
Advances and Obstacles in Using CRISPR/Cas9 Technology for Non-Coding RNA Gene Knockout in Human Mesenchymal Stromal Cells

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
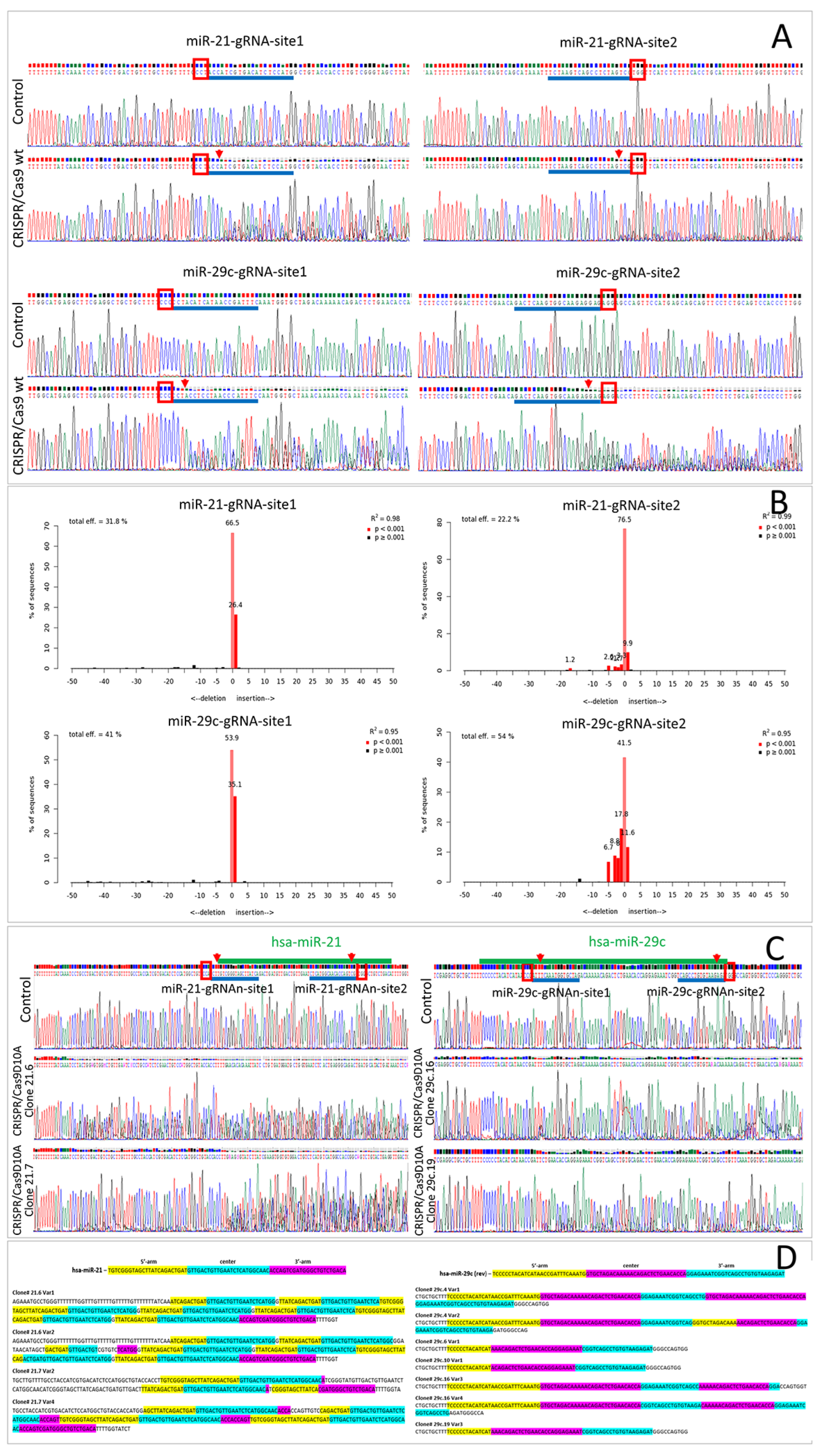
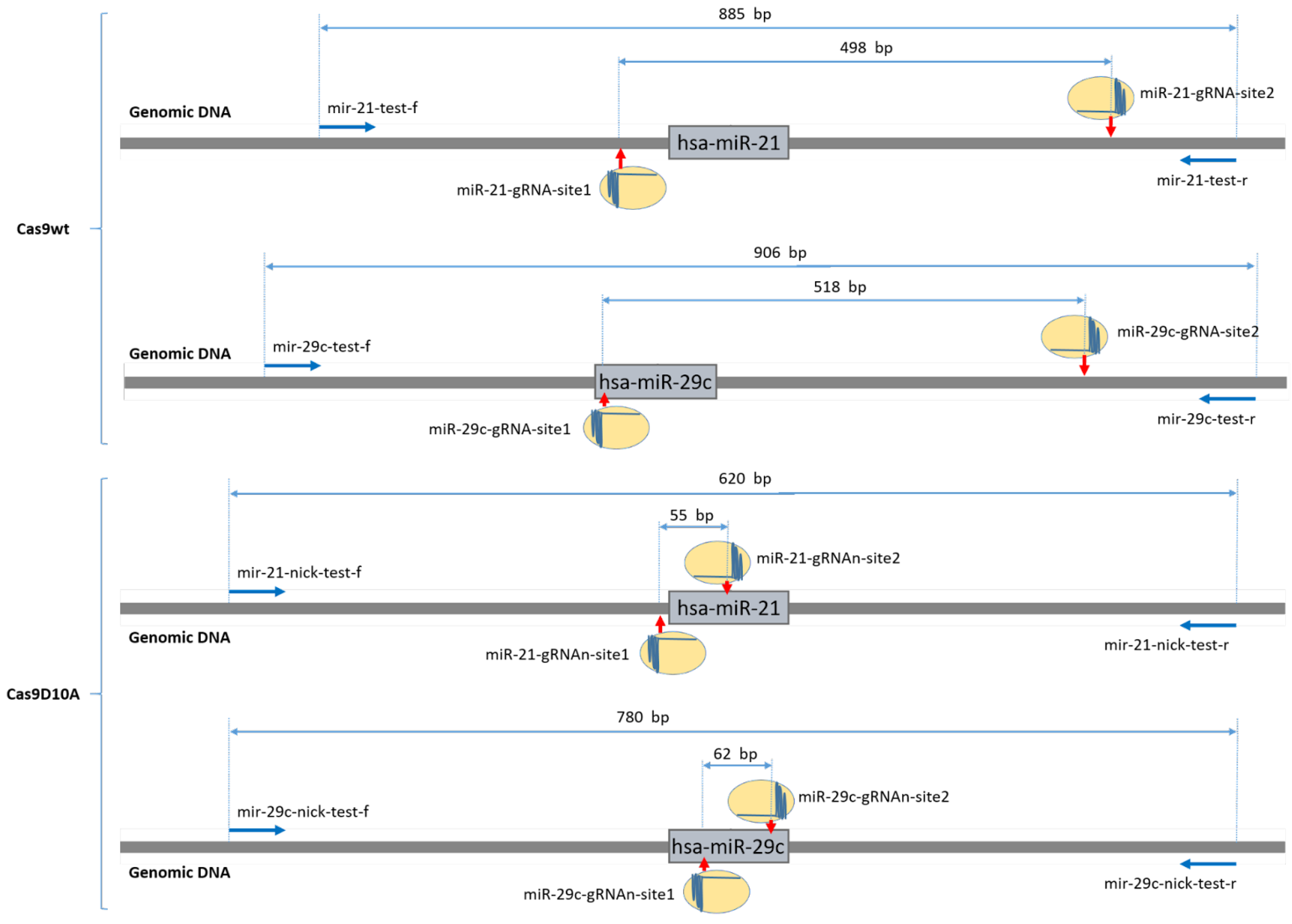
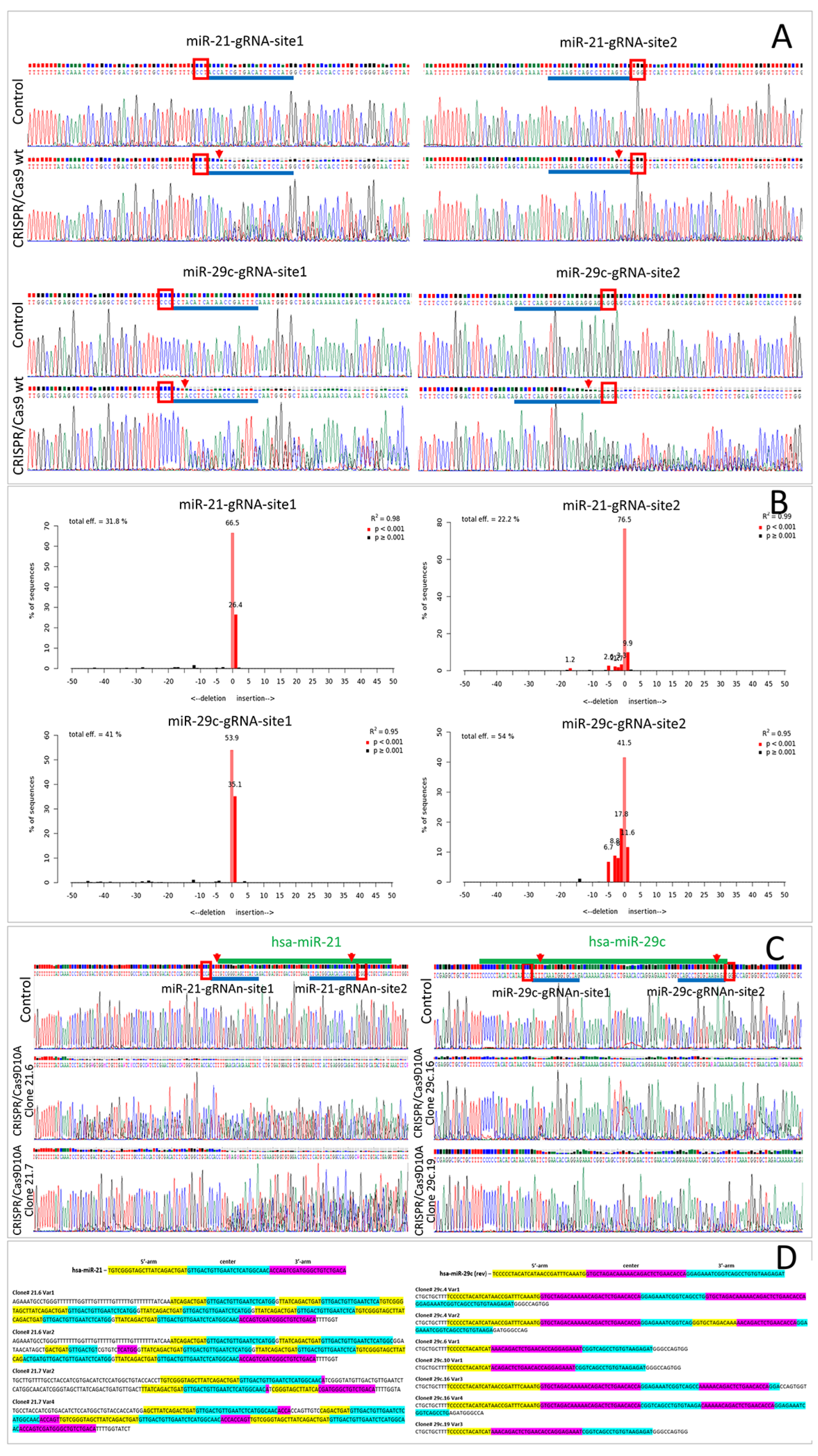
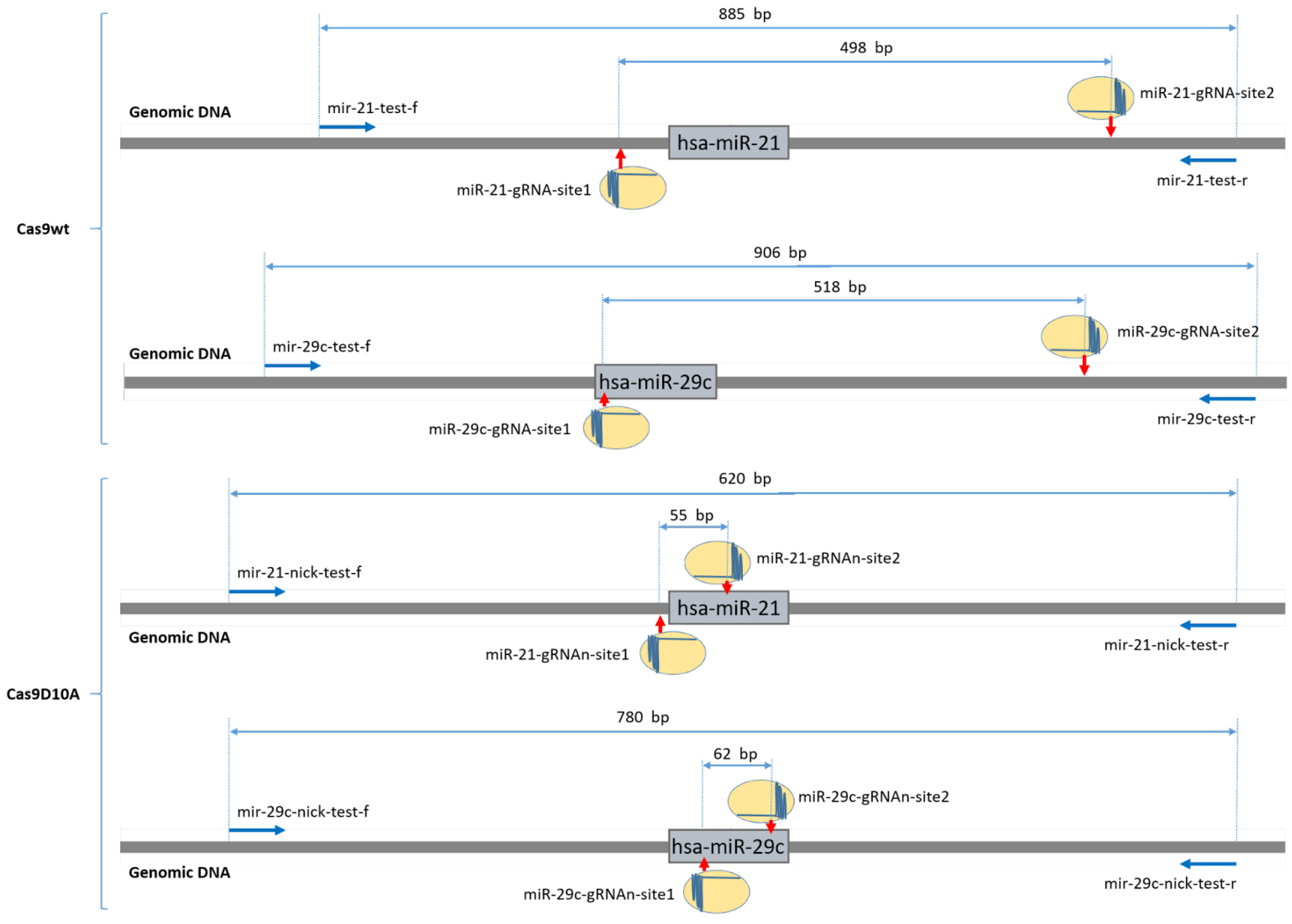
2.1. Wild-Type Cas9 Nuclease Is Found to Be Inefficient for the Deletion of Extended DNA Fragments in ASC52telo Cell Line
2.2. Cas9D10A Nickase Does Not Delete Extended DNA Fragments, but Disrupts miRNA Genes in ASC52telo Cell Line
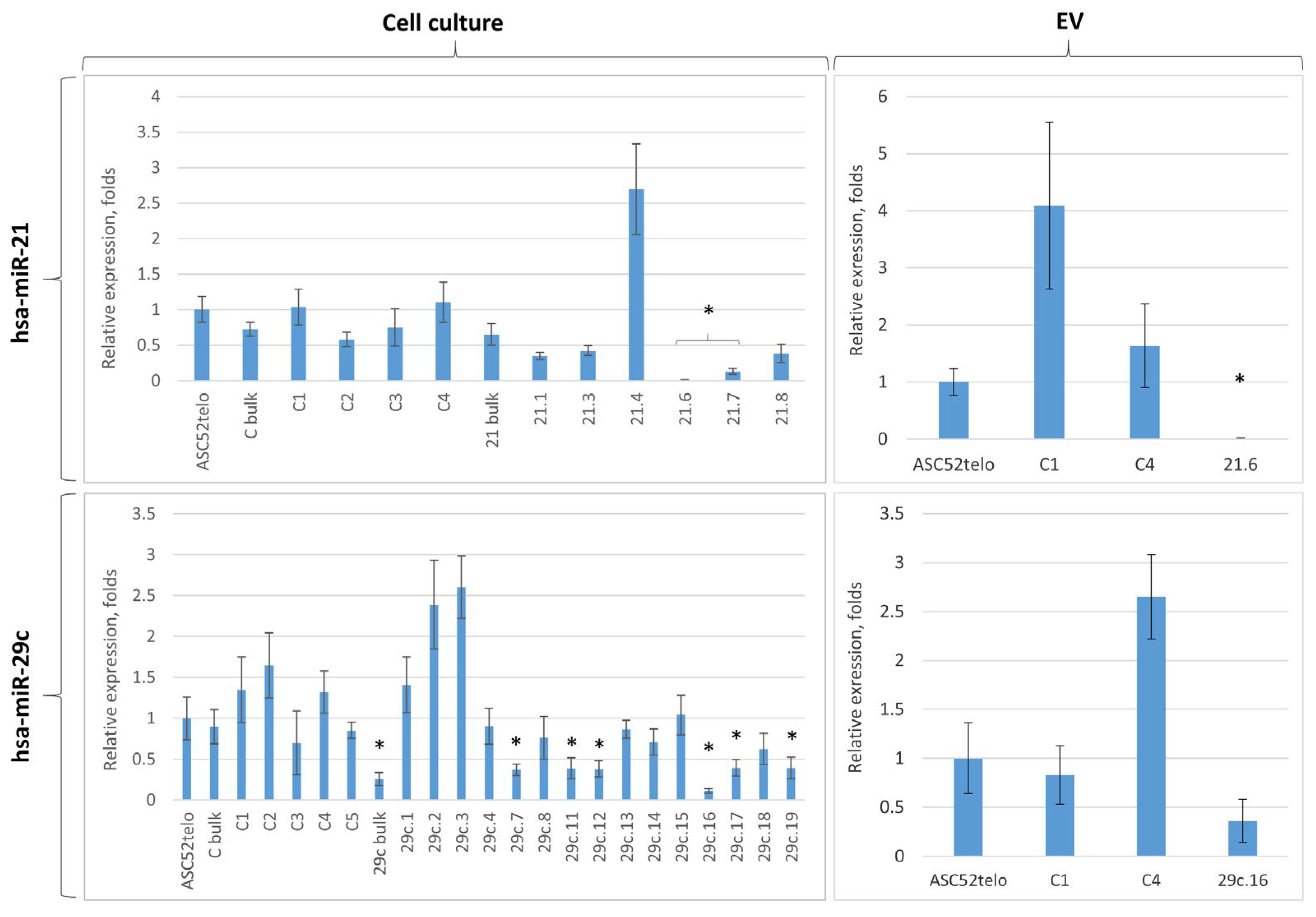
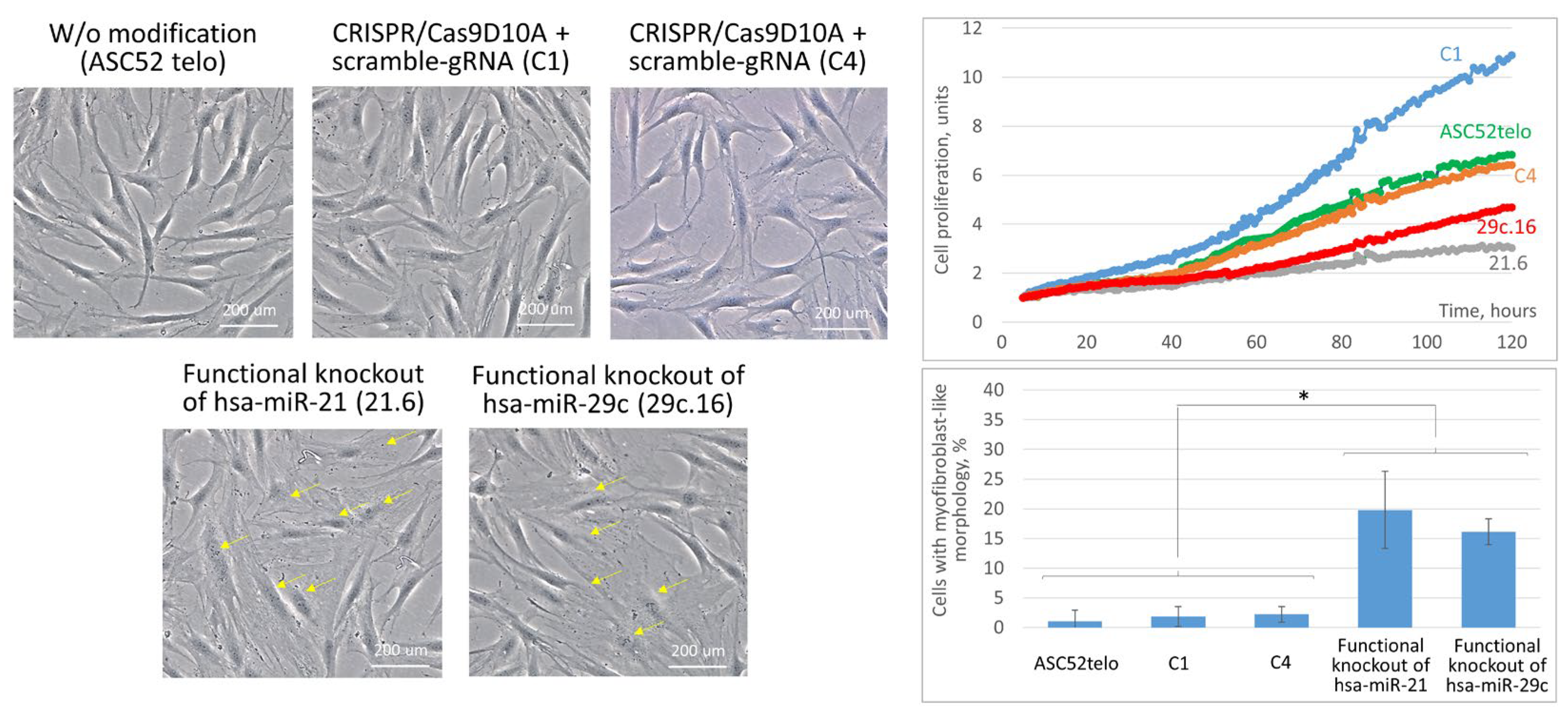
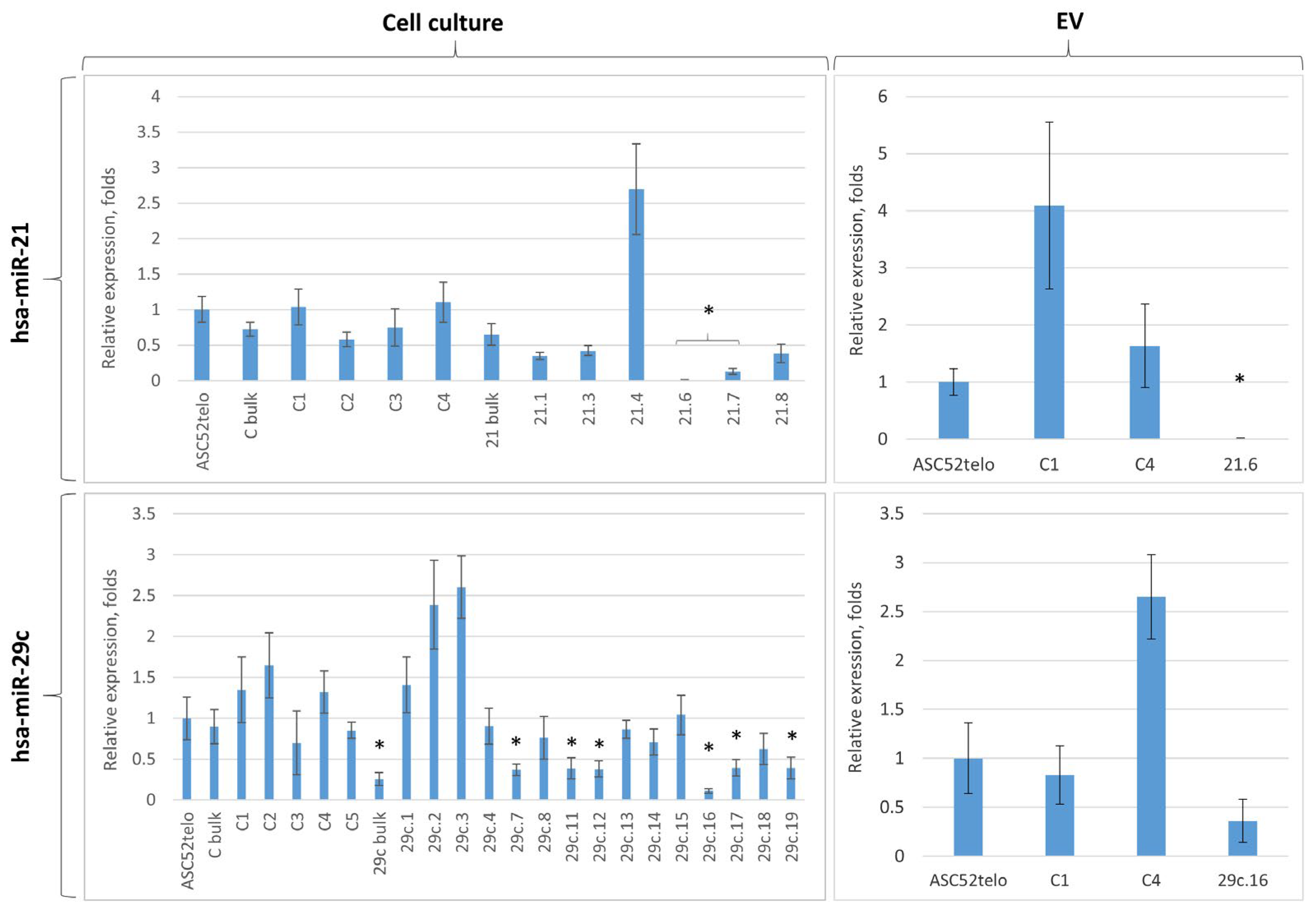
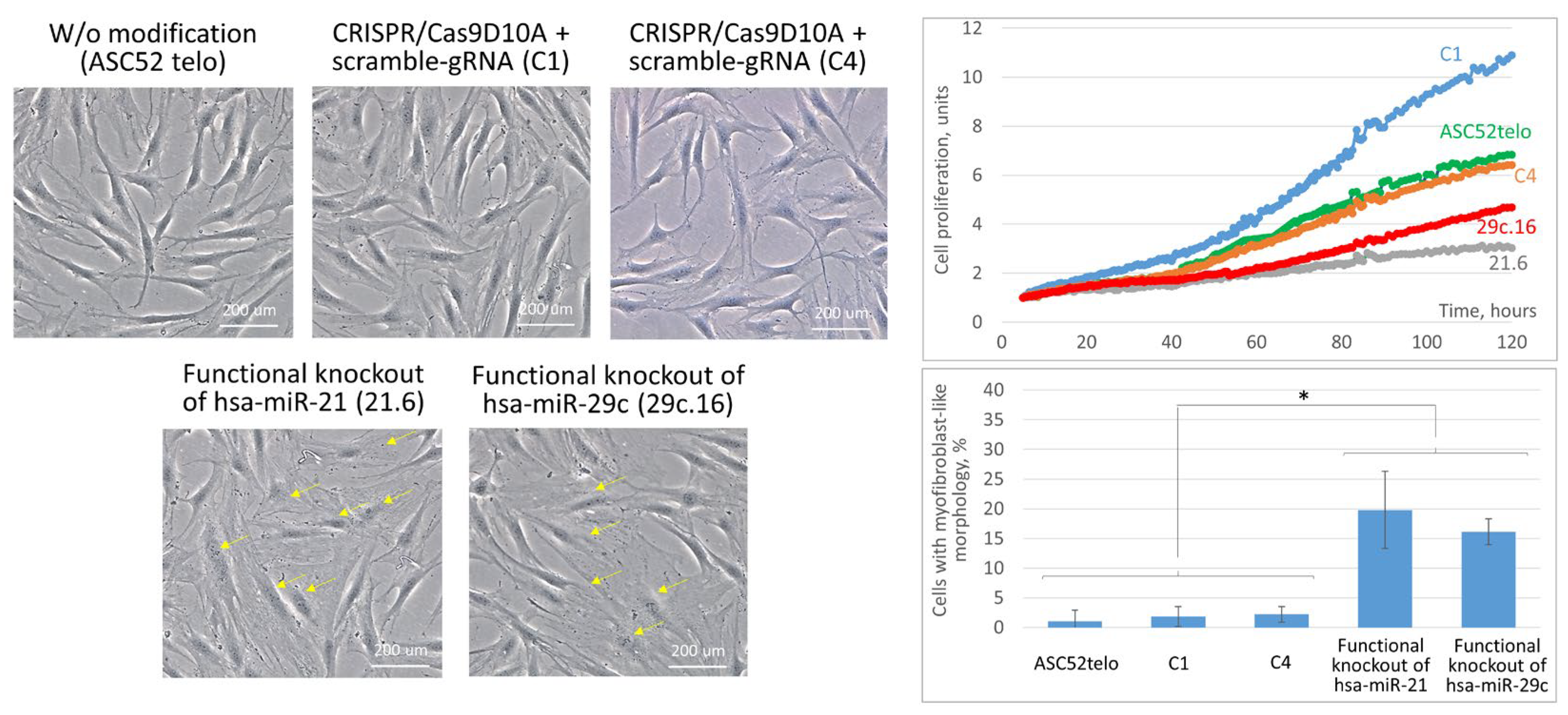
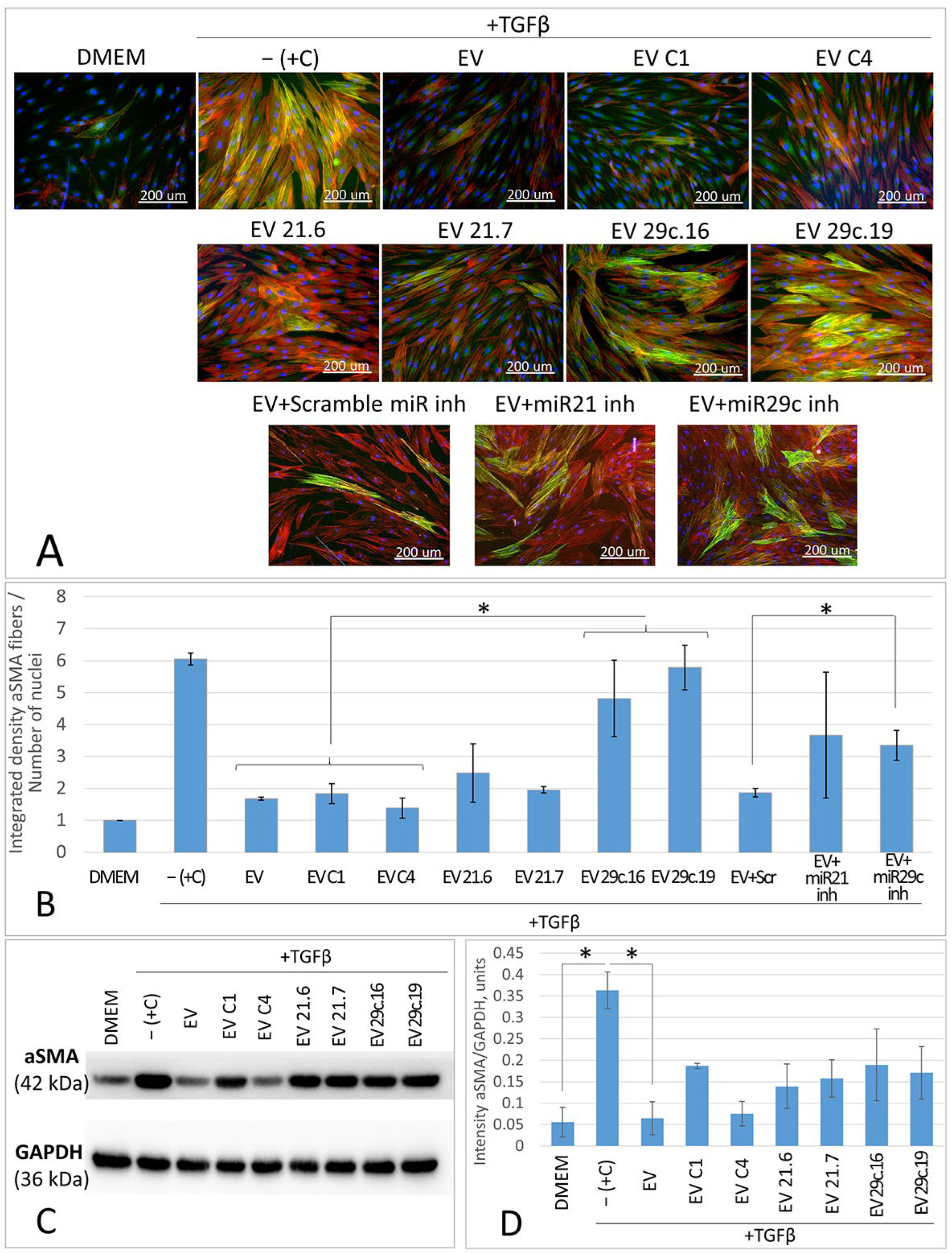
2.3. Cas9D10A Nickase System Allows Us to Establish Biological Functions of Certain miRNAs in ASC52telo Cells
3. Discussion
4. Materials and Methods
4.1. Design and Assembly of Genetic Constructs
4.2. Cell Cultures
