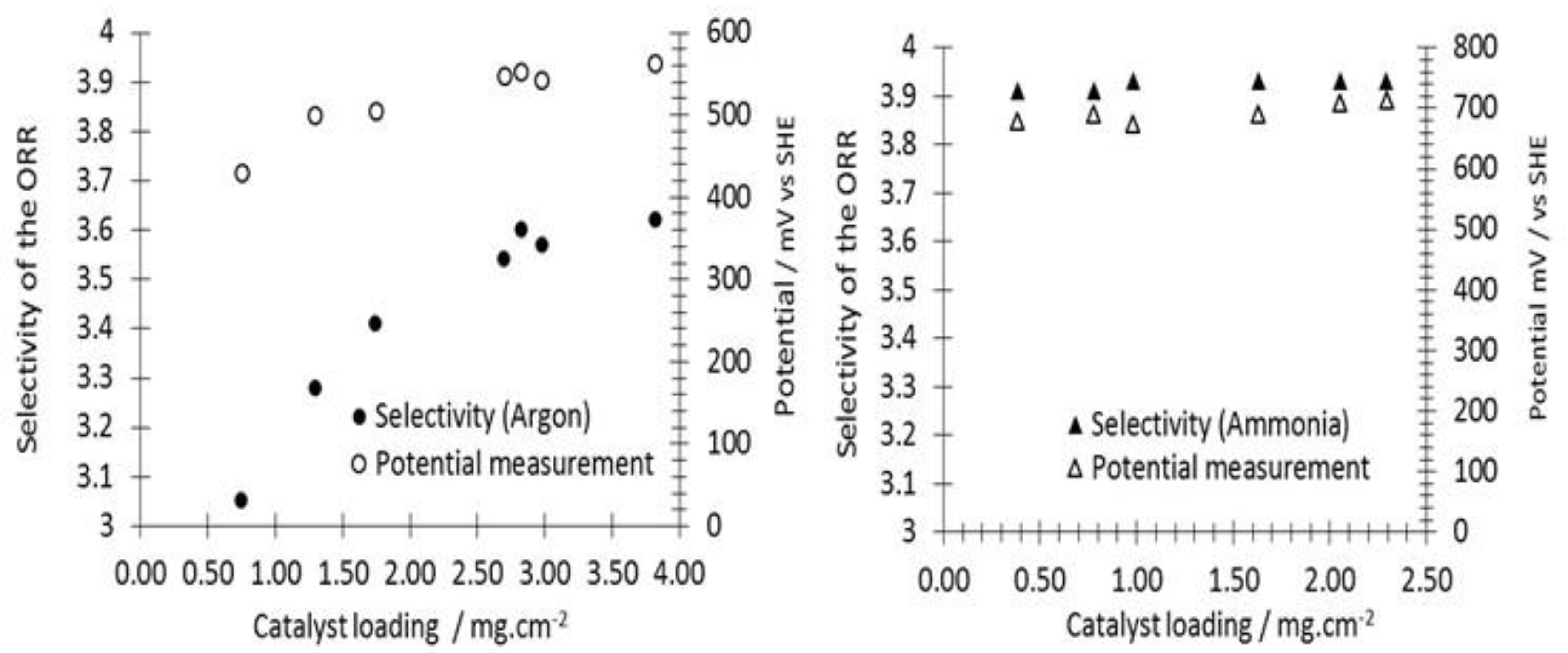
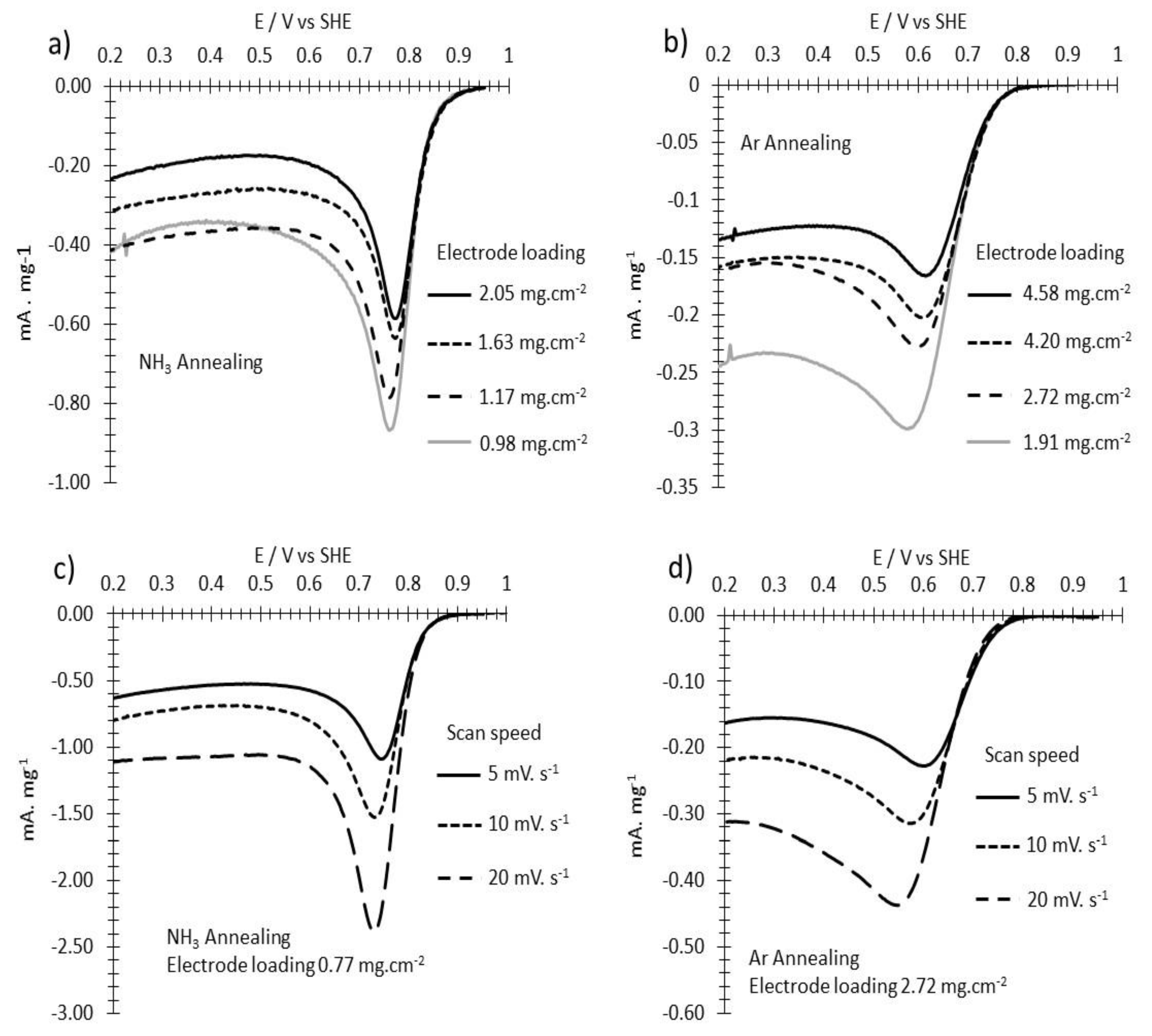
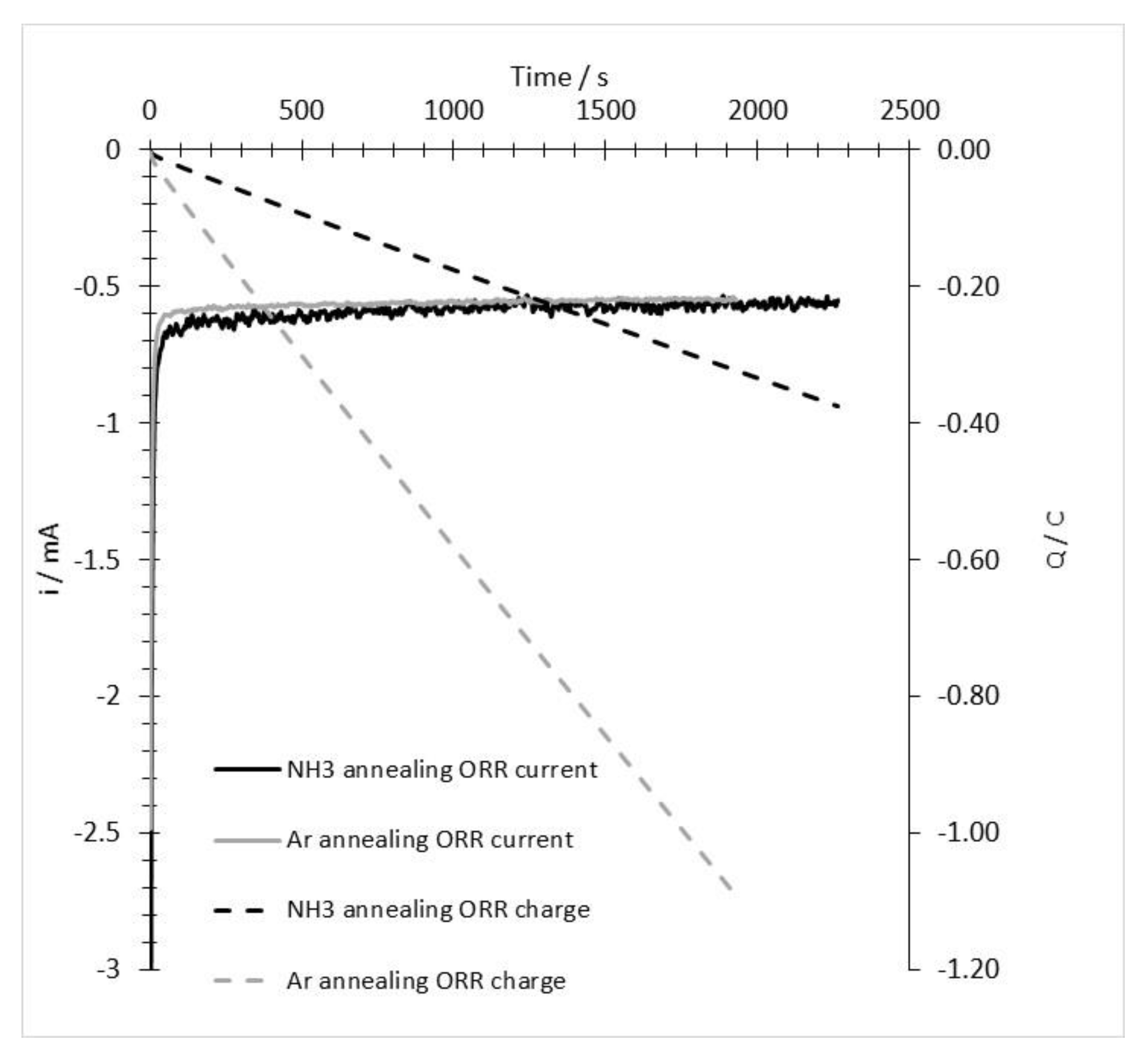
3.2. ORR Measurements
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
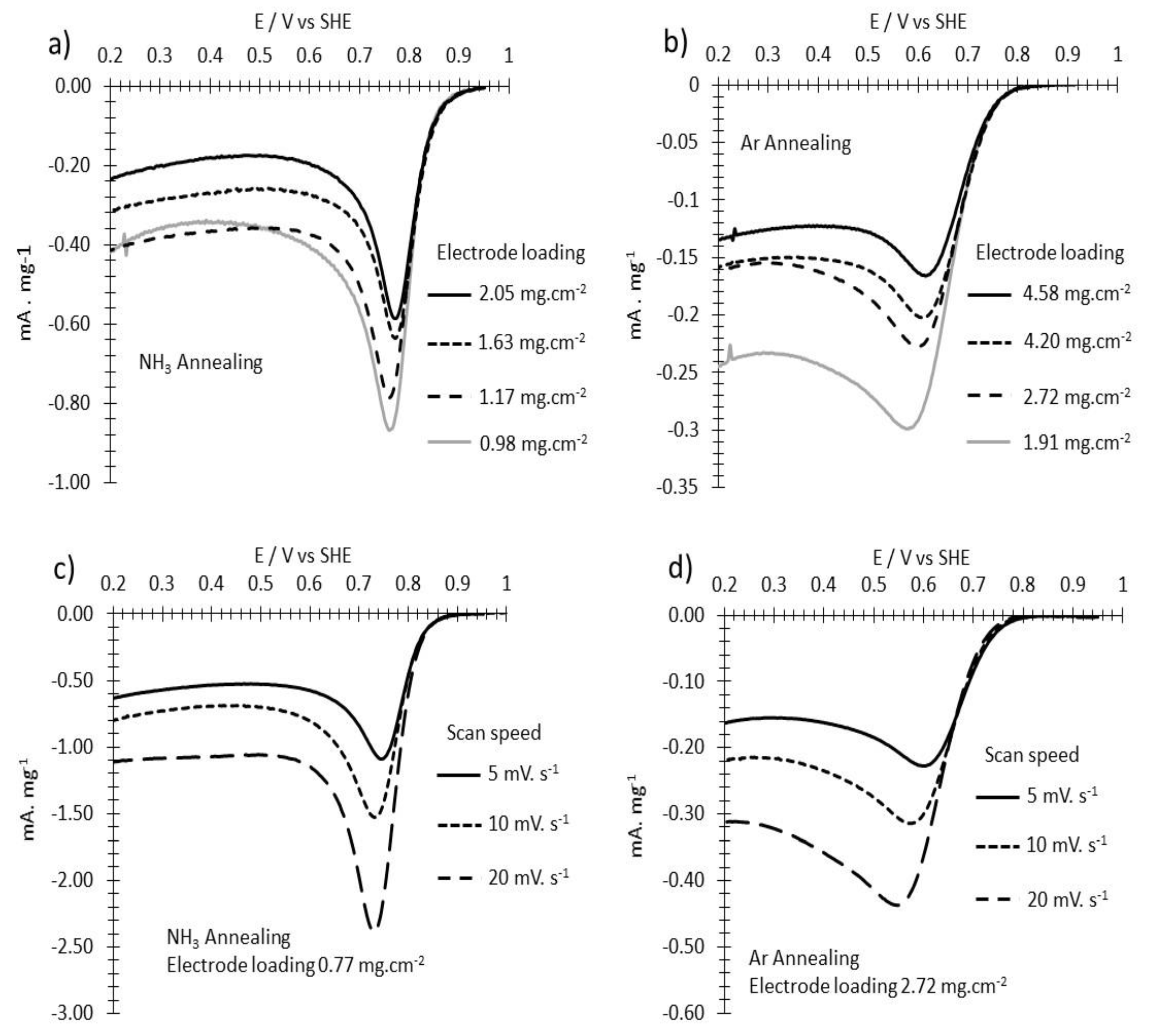
Appendix B
References
- Shao, M.; Chang, Q.; Dodelet, J.-P.; Chenitz, R. Recent advances in electrocatalysts for oxygen reduction. Chem. Rev. 2016, 116, 3594–3657. [Google Scholar] [CrossRef] [PubMed]
- Chokai, M.; Daidou, T.; Nabae, Y. Development of Pt-Free Carbon-Based Catalyst for PEFC Cathode Prepared from Polyacrylonitrile. ECS Trans. 2014, 64, 261–270. [Google Scholar] [CrossRef]
- Proietti, E.; Jaouen, F.; Lefevre, M.; Larouche, N.; Tian, J.; Herranz, J.; Dodelet, J.-P. Iron-based cathode catalyst with enhanced power density in polymer electrolyte membrane fuel cells. Nat. Commun. 2011, 2, 416. [Google Scholar] [CrossRef]
- Shuia, J.; Chena, C.; Grabstanowicza, L.D.; Zhao, D.; Di-Jia Liua, D.-J. Highly efficient nonprecious metal catalyst prepared with metal–organic framework in a continuous carbon nanofibrous network. 2015-PNAS 2015, 112, 10631–10634. [Google Scholar] [CrossRef] [PubMed]
- Serov, A.; Artyushkova, K.; Atanassov, P. Fe-N-C Oxygen Reduction Fuel Cell Catalyst Derived from Carbendazim: Synthesis, Structure, and Reactivity. Adv. Energy Mater. 2014, 4, 1301735. [Google Scholar] [CrossRef]
- Ratso, S.; Sahraie, N.R.; Sougrati, M.T.; Käärik, M.; Kook, M.; Saar, R.; Paiste, P.; Jia, Q.; Leis, J.; Mukerjee, S.; et al. Synthesis of highly-active Fe–N–C catalysts for PEMFC with carbide-derived carbons. Mat. Chem. 2018, 6, 14663–14674. [Google Scholar] [CrossRef]
- Perez, H.; Jorda, V.; Bonville, P.; Vigneron, J.; Frégnaux, M.; Etcheberry, A.; Quinsac, A.; Habert, A.; Leconte, Y. Synthesis and characterization of Carbon/Nitrogen/Iron based nanoparticles by laser pyrolysis as non-noble metal electrocatalysts for oxygen reduction. C 2018, 4, 43. [Google Scholar] [CrossRef]
- Jahnke, H.; Schönborn, M.; Zimmermann, G. Organic dyestuffs as catalysts for fuel cells. Top. Curr. Chem. 1976, 61, 133–181. [Google Scholar] [CrossRef] [PubMed]
- Osmieri, L. Transition Metal-Nitrogen-Carbon (M-N-C) catalysts for oxygen reduction reaction. Insights on synthesis and performance in polymer electrolyte fuel cells. Chem. Eng. 2019, 3, 16. [Google Scholar] [CrossRef]
- Cheng, X.; Than, X.-T.; Pinault, M.; Mayne, M.; Reynaud, C.; Vigneron, J.; Etcheberry, A.; Perez, H. Determination of selectivity and specific area related to oxygen reduction reaction as a function of catalyst loading on non-noble metal based electrocatalyst porous electrodes: An example on nitrogen doped carbon nanotube. Electrochim. Acta 2014, 135, 293–300. [Google Scholar] [CrossRef]
- Cheng, X.; Volatron, F.; Pardieu, E.; Borta, A.; Carrot, G.; Reynaud, C.; Mayne, M.; Pinault, M.; Etcheberry, A.; Perez, H. Nanocomposite electrodes based on pre-synthesized organically grafted platinum nanoparticles and carbon nanotubes III. Determination of oxygen reduction reaction selectivity and specific area of porous electrode related to the oxygen reduction reaction ranging from 2 m2.gPt−1 to 310 m2.gPt−1. Electrochim. Acta 2013, 89, 1–12. [Google Scholar] [CrossRef]
- Cheng, X.; Challier, L.; Etcheberry, A.; Noël, V.; Perez, H. The ABTS-HRP system as an alternative method to RRDE for the determination of the selectivity of the oxygen reduction reaction. Int. J. Electrochem. Sci. 2012, 7, 6247–6264. [Google Scholar]
- De la Puente, G.; Pis, J.J.; Menhdez, J.A.; Grange, P. Thermal stability of oxygenated carbons functions in activated carbons. J. Anal. Appl. Pyrolysis 1997, 43, 125–138. [Google Scholar] [CrossRef]
- Jansen, R.J.J.; Van Bekkum, H. XPS of nitrogen-containing functional groups on activated carbons. Carbon 1995, 33, 1021–1027. [Google Scholar] [CrossRef]
- Boehm, H.P. Some aspects of the surface chemistry of carbon blacks and other carbons. Carbon 1994, 32, 759–769. [Google Scholar] [CrossRef]
- Sherwood, T.K.; Gilliland, E.R.; Ing, S.W. Hydrogen Cyanide Synthesis from Elements and from Ammonia and Carbon. Ind. Eng. Chem. 1960, 52, 601–604. [Google Scholar] [CrossRef]
- Abotsi, G.M.K.; Scaroni, A.W. Reaction of carbons with ammonia: Effects on the surface charge and molybdenum adsorption. Carbon 1990, 28, 79–84. [Google Scholar] [CrossRef]
- Stöhr, B.; Boehm, H.P.; Schlögl, R. Enhancement of the catalytic activity of activated carbons in oxydation reactions by thermal treatment with ammonia or hydrogen cyanide and observation of a superoxide species as a possible intermediate. Carbon 1991, 29, 707–720. [Google Scholar] [CrossRef]
- Jaouen, F.; Lefèvre, M.; Dodelet, J.-P.; Cai, M. Heat-Treated Fe/N/C Catalysts for O2 Electroreduction: Are Active Sites Hosted in Micropores? J. Phys. Chem. B 2006, 110, 5553–5558. [Google Scholar] [CrossRef] [PubMed]
- Jaouen, F.; Charreteur, F.; Dodelet, J.-P. Fe-Based Catalysts for Oxygen Reduction in PEMFCs Importance of the Disordered Phase of the Carbon Support. J. Electrochem. Soc. 2006, 153, A689–A698. [Google Scholar] [CrossRef]
- Charreteur, F.; Jaouen, F.; Ruggeri, S.; Dodelet, J.-P. Fe/N/C non-precious catalysts for PEM fuel cells: Influence of the structural parameters of pristine commercial carbon blacks on their activity for oxygen reduction. Electrochim. Acta 2008, 53, 2925–2938. [Google Scholar] [CrossRef]
- Jaouen, F.; Dodelet, J.-P. Non-Noble Electrocatalysts for O2 Reduction: How Does Heat Treatment Affect Their Activity and Structure? Part, I. Model for Carbon Black Gasification by NH3: Parametric Calibration and Electrochemical Validation. J. Phys. Chem. C 2007, 111, 5963–5970. [Google Scholar] [CrossRef]
- Shafeeyan, M.S.; Wan Daud, W.M.A.; Houshmand, A.; Sharimi, A. A review on surface modification of activated carbon for carbon dioxide adsorption. J. Anal. Apll. Pyrolysis 2010, 89, 143–151. [Google Scholar] [CrossRef]
- Jimenez-Mateos, J.M.; Fiero, J.L.G. X-ray Photoelectron Spectroscopic Study of Petroleum Fuel Cokes. Surf. Interface Anal. 1996, 24, 223–226. [Google Scholar] [CrossRef]
- Susi, T.; Pichler, T.; Ayala, P. X-Ray photoelectron spectroscopy of graphitic carbon nanomaterials doped with heteroatoms. Beilstein J. Nanotechnol. 2015, 6, 177–191. [Google Scholar] [CrossRef]
- Pels, J.R.; Kapteijn, F.; Moulijn, J.A.; Zhu, Q.; Thomas, K.M. Evolution of nitrogen functionalities in carbonaceous materials during pyrolysis. Carbon 1995, 33, 1641–1653. [Google Scholar] [CrossRef]
- Casanovas, J.; Ricart, J.M.; Rubio, J.; Illas, F.; Jimenez-Mateos, J.M. Origin of the Large N 1s Binding Energy in X-ray Photoelectron Spectra of Calcined Carbonaceous Materials. J. Am. Chem. Soc. 1996, 118, 8071–8076. [Google Scholar] [CrossRef]
- Jansen, R.J.J.; Van Bekkum, H. Amination and ammoxidation of activated carbons. XPS of nitrogen-containing functional groups on activated carbons. Carbon 1994, 32, 1507–1516. [Google Scholar] [CrossRef]
- Kramm, U.I.; Herrman-Geppert, I.; Bogdanoff, P.; Fiechter, S. Effect of an ammonia treatment on structure, composition, and oxygen reduction reaction activity of Fe-N-C catalysts. J. Phys. Chem. C 2011, 115, 23417. [Google Scholar] [CrossRef]
- Sharifi, T.; Hu, G.; Jia, X.; Wagberg, T. Formation of Active Sites for Oxygen Reduction Reactions by Transformation of Nitrogen Functionalities in Nitrogen-Doped Carbon Nanotubes. ACS Nano 2012, 6, 8904–8912. [Google Scholar] [CrossRef] [PubMed]
- Stanczyk, K.; Dziembaj, R.; Piwowarska, Z.; Witkowski, S. Transformation of nitrogen structures in carbonization of model compounds determined by XPS. Carbon 1995, 33, 1383–1392. [Google Scholar] [CrossRef]
- Arrigo, R.; Hävecker, M.; Schlögl, R.; Su, D.S. Dynamic surface rearrangement and thermal stability of nitrogen functional groups on carbon nanotubes. Chem. Commun. 2008, 40, 4891–4893. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rate Production g·h−1 | Chemical Yield as-Formed wt % | Chemical Yield after Acetone Washing wt % | Specific Surface Area m2·g−1 | Iron Content wt % |
|---|---|---|---|---|
| 3.4 | 12.5 | 9.5 | 147 | 0.9 |
| Annealing Conditions | wt % Loss | Specific Surface Area m2·g−1 |
|---|---|---|
| Annealing under Ar | 8.0 | 153 |
| Annealing under NH3 | 80.2 | 1130 |
| Material | C1s At.% | O1s At.% | N1s At.% | N1s/C1s At.% |
|---|---|---|---|---|
| As-prepared | 92.0 | 4.6 | 3.4 | 0.037 |
| Argon annealing | 93.1 | 5.3 | 1.6 | 0.017 |
| Ammonia annealing | 96.2 | 2.3 | 1.5 | 0.016 |
| N1NH3/N1Ar | N2NH3/N2Ar | N3NH3/N3Ar | N4NH3/N4Ar | |
|---|---|---|---|---|
| Ratio | 1.47 | 0.86 | 0.82 | 1.21 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez, H.; Jorda, V.; Vigneron, J.; Frégnaux, M.; Etcheberry, A.; Quinsac, A.; Leconte, Y.; Sublemontier, O. Highly Active, High Specific Surface Area Fe/C/N ORR Electrocatalyst from Liquid Precursors by Combination of CO2 Laser Pyrolysis and Single NH3 Thermal Post-Treatment. C 2019, 5, 26. https://doi.org/10.3390/c5020026
Perez H, Jorda V, Vigneron J, Frégnaux M, Etcheberry A, Quinsac A, Leconte Y, Sublemontier O. Highly Active, High Specific Surface Area Fe/C/N ORR Electrocatalyst from Liquid Precursors by Combination of CO2 Laser Pyrolysis and Single NH3 Thermal Post-Treatment. C. 2019; 5(2):26. https://doi.org/10.3390/c5020026
Chicago/Turabian StylePerez, Henri, Virginie Jorda, Jackie Vigneron, Mathieu Frégnaux, Arnaud Etcheberry, Axelle Quinsac, Yann Leconte, and Olivier Sublemontier. 2019. "Highly Active, High Specific Surface Area Fe/C/N ORR Electrocatalyst from Liquid Precursors by Combination of CO2 Laser Pyrolysis and Single NH3 Thermal Post-Treatment" C 5, no. 2: 26. https://doi.org/10.3390/c5020026
APA StylePerez, H., Jorda, V., Vigneron, J., Frégnaux, M., Etcheberry, A., Quinsac, A., Leconte, Y., & Sublemontier, O. (2019). Highly Active, High Specific Surface Area Fe/C/N ORR Electrocatalyst from Liquid Precursors by Combination of CO2 Laser Pyrolysis and Single NH3 Thermal Post-Treatment. C, 5(2), 26. https://doi.org/10.3390/c5020026