Controlling the Surface Oxygen Groups of Polyacrylonitrile-Based Carbon Nanofiber Membranes While Limiting Fiber Degradation


Abstract
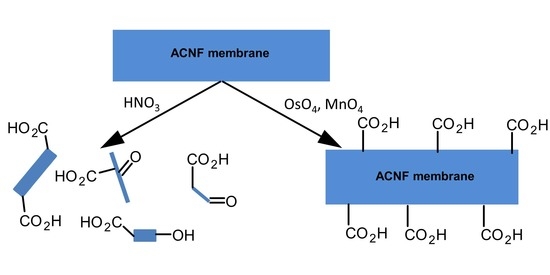
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Oxidation Experiments
2.2.1. Control Method 1: HNO3 Oxidation
2.2.2. Control Method 2: HNO3/H2SO4 Oxidation
2.2.3. Low Concentration MnO4 Oxidation (MnO4-L)
2.2.4. High Concentration MnO4 Oxidation (MnO4-H)
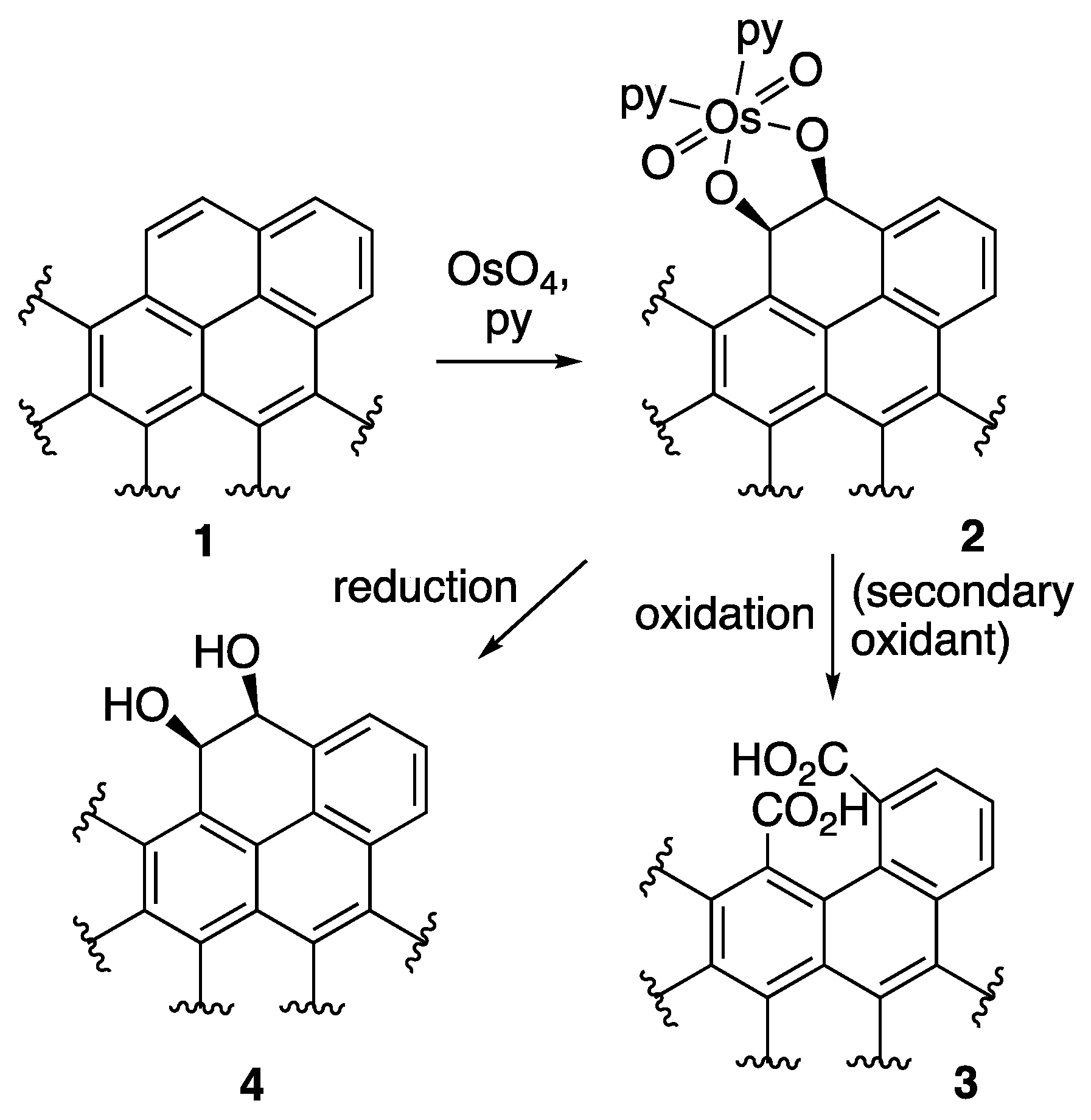
2.2.5. OsO4 + Oxone® Oxidation
2.2.6. OsO4 + MnO4-L Oxidation
2.2.7. OsO4 + MnO4-H Oxidation
2.2.8. RuO4 Oxidation
2.3. Physical Characterization of Oxidized ACNF Samples
2.4. Chemical Characterization of Oxidized ACNF Samples
3. Results and Discussion
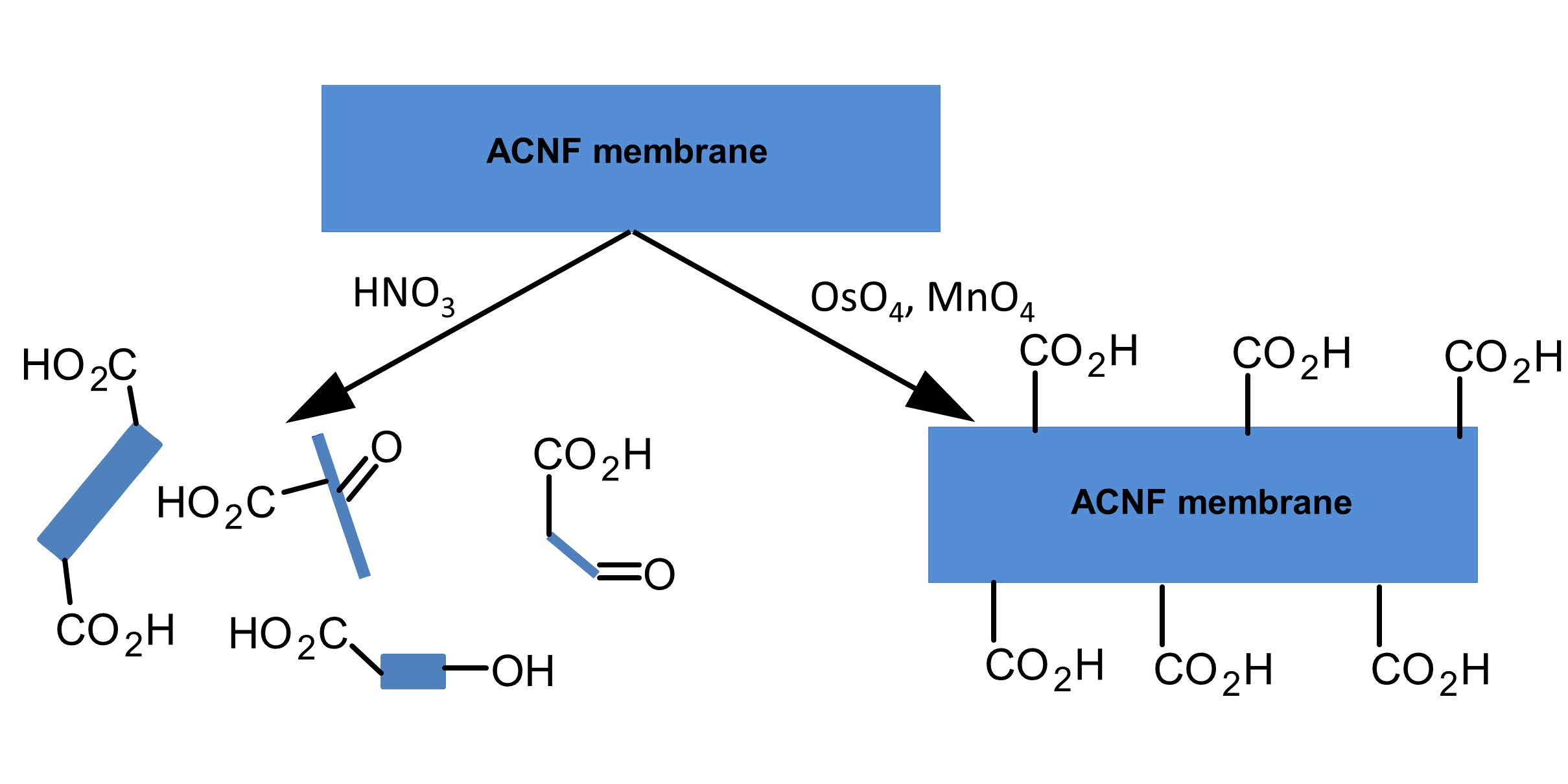
3.1. Physical Characterization of the Oxidation Products
3.2. FTIR Characterization of the Oxidation Products
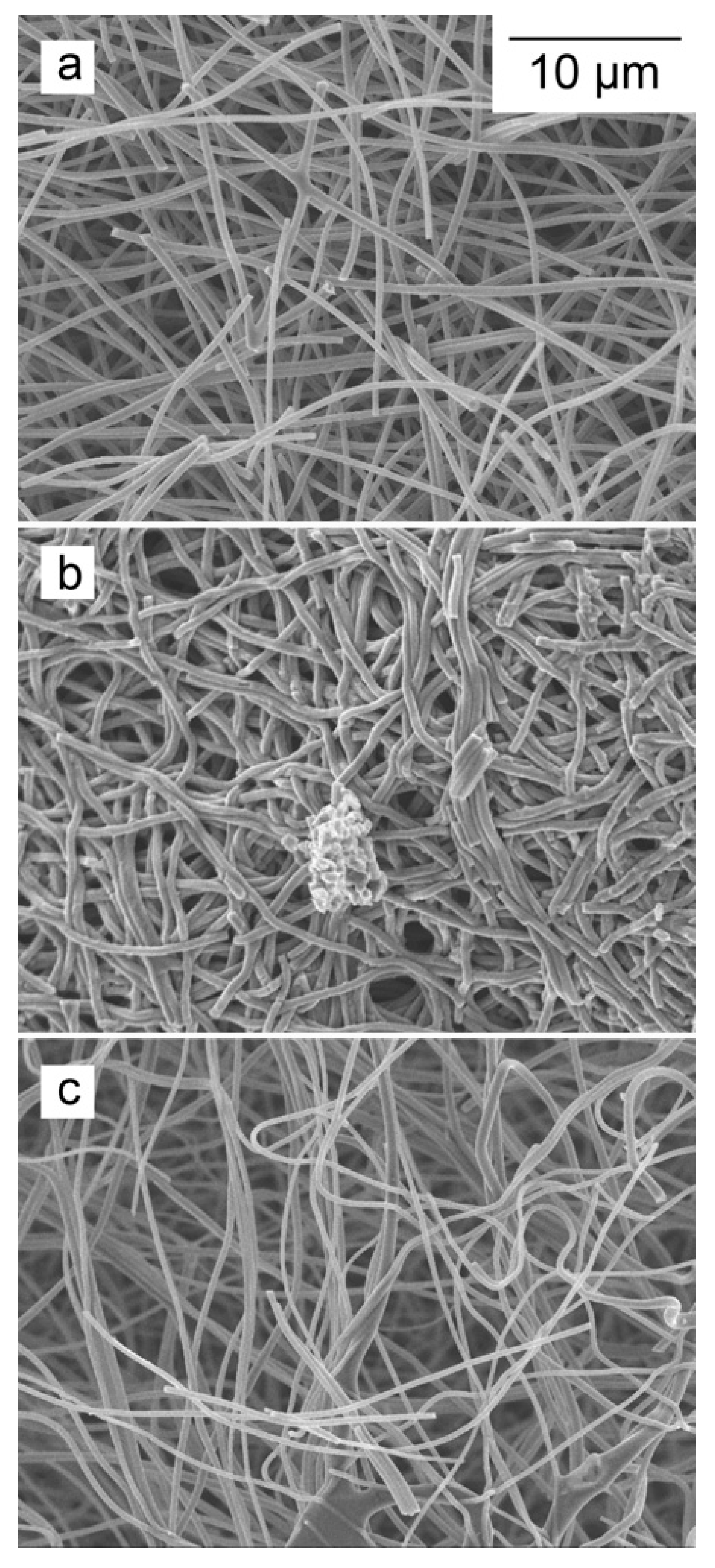
3.3. Specification of the Active Hydrogen Functionalities in the Oxidized ACNFs Using Boehm Titrations
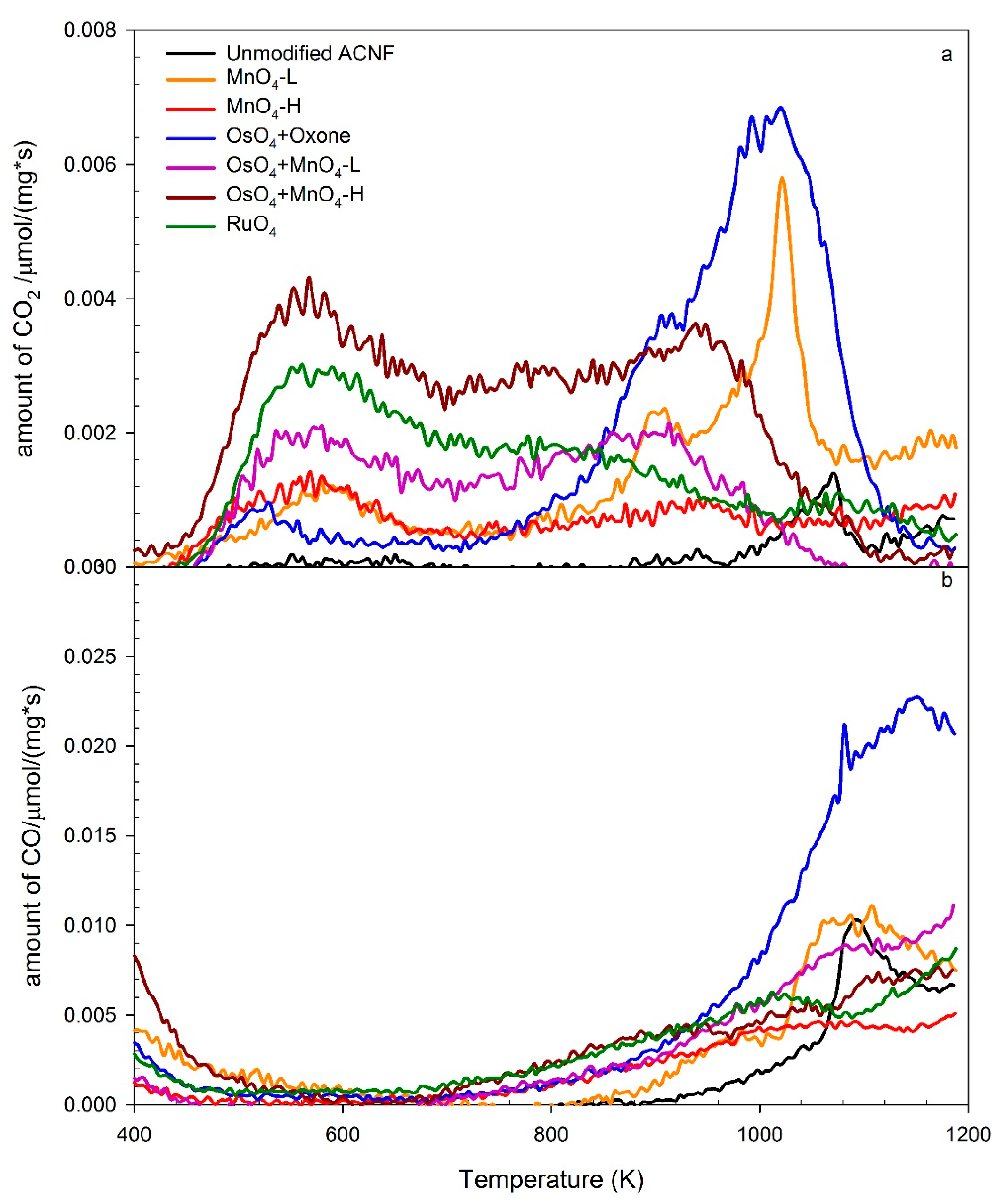
3.4. TPD-MS Analyses of the Oxidized ACNFs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mauter, M.S.; Elimelech, M. Environmental applications of carbon-based nanomaterials. Environ. Sci. Technol. 2008, 42, 5843–5859. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.P.; Lu, C.; Su, F. Sorption of divalent metal ions from aqueous solution by carbon nanotubes: A review. Sep. Purif. Technol. 2007, 58, 224–231. [Google Scholar] [CrossRef]
- Hummer, G.; Rasaiah, J.C.; Noworyta, J.P. Water conduction through the hydrophobic channel of a carbon nanotube. Nature 2001, 414, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Vecitis, C.D.; Schnoor, M.H.; Rahaman, M.S.; Schiffman, J.D.; Elimelech, M. Electrochemical multiwalled carbon nanotube filter for viral and bacterial removal and inactivation. Environ. Sci. Technol. 2011, 45, 3672–3679. [Google Scholar] [CrossRef] [PubMed]