4.3. Lentiviral Particle Assembly and Genetic Modification
4.4. Analysis of the Genome Editing Efficiency
4.5. Medium Conditioning
4.6. PCR Analysis of miRNAs in Cell Culture and Extracellular Vesicles
4.7. Cellular Model of Fibrosis
4.8. Immunocytochemical Analysis
4.9. Western Blotting
4.10. Evaluation of the Proliferative Activity of ASC52telo Clones
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef]
- Cai, Y.; Yu, X.; Hu, S.; Yu, J. A Brief Review on the Mechanisms of MiRNA Regulation. Genom. Proteom. Bioinform. 2009, 7, 147–154. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The Role of MicroRNAs in Human Cancer. Sig. Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Zhao, C.; Sun, X.; Li, L. Biogenesis and Function of Extracellular MiRNAs. ExRNA 2019, 1, 38. [Google Scholar] [CrossRef]
- Karagyaur, M.; Primak, A.; Efimenko, A.; Skryabina, M.; Tkachuk, V. The Power of Gene Technologies: 1001 Ways to Create a Cell Model. Cells 2022, 11, 3235. [Google Scholar] [CrossRef] [PubMed]
- Pushparaj, P.N.; Aarthi, J.J.; Manikandan, J.; Kumar, S.D. siRNA, miRNA, and shRNA: In vivo applications. J. Dent. Res. 2008, 87, 992–1003. [Google Scholar] [CrossRef]
- Mani, I. Genome editing in cardiovascular diseases. Prog. Mol. Biol. Transl. Sci. 2021, 181, 289–308. [Google Scholar] [CrossRef] [PubMed]
- Velu, C.S.; Grimes, H.L. Utilizing AntagomiR (Antisense MicroRNA) to Knock Down MicroRNA in Murine Bone Marrow Cells. In Rational Drug Design; Zheng, Y., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 185–195. ISBN 978-1-62703-007-6. [Google Scholar]
- Abreu, R.C.; Ramos, C.V.; Becher, C.; Lino, M.; Jesus, C.; Costa Martins, P.A.; Martins, P.A.T.; Moreno, M.J.; Fernandes, H.; Ferreira, L. Exogenous Loading of MiRNAs into Small Extracellular Vesicles. J. Extracell. Vesicles 2021, 10, e12111. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef]
- Kurata, J.S.; Lin, R.J. MicroRNA-focused CRISPR-Cas9 library screen reveals fitness-associated miRNAs. RNA 2018, 24, 966–981. [Google Scholar] [CrossRef]
- Basalova, N.; Sagaradze, G.; Arbatskiy, M.; Evtushenko, E.; Kulebyakin, K.; Grigorieva, O.; Akopyan, Z.; Kalinina, N.; Efimenko, A. Secretome of Mesenchymal Stromal Cells Prevents Myofibroblasts Differentiation by Transferring Fibrosis-Associated microRNAs within Extracellular Vesicles. Cells 2020, 9, 1272. [Google Scholar] [CrossRef]
- Basalova, N.; Arbatskiy, M.; Popov, V.; Grigorieva, O.; Vigovskiy, M.; Zaytsev, I.; Novoseletskaya, E.; Sagaradze, G.; Danilova, N.; Malkov, P.; et al. Mesenchymal stromal cells facilitate resolution of pulmonary fibrosis by miR-29c and miR-129 intercellular transfer. Exp. Mol. Med. 2023, 55, 1399–1412. [Google Scholar] [CrossRef]
- Trevino, A.E.; Zhang, F. Genome Editing Using Cas9 Nickases. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 546, pp. 161–174. ISBN 978-0-12-801185-0. [Google Scholar]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013, 3, 2510. [Google Scholar] [CrossRef]
- Munoz, I.M.; Szyniarowski, P.; Toth, R.; Rouse, J.; Lachaud, C. Improved genome editing in human cell lines using the CRISPR method. PLoS ONE 2014, 9, e109752. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Feng, Y.L.; Xiao, J.J.; Liu, Q.; Sun, X.N.; Xiang, J.F.; Kong, N.; Liu, S.C.; Chen, G.Q.; Wang, Y.; et al. Harnessing accurate non-homologous end joining for efficient precise deletion in CRISPR/Cas9-mediated genome editing. Genome Biol. 2018, 19, 170. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, G.K.; Khullar, N.; Sidhu, I.S.; Navik, U.S.; Reddy, A.P.; Reddy, P.H.; Bhatti, J.S. Emerging Role of Non-coding RNA in Health and Disease. Metab. Brain Dis. 2021, 36, 1119–1134. [Google Scholar] [CrossRef] [PubMed]
- Ramón y Cajal, S.; Segura, M.F.; Hümmer, S. Interplay Between NcRNAs and Cellular Communication: A Proposal for Understanding Cell-Specific Signaling Pathways. Front. Genet. 2019, 10, 281. [Google Scholar] [CrossRef]
- Shi, J.; Zhou, T.; Chen, Q. Exploring the Expanding Universe of Small RNAs. Nat. Cell Biol. 2022, 24, 415–423. [Google Scholar] [CrossRef]
- Shukla, G.C.; Singh, J.; Barik, S. MicroRNAs: Processing, Maturation, Target Recognition and Regulatory Functions. Mol. Cell Pharmacol. 2011, 3, 83–92. [Google Scholar]
- Chang, H.; Yi, B.; Ma, R.; Zhang, X.; Zhao, H.; Xi, Y. CRISPR/Cas9, a Novel Genomic Tool to Knock down MicroRNA in Vitro and in Vivo. Sci. Rep. 2016, 6, 22312. [Google Scholar] [CrossRef]
- Bae, S.; Kweon, J.; Kim, H.S.; Kim, J.-S. Microhomology-Based Choice of Cas9 Nuclease Target Sites. Nat. Methods 2014, 11, 705–706. [Google Scholar] [CrossRef]
- Ellwanger, D.C.; Büttner, F.A.; Mewes, H.W.; Stümpflen, V. The sufficient minimal set of miRNA seed types. Bioinformatics 2011, 27, 1346–1350. [Google Scholar] [CrossRef]
- Joy, N.; Maimoonath Beevi, Y.P.; Soniya, E.V. A deeper view into the significance of simple sequence repeats in pre-miRNAs provides clues for its possible roles in determining the function of microRNAs. BMC Genet. 2018, 19, 29c. [Google Scholar] [CrossRef]
- Witkos, T.M.; Krzyzosiak, W.J.; Fiszer, A.; Koscianska, E. A potential role of extended simple sequence repeats in competing endogenous RNA crosstalk. RNA Biol. 2018, 15, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Tan, Z.; Zeng, G.; Peng, J. Comprehensive analysis of simple sequence repeats in pre-miRNAs. Mol. Biol. Evol. 2010, 27, 2227–2232. [Google Scholar] [CrossRef]
- Peterson, S.M.; Thompson, J.A.; Ufkin, M.L.; Sathyanarayana, P.; Liaw, L.; Congdon, C.B. Common Features of MicroRNA Target Prediction Tools. Front. Genet. 2014, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Kehl, T.; Backes, C.; Kern, F.; Fehlmann, T.; Ludwig, N.; Meese, E.; Lenhof, H.-P.; Keller, A. About MiRNAs, MiRNA Seeds, Target Genes and Target Pathways. Oncotarget 2017, 8, 107167–107175. [Google Scholar] [CrossRef] [PubMed]
- Kosicki, M.; Allen, F.; Steward, F.; Tomberg, K.; Pan, Y.; Bradley, A. Cas9-Induced Large Deletions and Small Indels Are Controlled in a Convergent Fashion. Nat. Commun. 2022, 13, 3422. [Google Scholar] [CrossRef]
- Bauer, D.E.; Canver, M.C.; Orkin, S.H. Generation of Genomic Deletions in Mammalian Cell Lines via CRISPR/Cas9. JoVE 2014, 52118. [Google Scholar] [CrossRef]
- Zheng, C.; Baum, B.J. Evaluation of Promoters for Use in Tissue-Specific Gene Delivery. In Gene Therapy Protocols; Doux, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 205–219. ISBN 978-1-60327-247-6. [Google Scholar]
- Nicklin, S.A.; Reynolds, P.N.; Brosnan, M.J.; White, S.J.; Curiel, D.T.; Dominiczak, A.F.; Baker, A.H. Analysis of Cell-Specific Promoters for Viral Gene Therapy Targeted at the Vascular Endothelium. Hypertension 2001, 38, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Seki, A.; Rutz, S. Optimized RNP Transfection for Highly Efficient CRISPR/Cas9-Mediated Gene Knockout in Primary T Cells. J. Exp. Med. 2018, 215, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Shen, J.; Li, D.; Cheng, Y. Strategies in the Delivery of Cas9 Ribonucleoprotein for CRISPR/Cas9 Genome Editing. Theranostics 2021, 11, 614–648. [Google Scholar] [CrossRef]
- Ferreira da Silva, J.; