
Carbon Materials from Lignin and Sodium Lignosulfonate via Diisocyanate Cross-Linking and Subsequent Carbonization

Abstract
:
1. Introduction
2. Results and Discussion
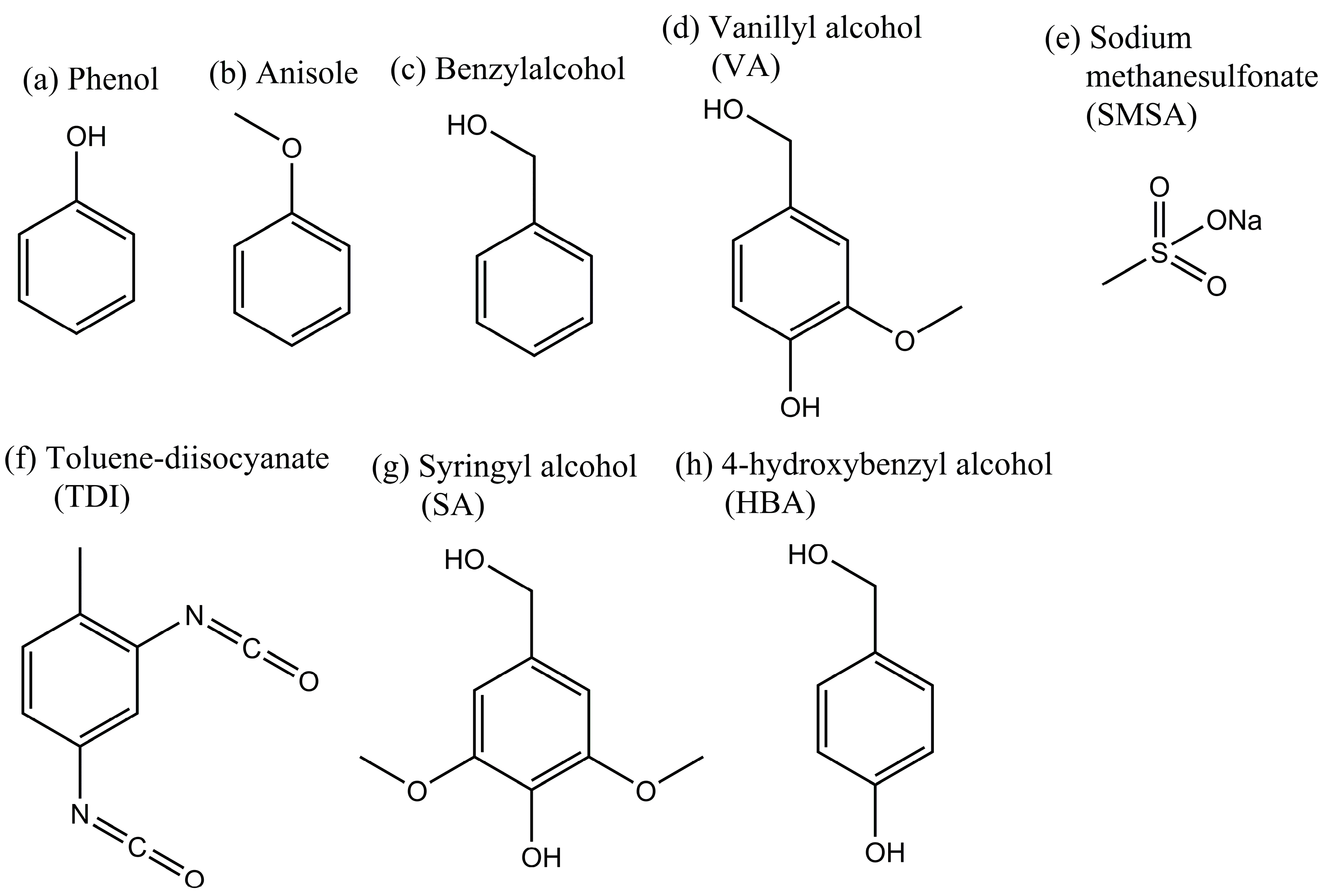
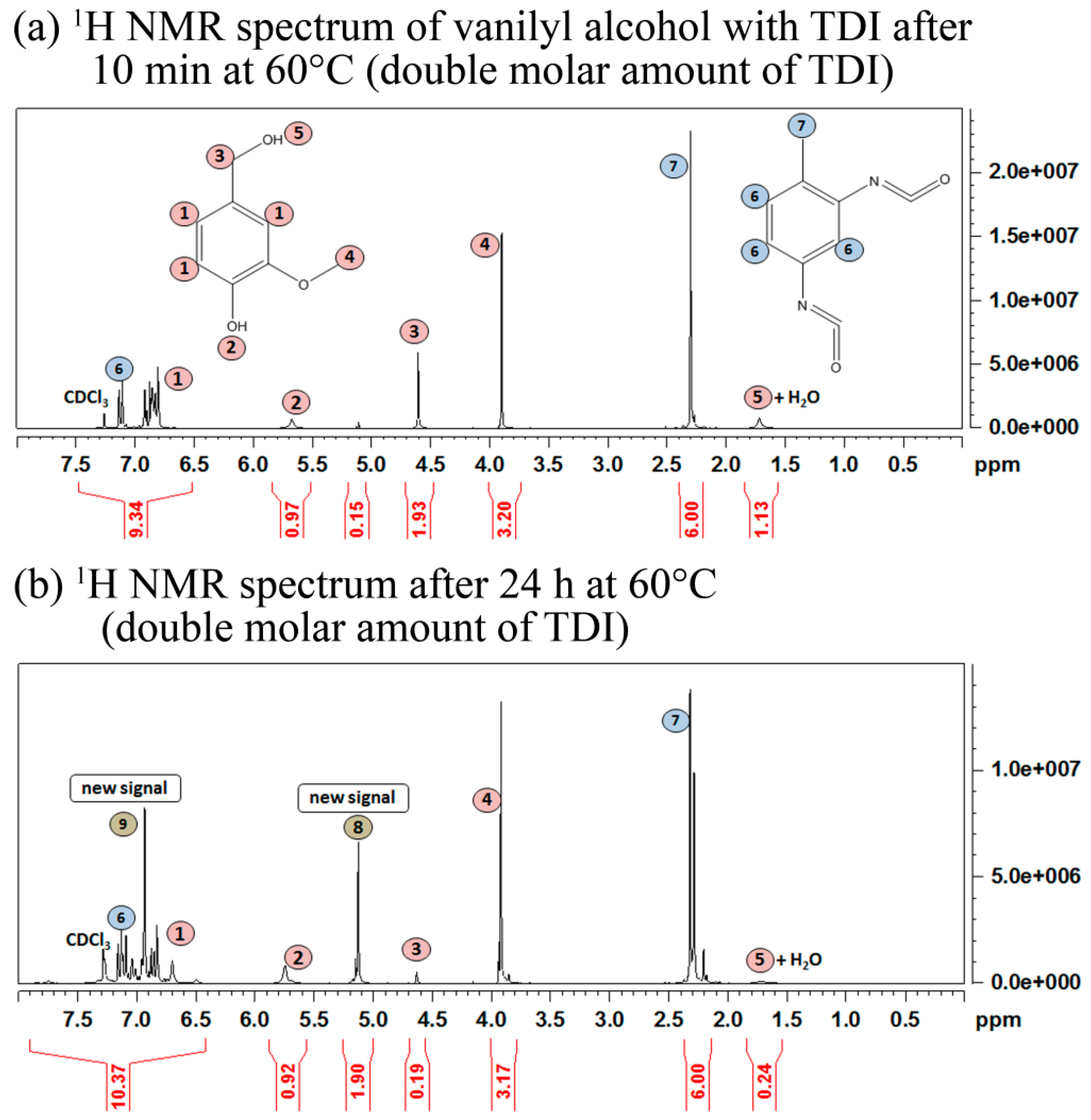
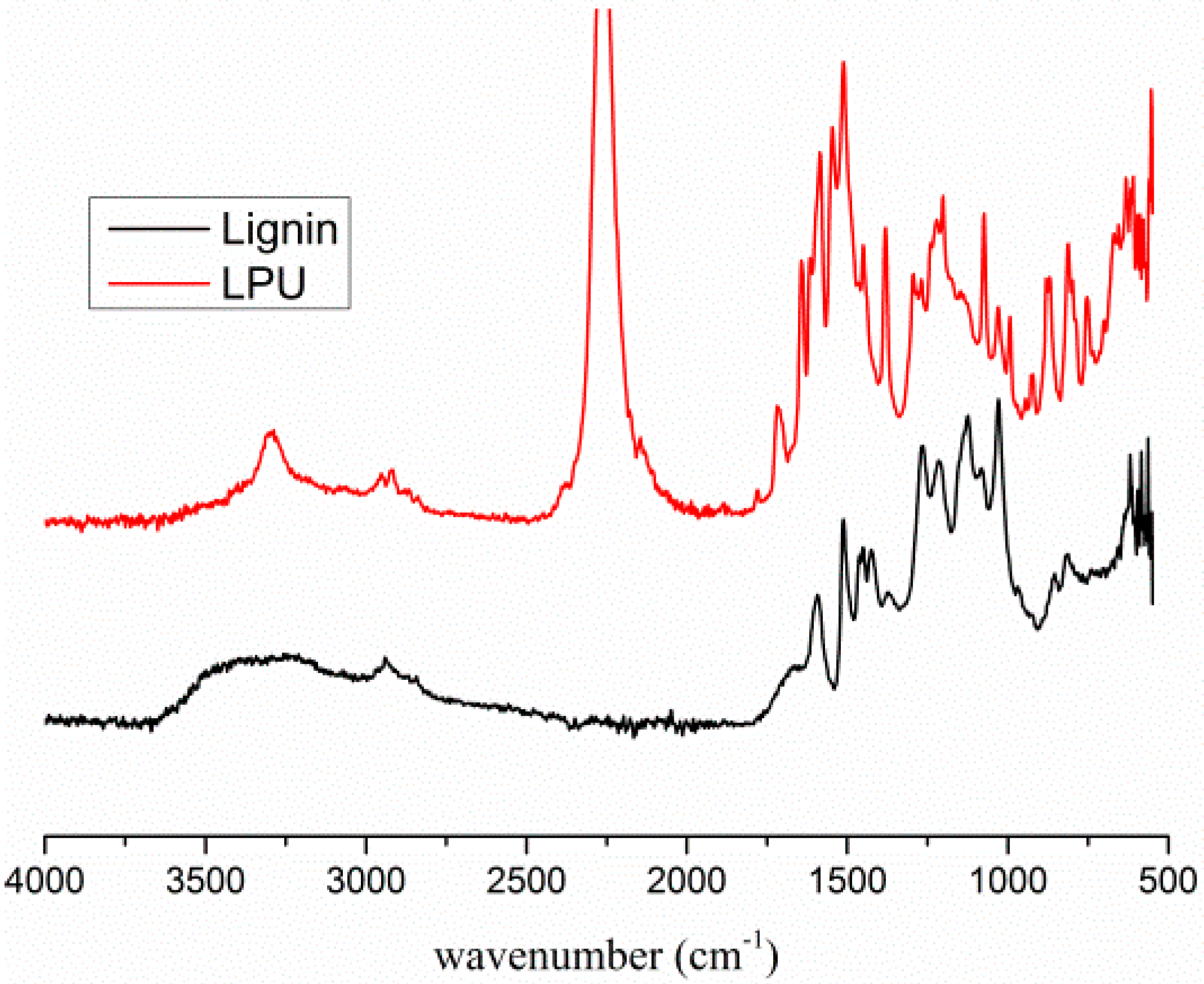
2.1. Cross-Linking Reaction Study with Model Substances
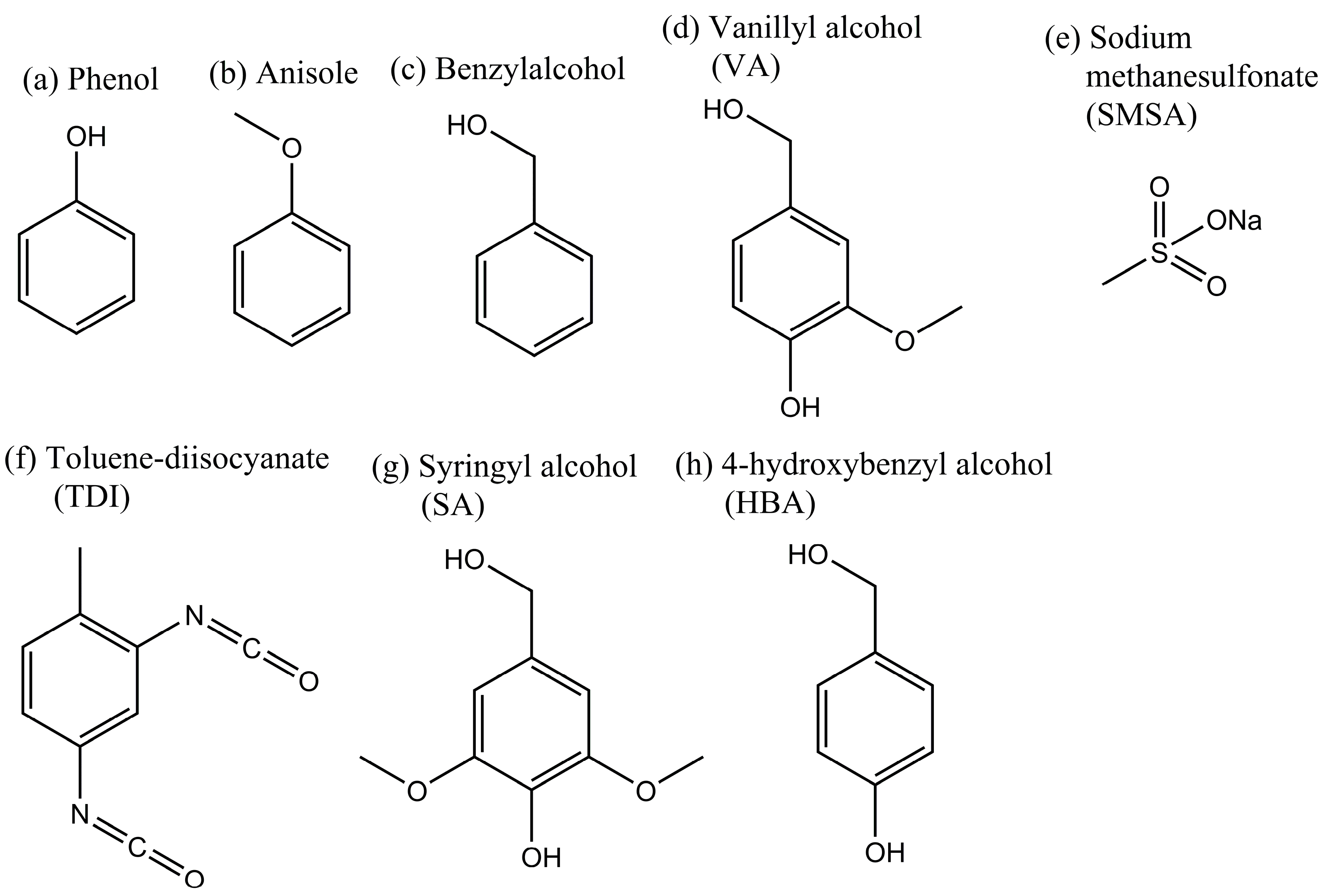
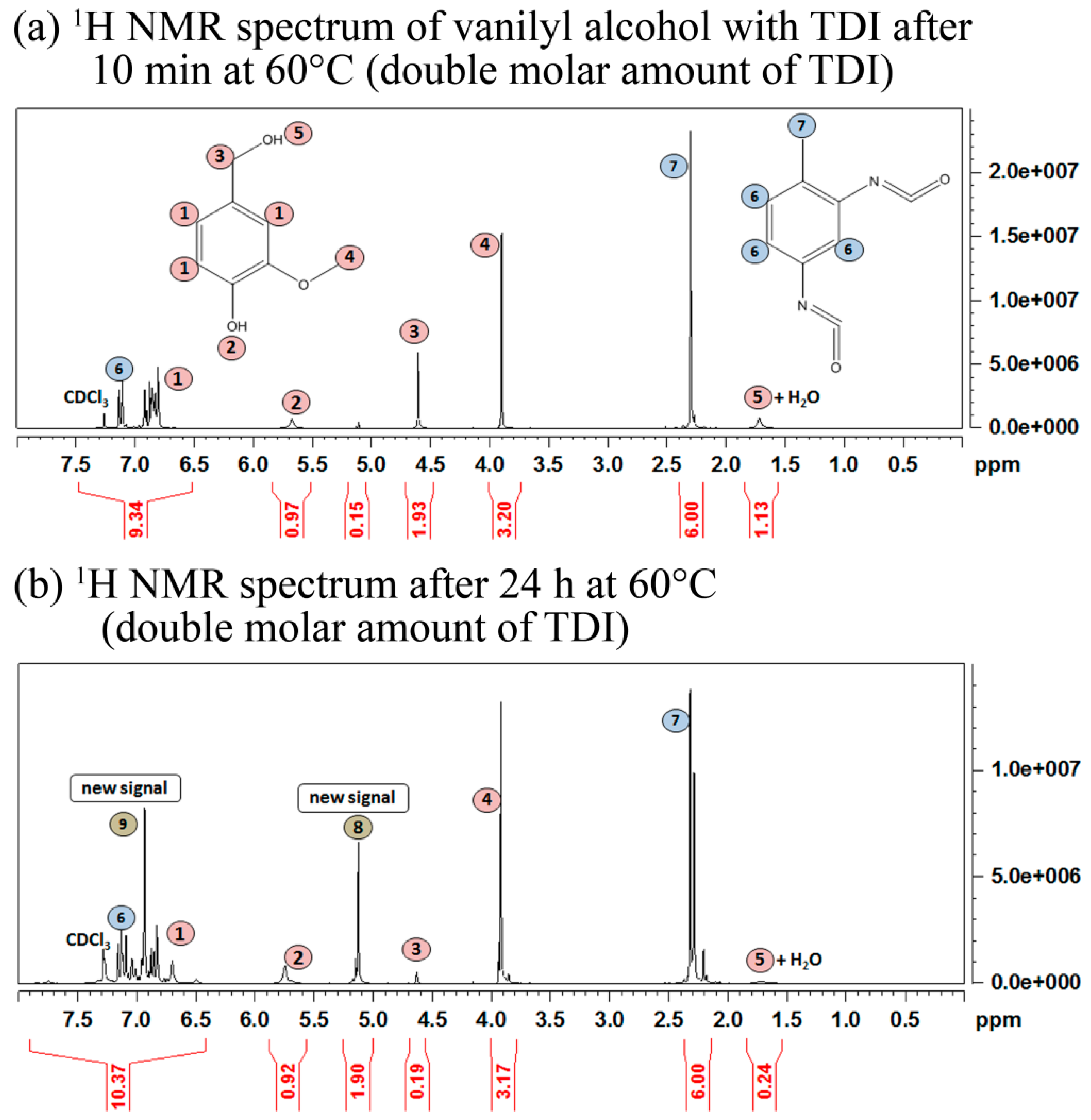
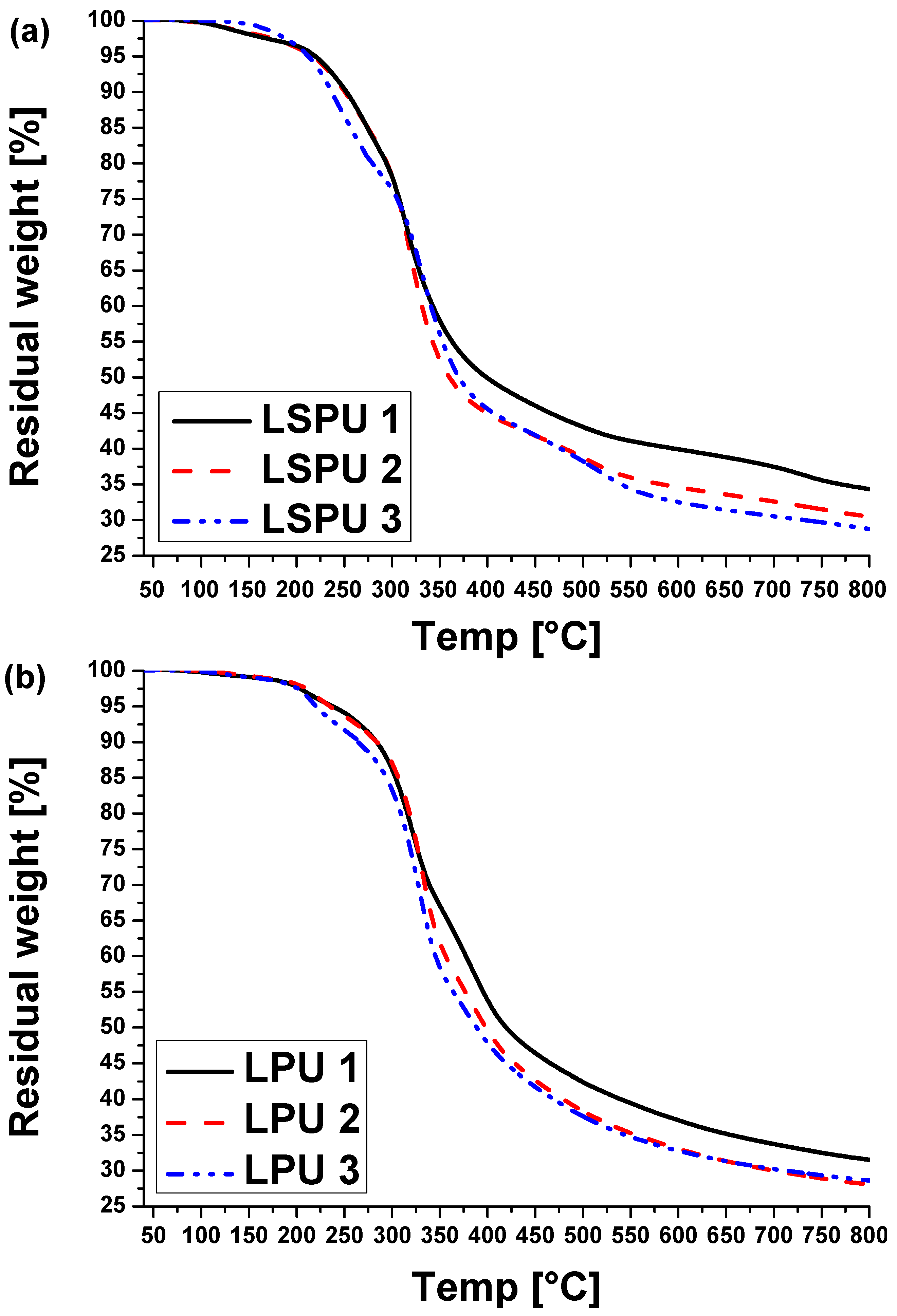
2.2. Carbonization of Cross-Linked Kraft Lignin or Lignosulfonates


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Initial Composition TDI:Lignin or TDI:Lignosulfonate (g:g) | Elemental Analysis | Residue after TGA-Carbonization (wt%) | |||
|---|---|---|---|---|---|---|
| C (Ccalc) * (wt%) | H (Hcalc) * (wt%) | N (Ncalc) * (wt%) | S (Scalc) * (wt%) | |||
| LPU 1 | 0.5 | 62 (61) | 4.9 (4.7) | 6.8 (5.4) | - | 32 |
| LPU 2 | 1.0 | 62 (62) | 4.8 (4.4) | 9.2 (8.0) | - | 28 |
| LPU 3 | 1.5 | 62 (62) | 4.7 (4.2) | 10 (9.7) | - | 28 |
| LSPU 1 | 0.5 | 48 (48) | 4.3 (4.1) | 5.5 (5.4) | 2.9 (2.6) | 34 |
| LSPU 2 | 1.0 | 53 (51) | 4.5 (3.9) | 9.1 (8.0) | 2.6 (1.9) | 30 |
| LSPU 3 | 1.5 | 56 (53) | 4.2 (3.8) | 11 (9.7) | 2.2 (1.5) | 29 |

2.3. Carbon Material
| Carbonized Material | Elemental Analysis | Bulk Density (g·cm−3) | BET Surface Areas (m²·g−1) | ||
|---|---|---|---|---|---|
| C (wt%) | H (wt%) | N (wt%) | |||
| LPU 1 | 85 | 0.8 | 2.9 | 0.145 | 9.64 |
| LPU 2 | 87 | 0.9 | 3.1 | 0.153 | 80.3 |
| LPU 3 | 85 | 0.8 | 3.3 | 0.132 | 0.86 |
| LSPU 1 | 68 | 0.8 | 1.1 | 0.520 | 42.8 |
| LSPU 2 | 65 | 0.8 | 0.9 | 0.492 | 70.8 |
| LSPU 3 | 67 | 0.9 | 1.1 | 0.501 | 1.51 |
| Name | Elemental Analysis | ||
|---|---|---|---|
| C (wt%) | H (wt%) | S (wt%) | |
| Lignin | 61 | 5.3 | - |
| Lignosulfonate | 40 | 4.4 | 3.8 |


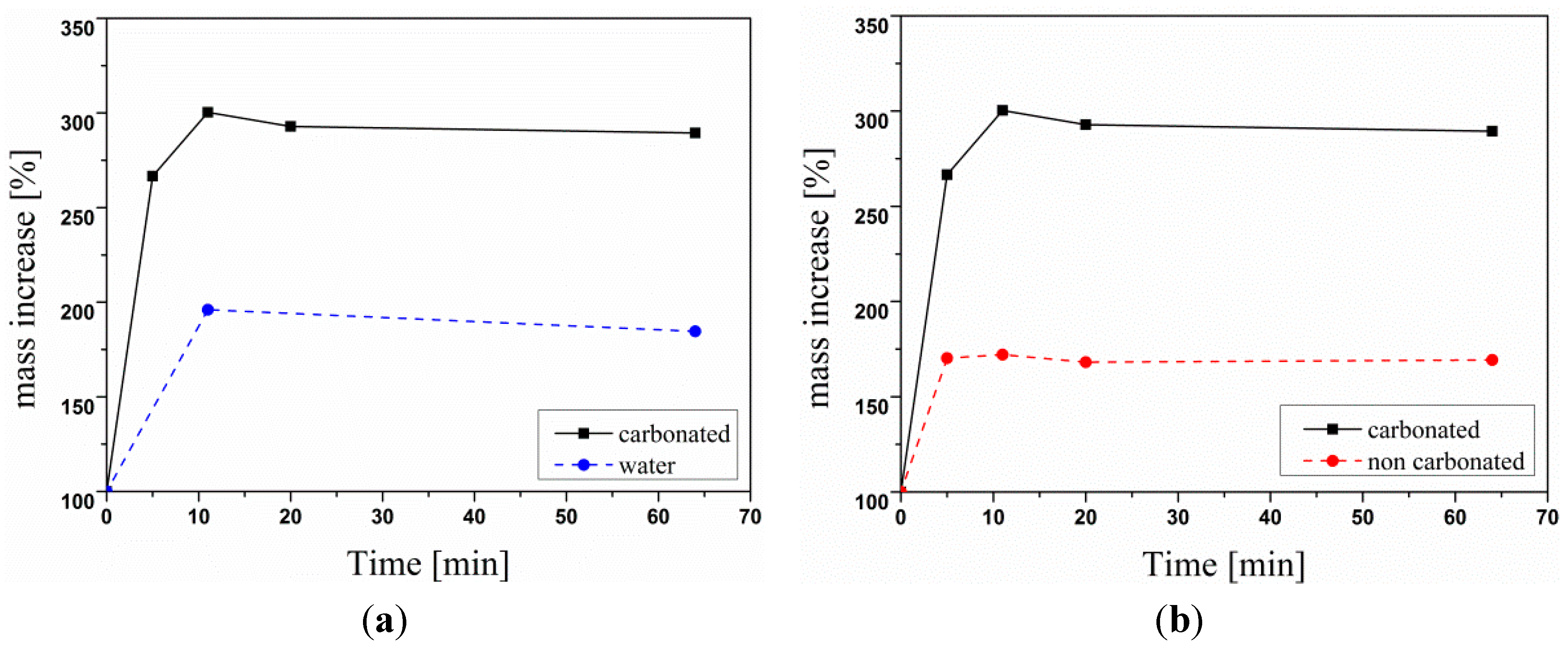
3. Experimental Section
3.1. Material
3.1.1. Cross-Linking Reaction Study
3.1.2. Production of LPUs and LSPUs
3.1.3. Carbonization Reaction
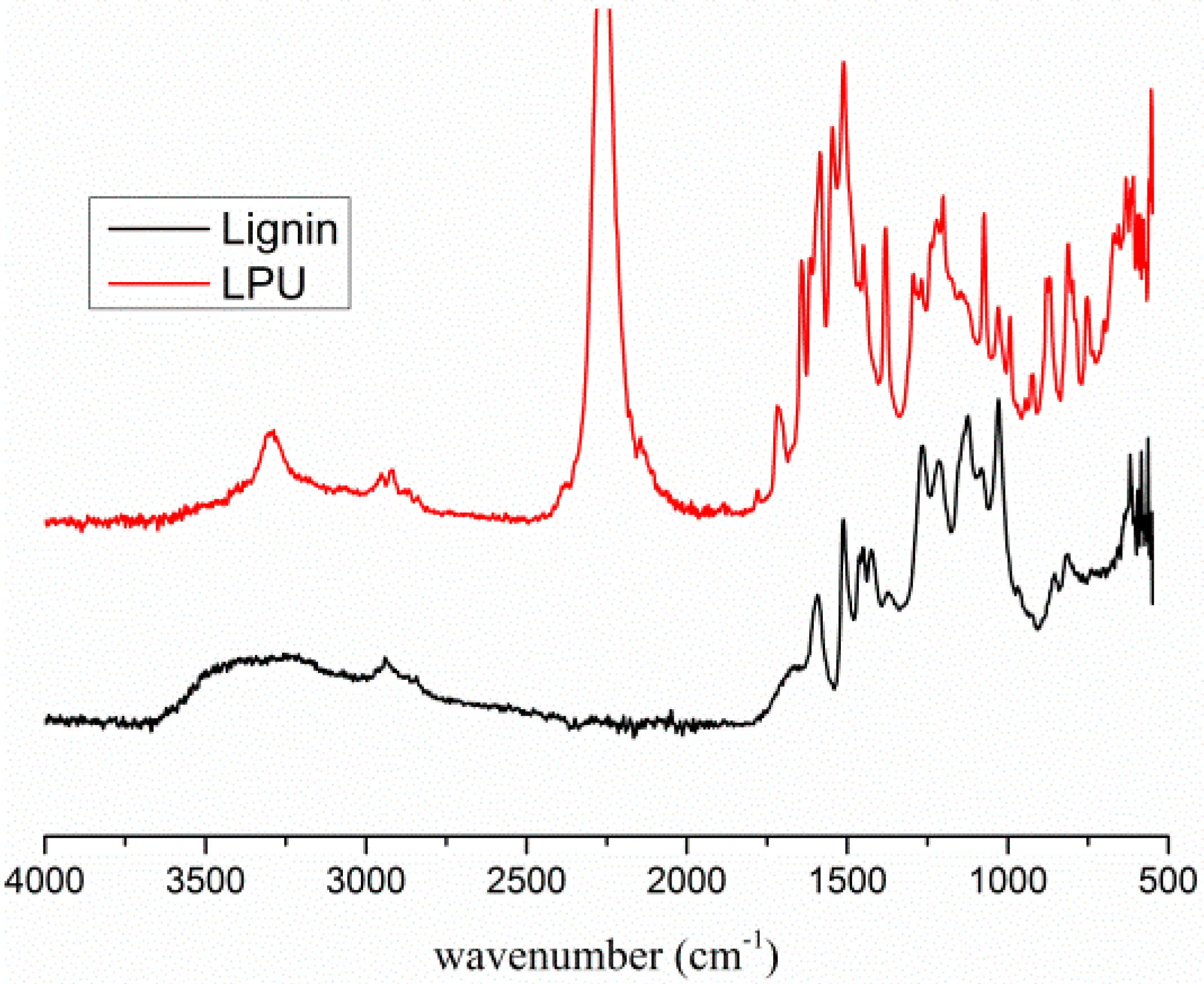
3.2. General Measurements
3.2.1. NMR Spectroscopy
3.2.2. Elemental Analysis
3.2.3. Scanning Electron Microscopy
3.2.4. BET Surface Area Determination
3.2.5. FT-IR-Spectroscopy
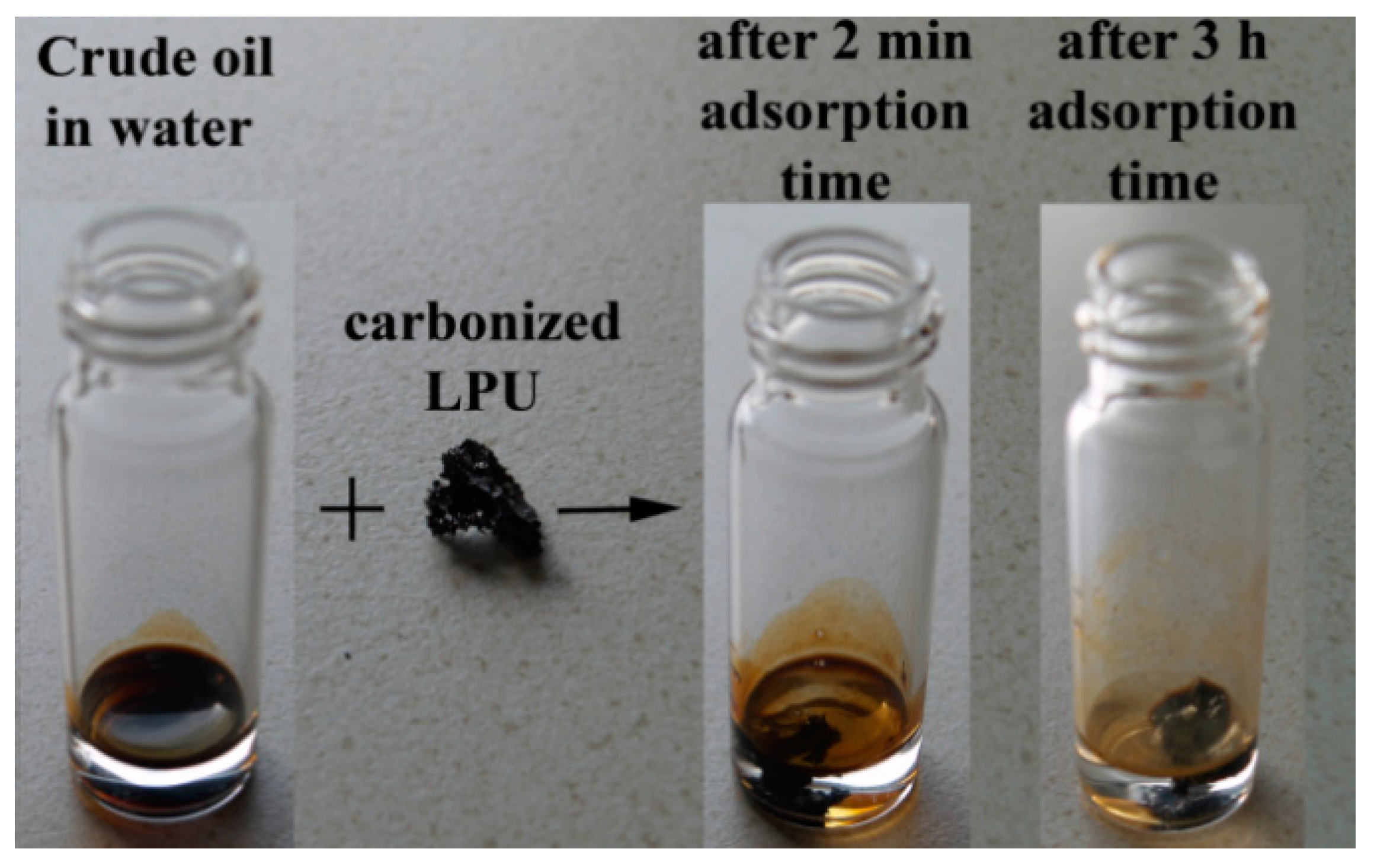
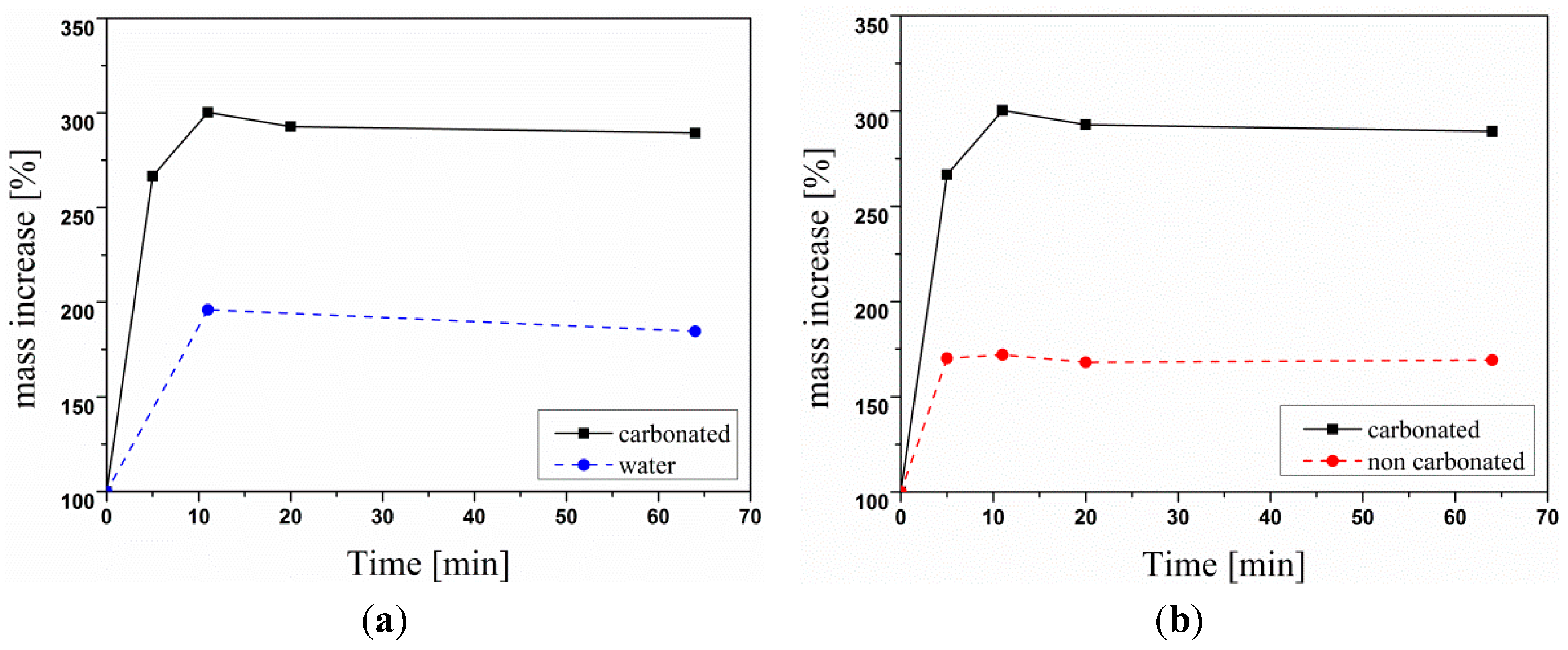
3.2.6. Crude Oil Adsorption Test
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Laurichesse, S.; Avérous, L. Chemical modification of lignins: Towards biobased polymers. Prog. Polym. Sci. 2014, 39, 1266–1290. [Google Scholar] [CrossRef]
- Ragauskas, A.J.; Beckham, G.T.; Biddy, M.J.; Chandra, R.; Chen, F.; Davis, M.F.; Davison, B.H.; Dixon, R.A.; Gilna, P.; Keller, M.; et al. Lignin Valorization: Improving Lignin Processing in the Biorefinery. Science 2014, 344. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.A.; Rials, T.G. Recent advances in low-cost carbon fiber manufacture from lignin. J. Appl. Polym. Sci. 2013, 130, 713–728. [Google Scholar] [CrossRef]
- Maradur, S.P.; Kim, C.H.; Kim, S.Y.; Kim, B.-H.; Kim, W.C.; Yang, K.S. Preparation of carbon fibers from a lignin copolymer with polyacrylonitrile. Synth. Met. 2012, 162, 453–459. [Google Scholar] [CrossRef]
- Hu, K.; Kulkarni, D.D.; Choi, I.; Tsukruk, V.V. Graphene-polymer nanocomposites for structural and functional applications. Prog. Polym. Sci. 2014, 39, 1934–1972. [Google Scholar] [CrossRef]
- Trigueiro, J.P.; Lavall, R.L.; Silva, G.G. Supercapacitors based on modified graphene electrodes with poly(ionic liquid). J. Power Sources 2014, 256, 264–273. [Google Scholar] [CrossRef]
- German, R.; Venet, P.; Sari, A.; Briat, O.; Vinassa, J.-M. Improved Supercapacitor Floating Ageing Interpretation Through Multipore Impedance Model Parameters Evolution. IEEE Trans. Power Electron. 2014, 29, 3669–3678. [Google Scholar] [CrossRef]
- Worsley, M.A.; Pauzauskie, P.J.; Olson, T.; Biener, J.; Satcher, J.H., Jr.; Baumann, T.F. Synthesis of Graphene Aerogel with High Electrical Conductivity. J. Am. Chem. Soc. 2010, 132, 14067–14069. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.-J.; Peng, C.; Lang, J.-W.; Zhu, D.-D.; Yan, X.-B. Hierarchical porous activated carbon produced from spinach leaves as an electrode material for an electric double layer capacitor. New Carbon Mater. 2014, 29, 209–215. [Google Scholar] [CrossRef]
- Torres, J.A.; Kaner, R.B. Graphene synthesis: Graphene closer to fruition. Nat. Mater. 2014, 13, 328–329. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.S.; Shoaibi, A.A.; Srinivasakannan, C. A comparison of microstructure and adsorption characteristics of activated carbons by CO2 and H3PO4 activation from date palm pits. New Carbon Mater. 2012, 27, 344–351. [Google Scholar] [CrossRef]
- Fraccarollo, A.; Canti, L.; Marchese, L.; Cossi, M. Monte Carlo Modeling of Carbon Dioxide Adsorption in Porous Aromatic Frameworks. Langmuir 2014, 30, 4147–4156. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Liu, Y.; Yu, G.; Guan, J.; Pan, C.; Du, Y.; Xiong, X.; Wang, Z. Facile Preparation of Dibenzoheterocycle-Functional Nanoporous Polymeric Networks with High Gas Uptake Capacities. Macromolecules 2014, 47, 2875–2882. [Google Scholar] [CrossRef]
- Kuzmicz, D.; Coupillaud, P.; Men, Y.; Vignolle, J.; Vendraminetto, G.; Ambrogi, M.; Taton, D.; Yuan, J. Functional mesoporous poly(ionic liquid)-based copolymer monoliths: From synthesis to catalysis and microporous carbon production. Polymer 2014, 55, 3423–3430. [Google Scholar] [CrossRef]
- Duan, L.-Q.; Ma, Q.-S.; Chen, Z.-H. The production of high surface area porous carbonaceous materials from polysiloxane. New Carbon Mater. 2013, 28, 235–240. [Google Scholar] [CrossRef]
- Mansouri, N.-E.E.; Yuan, Q.; Huang, F. Characterization of alkaline lignins for use in phenol-formaldehyde and epoxy resins. Bioresources 2011, 6, 2647–2662. [Google Scholar]
- Shen, Q.; Zhang, T.; Zhang, W.-X.; Chen, S.; Mezgebe, M. Lignin-based activated carbon fibers and controllable pore size and properties. J. Appl. Polym. Sci. 2011, 121, 989–994. [Google Scholar] [CrossRef]
- Mansouri, N.-E.E.; Pizzi, A.; Salvado, J. Lignin-based polycondensation resins for wood adhesives. J. Appl. Polym. Sci. 2007, 103, 1690–1699. [Google Scholar] [CrossRef]
- Muller, P.C.; Kelley, S.S.; Glasser, W.G. Engineering Plastics from Lignin. IX. Phenolic Resin Synthesis and Characterization. J. Adhes. 1984, 17, 185–206. [Google Scholar] [CrossRef]
- Hatakeyama, H.; Nakayachi, A.; Hatakeyama, T. Thermal and mechanical properties of polyurethane-based geocomposites derived from lignin and molasses. Compos. Part A—Appl. Sci. 2005, 36, 698–704. [Google Scholar] [CrossRef]
- Hatakeyama, T.; Matsumoto, Y.; Asano, Y.; Hatakeyama, H. Glass transition of rigid polyurethane foams derived from sodium lignosulfonate mixed with diethylene, triethylene and polyethylene glycols. Thermochim. Acta 2004, 416, 29–33. [Google Scholar] [CrossRef]
- Hatakeyama, T.; Asano, Y.; Hatakeyama, H. Mechanical and thermal properties of rigid polyurethane foams derived from sodium lignosulfonate mixed with diethylene-, triethylene- and polyethylene glycols. Macromol. Symp. 2003, 197, 171–180. [Google Scholar] [CrossRef]
- Chung, H.; Washburn, N.R. Improved Lignin Polyurethane Properties with Lewis Acid Treatment. ACS Appl. Mater. Interfaces 2012, 4, 2840–2846. [Google Scholar] [CrossRef] [PubMed]
- Ismail, T.N.; Hassan, H.A.; Hirose, S.; Taguchi, Y.; Hatakeyama, T.; Hatakeyama, H. Synthesis and thermal properties of ester-type crosslinked epoxy resins derived from lignosulfonate and glycerol. Polym. Int. 2009, 59, 181–186. [Google Scholar] [CrossRef]
- Wohlmann, B.; Woelki, M.; Ebert, A.; Engelmann, G.; Fink, H.P. Lignin Derivative, Shaped Body Comprising the Derivative, and Carbon Fibers Produced from the Shaped Body. WO 2010081775 A1, 22 July 2010. [Google Scholar]
- Yamaji, T.; Saito, T.; Hayamizu, K.; Yanagisawa, M.; Yamamoto, O. Spectral Database for Organic Compounds, SDBS. Available online: http://sdbs.db.aist.go.jp (accessed on 25 September 2014).
- Zhuang, J.M.; Steiner, P.R. Thermal reactions of diisocyanate (MDI) with phenols and benzylalcohols: DSC study and synthesis of MDI adducts. Holzforsch.—Int. J. Biol. Chem. Phys. Technol. Wood 1993, 47, 425–434. [Google Scholar] [CrossRef]
- Brudin, S.; Schoenmakers, P. Analytical methodology for sulfonated lignins. J. Sep. Sci. 2010, 33, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Y.; Zhou, Q.Q. The production of porous carbon from calcium lignosulfonate without activation process and the capacitive performance. Electrochim. Acta 2012, 71, 92–99. [Google Scholar] [CrossRef]
- Zhang, W.-J.; Li, T.-H.; Lu, M.; Hou, C.-L. A comparative study of the characteristics and carbonization behaviors of three modified coal tar pitches. New Carbon Mater. 2013, 28, 140–144. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, S.; Zheng, Y.; Luo, Z.; Cen, K. Mechanism study of wood lignin pyrolysis by using TG-FTIR analysis. J. Anal. Appl. Pyrolysis 2008, 82, 170–177. [Google Scholar] [CrossRef]
- Grønli, M.G.; Várhegyi, G.; di Blasi, C. Thermogravimetric Analysis and Devolatilization Kinetics of Wood. Ind. Eng. Chem. Res. 2002, 41, 4201–4208. [Google Scholar] [CrossRef]
- Brebu, M.; Tamminen, T.; Spiridon, I. Thermal degradation of various lignins by TG-MS/FTIR and Py-GC-MS. J. Anal. Appl. Pyrolysis 2013, 104, 531–539. [Google Scholar] [CrossRef]
- Li, B.; Lv, W.; Zhang, Q.; Wang, T.; Ma, L. Pyrolysis and catalytic pyrolysis of industrial lignins by TG-FTIR: Kinetics and products. J. Anal. Appl. Pyrolysis 2014, 108, 295–300. [Google Scholar] [CrossRef]
- Wu, H.; Fan, S.-W.; Yuan, X.-W.; Chen, L.-F.; Deng, J.-L. Fabrication of carbon fibers from jute fibers by pre-oxidation and carbonization. New Carbon Mater. 2013, 28, 448–453. [Google Scholar] [CrossRef]
- Okiel, K.; El-Sayed, M.; El-Kady, M.Y. Treatment of oil-water emulsions by adsorption onto activated carbon, bentonite and deposited carbon. Egypt. J. Pet. 2011, 20, 9–15. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, Y.; Zhang, Y.; Sun, X.; Ye, H.; Li, W. Synthesis and characterization of polyurethane elastomers. J. Elastom. Plast. 2008, 40, 161–177. [Google Scholar] [CrossRef]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leitner, S.P.; Gratzl, G.; Paulik, C.; Weber, H.K. Carbon Materials from Lignin and Sodium Lignosulfonate via Diisocyanate Cross-Linking and Subsequent Carbonization. C 2015, 1, 43-57. https://doi.org/10.3390/c1010043
Leitner SP, Gratzl G, Paulik C, Weber HK. Carbon Materials from Lignin and Sodium Lignosulfonate via Diisocyanate Cross-Linking and Subsequent Carbonization. C. 2015; 1(1):43-57. https://doi.org/10.3390/c1010043
Chicago/Turabian StyleLeitner, Sebastian P., Günther Gratzl, Christian Paulik, and Hedda K. Weber. 2015. "Carbon Materials from Lignin and Sodium Lignosulfonate via Diisocyanate Cross-Linking and Subsequent Carbonization" C 1, no. 1: 43-57. https://doi.org/10.3390/c1010043
APA StyleLeitner, S. P., Gratzl, G., Paulik, C., & Weber, H. K. (2015). Carbon Materials from Lignin and Sodium Lignosulfonate via Diisocyanate Cross-Linking and Subsequent Carbonization. C, 1(1), 43-57. https://doi.org/10.3390/c1010043