- Al-Saleh, M.H.; Sundararaj, U. A review of vapor grown carbon nanofiber/polymer conductive composites. Carbon 2009, 47, 2–22. [Google Scholar] [CrossRef]
- Jang, J.; Bae, J. Carbon nanofiber/polypyrrole nanocable as toxic gas sensor. Sens. Actuators B Chem. 2007, 122, 7–13. [Google Scholar] [CrossRef]
- Mitchell, R.R.; Gallant, B.M.; Thompson, C.V.; Shao-Horn, Y. All-carbon-nanofiber electrodes for high-energy rechargeable Li-O2 batteries. Energy Environ. Sci. 2011, 4, 2952–2958. [Google Scholar] [CrossRef]
- Faur-Brasquet, C.; Kadirvelu, K.; Le Cloirec, P. Removal of metal ions from aqueous solution by adsorption onto activated carbon cloths: Adsorption competition with organic matter. Carbon 2002, 40, 2387–2392. [Google Scholar] [CrossRef]
- Gao, L.; Wang, X.; Xie, Z.; Song, W.; Wang, L.; Wu, X.; Qu, F.; Chen, D.; Shen, G. High-performance energy-storage devices based on WO3 nanowire arrays/carbon cloth integrated electrodes. J. Mater. Chem. A 2013, 1, 7167–7173. [Google Scholar] [CrossRef]
- Zhao, F.; Rahunen, N.; Varcoe, J.R.; Chandra, A.; Avignone-Rossa, C.; Thumser, A.E.; Slade, R.C.T. Activated carbon cloth as anode for sulfate removal in a microbial fuel cell. Environ. Sci. Technol. 2008, 42, 4971–4976. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Shiratori, N.; Lee, G.H.; Miyawaki, J.; Mochida, I.; Yoon, S.-H.; Jang, J. Activated carbon nanofiber produced from electrospun polyacrylonitrile nanofiber as a highly efficient formaldehyde adsorbent. Carbon 2010, 48, 4248–4255. [Google Scholar] [CrossRef]
- Fan, Z.; Yan, J.; Wei, T.; Zhi, L.; Ning, G.; Li, T.; Wei, F. Asymmetric supercapacitors based on graphene/MnO2 and activated carbon nanofiber electrodes with high power and energy density. Adv. Funct. Mater. 2011, 21, 2366–2375. [Google Scholar] [CrossRef]
- Kim, C.; Yang, K.S. Electrochemical properties of carbon nanofiber web as an electrode for supercapacitor prepared by electrospinning. Appl. Phys. Lett. 2003, 83, 1216–1218. [Google Scholar] [CrossRef]
- Qu, X.; Brame, J.; LI, Q.; Alvarez, P.J.J. Nanotechnology for a safe and sustainable water supply: Enabling integrated water treatment and reuse. Acc. Chem. Res. 2012, 46, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Dai, L.; Gao, M.; Huang, S.; Mau, A. Plasma activation of carbon nanotubes for chemical modification. J. Phys. Chem. B 2001, 105, 618–622. [Google Scholar] [CrossRef]
- Karousis, N.; Tagmatarchis, N.; Tasis, D. Current progress on the chemical modification of carbon nanotubes. Chem. Rev. 2010, 110, 5366–5397. [Google Scholar] [CrossRef] [PubMed]
- Morales-Lara, F.; Pérez-Mendoza, M.J.; Altmajer-Vaz, D.; García-Román, M.; Melguizo, M.; López-Garzón, F.J.; Domingo-García, M. Functionalization of multiwall carbon nanotubes by ozone at basic pH. Comparison with oxygen plasma and ozone in gas phase. J. Phys. Chem. C 2013, 117, 11647–11655. [Google Scholar] [CrossRef]
- Rasheed, A.; Howe, J.Y.; Dadmun, M.D.; Britt, P.F. The efficiency of the oxidation of carbon nanofibers with various oxidizing agents. Carbon 2007, 45, 1072–1080. [Google Scholar] [CrossRef]
- Saito, T.; Matsushige, K.; Tanaka, K. Chemical treatment and modification of multi-walled carbon nanotubes. Phys. B Condens. Matter 2002, 323, 280–283. [Google Scholar] [CrossRef]
- Kumar, S.; Rath, T.; Mahaling, R.N.; Reddy, C.S.; Das, C.K.; Pandey, K.N.; Srivastava, R.B.; Yadaw, S.B. Study on mechanical, morphological and electrical properties of carbon nanofiber/polyetherimide composites. Mater. Sci. Eng. B Solid State Adv. Technol. 2007, 141, 61–70. [Google Scholar] [CrossRef]
- Wang, J.; Lin, Y. Functionalized carbon nanotubes and nanofibers for biosensing applications. TrAC Trends Anal. Chem. 2008, 27, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Kampalanonwat, P.; Supaphol, P. Preparation and adsorption behavior of aminated electrospun polyacrylonitrile nanofiber mats for heavy metal ion removal. ACS Appl. Mater. Interfaces 2010, 2, 3619–3627. [Google Scholar] [CrossRef] [PubMed]
- Toebes, M.L.; van Heeswijk, J.M.P.; Bitter, J.H.; Jos van Dillen, A.; de Jong, K.P. The influence of oxidation on the texture and the number of oxygen-containing surface groups of carbon nanofibers. Carbon 2004, 42, 307–315. [Google Scholar] [CrossRef]
- Zhou, J.-H.; Sui, Z.-J.; Zhu, J.; Li, P.; Chen, D.; Dai, Y.-C.; Yuan, W.-K. Characterization of surface oxygen complexes on carbon nanofibers by TPD, XPS and FT-IR. Carbon 2007, 45, 785–796. [Google Scholar] [CrossRef]
- Pradhan, B.K.; Sandle, N.K. Effect of different oxidizing agent treatments on the surface properties of activated carbons. Carbon 1999, 37, 1323–1332. [Google Scholar] [CrossRef]
- Kyzas, G.Z.; Lazaridis, N.K.; Deliyanni, E.A. Oxidation time effect of activated carbons for drug adsorption. Chem. Eng. J. 2013, 234, 491–499. [Google Scholar] [CrossRef]
- Datsyuk, V.; Kalyva, M.; Papagelis, K.; Parthenios, J.; Tasis, D.; Siokou, A.; Kallitsis, I.; Galiotis, C. Chemical oxidation of multiwalled carbon nanotubes. Carbon 2008, 46, 833–840. [Google Scholar] [CrossRef]
- Rosca, I.D.; Watari, F.; Uo, M.; Akasaka, T. Oxidation of multiwalled carbon nanotubes by nitric acid. Carbon 2005, 43, 3124–3131. [Google Scholar] [CrossRef]
- Moreno-Castilla, C.; Ferro-Garcia, M.; Joly, J.; Bautista-Toledo, I.; Carrasco-Marin, F.; Rivera-Utrilla, J. Activated carbon surface modifications by nitric acid, hydrogen peroxide, and ammonium peroxydisulfate treatments. Langmuir 1995, 11, 4386–4392. [Google Scholar] [CrossRef]
- Hernadi, K.; Siska, A.; Thiên-Nga, L.; Forró, L.; Kiricsi, I. Reactivity of different kinds of carbon during oxidative purification of catalytically prepared carbon nanotubes. Solid State Ion. 2001, 141–142, 203–209. [Google Scholar] [CrossRef]
- Pereira, M.F.R.; Soares, S.F.; Órfão, J.J.M.; Figueiredo, J.L. Adsorption of dyes on activated carbons: Influence of surface chemical groups. Carbon 2003, 41, 811–821. [Google Scholar] [CrossRef]
- Sullivan, P.D.; Stone, B.R.; Hashisho, Z.; Rood, M.J. Water adsorption with hysteresis effect onto microporous activated carbon fabrics. Adsorption 2007, 13, 173–189. [Google Scholar] [CrossRef]
- Li, R.; Zhang, L.; Wang, P. Rational design of nanomaterials for water treatment. Nanoscale 2015, 7, 17167–17194. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Utrilla, J.; Sánchez-Polo, M.; Gómez-Serrano, V.; Alvarez, P.; Alvim-Ferraz, M.; Dias, J. Activated carbon modifications to enhance its water treatment applications. An overview. J. Hazard. Mater. 2011, 187, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M. Osmium tetroxide cis-hydroxylation of unsaturated substrates. Chem. Rev. 1980, 80, 187–213. [Google Scholar] [CrossRef]
- Hwang, K.C. Efficient cleavage of carbon graphene layers by oxidants. J. Chem. Soc. Chem. Commun. 1995, 2, 173–174. [Google Scholar] [CrossRef]
- Berkowitz, L.M.; Rylander, P.N. Use of ruthenium tetroxide as a multi-purpose oxidant. J. Am. Chem. Soc. 1958, 80, 6682–6684. [Google Scholar] [CrossRef]
- Manickam, S.S.; Karra, U.; Huang, L.; Bui, N.-N.; Li, B.; McCutcheon, J.R. Activated carbon nanofiber anodes for microbial fuel cells. Carbon 2013, 53, 19–28. [Google Scholar] [CrossRef]
- Bai, Y.; Huang, Z.-H.; Kang, F. Surface oxidation of activated electrospun carbon nanofibers and their adsorption performance for benzene, butanone and ethanol. Colloids Surf. A 2014, 443, 66–71. [Google Scholar] [CrossRef]
- Collin, R.J.; Jones, J.; Griffith, W.P. Reaction of osmium tetroxide with alkenes, glycols, and alkynes; oxo-osmium(VI) esters and their structures. J. Chem. Soc. Dalton Trans. 1974, 10, 1094–1097. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Eder, S.J.; Scherer, W. Mehrfachbindungen zwischen Hauptgruppenelementen und Übergangsmetallen, CIX. Strukturchemische Aspekte der Fluorolefinchemie von Osmiumtetraoxid: Halogenierte Osmatester und Folgeprodukte. Chem. Ber. 1993, 126, 39–43. [Google Scholar] [CrossRef]
- Jagiello, J.; Olivier, J.P. A simple two-dimensional NLDFT model of gas adsorption in finite carbon pores. Application to pore structure analysis. J. Phys. Chem. C 2009, 113, 19382–19385. [Google Scholar] [CrossRef]
- Culmo, R.F. The Elemental Analysis of Various Classes of Chemical Compounds Using CHN; PerkinElmer, Inc.: Shelton, CT, USA, 2013. [Google Scholar]
- Balcerzak, M.; Świecicka, E.; Bystrońska, D. Simple selective spectrophotometric method for the determination of ruthenium in carbon supported Pt-Ru-Ge catalyst. Anal. Lett. 1999, 32, 1799–1805. [Google Scholar] [CrossRef]
- Chahrour, O.; Malone, J.; Collins, M.; Salmon, V.; Greenan, C.; Bombardier, A.; Ma, Z.; Dunwoody, N. Development and validation of an ICP-MS method for the determination of elemental impurities in TP-6076 active pharmaceutical ingredient (API) according to USP 〈232〉/〈233〉. J. Pharm. Biomed. Anal. 2017, 145, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Boehm, H. Some aspects of the surface chemistry of carbon blacks and other carbons. Carbon 1994, 32, 759–769. [Google Scholar] [CrossRef]
- Boehm, H. Chemical identification of surface groups. Adv. Catal. 1966, 16, 179–274. [Google Scholar]
- Shafeeyan, M.S.; Daud, W.M.A.W.; Houshmand, A.; Shamiri, A. A review on surface modification of activated carbon for carbon dioxide adsorption. J. Anal. Appl. Pyrolysis 2010, 89, 143–151. [Google Scholar] [CrossRef]
- Stuart, B. Infrared Spectroscopy: Fundamentals and Applications; John Wiley & Sons: New York, NY, USA, 2004. [Google Scholar]
- McGann, J.P.; Zhong, M.; Kim, E.K.; Natesakhawat, S.; Jaroniec, M.; Whitacre, J.F.; Matyjasqewski, K.; Kowalewski, T. Block copolymer templating as a path to porous nanostructured carbons with highly accessible nitrogens for enhanced (electro)chemical performance. Macromol. Chem. Phys. 2012, 213, 1078–1090. [Google Scholar] [CrossRef]
- Dandekar, A.; Baker, R.; Vannice, M. Characterization of activated carbon, graphitized carbon fibers and synthetic diamond powder using tpd and drifts. Carbon 1998, 36, 1821–1831. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R.; Freitas, M.M.A.; Órfão, J.J.M. Modification of the surface chemistry of activated carbons. Carbon 1999, 37, 1379–1389. [Google Scholar] [CrossRef]
- Kalijadis, A.M.; Vukčević, M.M.; Jovanović, Z.M.; Laušević, Z.V.; Laušević, M.D. Characterization of surface oxygen groups on different carbon materials by the Boehm method and temperature programmed desorption. J. Serbian Chem. Soc. 2011, 76, 757–768. [Google Scholar] [CrossRef]
- Mangun, C.L.; Benak, K.R.; Daley, M.A.; Economy, J. Oxidation of activated carbon fibers: Effect on pore size, surface chemistry, and adsorption properties. Chem. Mater. 1999, 11, 3476–3483. [Google Scholar] [CrossRef]
- Parikh, S.J.; Chorover, J. FTIR spectroscopic study of biogenic Mn-oxide formation by Pseudomonas putida GB-1. Geomicrobiol. J. 2005, 22, 207–218. [Google Scholar] [CrossRef]
- Bhadra, B.N.; Seo, P.W.; Jhung, S.H. Adsorption of Diclofenac sodium from water using oxidized activated carbon. Chem. Eng. J. 2016, 301, 27–34. [Google Scholar] [CrossRef]
- Travis, B.R.; Narayan, R.S.; Borhan, B. Osmium tetroxide-promoted catalytic oxidative cleavage of olefins: An organometallic ozonolysis. J. Am. Chem. Soc. 2002, 124, 3824–3825. [Google Scholar] [CrossRef] [PubMed]
- Schomake, J.M.; Travis, B.R.; Borhan, B. Direct lactonization of alkenols via osmium tetroxide-mediated oxidative cleavage. Org. Lett. 2003, 5, 3089–3092. [Google Scholar] [CrossRef] [PubMed]
- De la Puente, G.; Pis, J.J.; Menéndez, J.A.; Grange, P. Thermal stability of oxygenated functions in activated carbons. J. Anal. Appl. Pyrolysis 1997, 43, 125–138. [Google Scholar] [CrossRef]
- Schönherr, J.; Buchheim, J.R.; Scholz, P.; Adelhelm, P. Boehm titration revisited (part II): A comparison of Boehm titration with other analytical techniques on the quantification of oxygen-containing surface groups for a variety of carbon materials. C 2018, 4, 22. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Mass Yield (%) | Average Fiber Diameter (nm) | Visible Physical Change of Membrane |
|---|---|---|---|
| Unmodified | – | 430 ± 65 | – |
| HNO3 | 82.4 | 285 ± 41 | powdered material |
| HNO3/H2SO4 | 30.0 | 529 ± 73 | powdered material |
| MnO4-L | 99.1 | 534 ± 120 | none |
| MnO4-H | 105 1 | 345 ± 58 | none |
| OsO4 + Oxone | 97.2 | 300 ± 39 | none |
| OsO4 + MnO4-L | 98.8 | 384 ± 73 | none |
| OsO4 + MnO4-H | 96.5 | 359 ± 94 | none |
| RuO4 | 49.6 | 499 ± 66 | none |
| Sample | BET Specific Surface Area (m2/g) | Average Pore Width (Å) | Micropore Volume (cm3/g) | Total Pore Volume (cm3) | Microporosity (%) |
|---|---|---|---|---|---|
| Unmodified | 604 | 20.5 | 0.24 | 0.25 | 96 |
| MnO4-L | 134 | 22.4 | 0.048 | 0.066 | 73 |
| MnO4-H | 457 | 24.9 | 0.17 | 0.17 | 100 |
| OsO4-Oxone® | 21.9 | 56.8 | 0.001 | 0.067 | 1.5 |
| OsO4-MnO4-L | 8.44 | 65.2 | 0.0003 | 0.017 | 1.8 |
| OsO4-MnO4-H | 294 | 21.7 | 0.11 | 0.13 | 85 |
| RuO4 | 7.62 | 83.9 | 0.002 | 0.015 | 13 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, Y.; Li, R.; Brückner, C.; Vadas, T.M. Controlling the Surface Oxygen Groups of Polyacrylonitrile-Based Carbon Nanofiber Membranes While Limiting Fiber Degradation. C 2018, 4, 40. https://doi.org/10.3390/c4030040
Han Y, Li R, Brückner C, Vadas TM. Controlling the Surface Oxygen Groups of Polyacrylonitrile-Based Carbon Nanofiber Membranes While Limiting Fiber Degradation. C. 2018; 4(3):40. https://doi.org/10.3390/c4030040
Chicago/Turabian StyleHan, Yi, Ruoshi Li, Christian Brückner, and Timothy M. Vadas. 2018. "Controlling the Surface Oxygen Groups of Polyacrylonitrile-Based Carbon Nanofiber Membranes While Limiting Fiber Degradation" C 4, no. 3: 40. https://doi.org/10.3390/c4030040
APA StyleHan, Y., Li, R., Brückner, C., & Vadas, T. M. (2018). Controlling the Surface Oxygen Groups of Polyacrylonitrile-Based Carbon Nanofiber Membranes While Limiting Fiber Degradation. C, 4(3), 40. https://doi.org/10.3390/c4030040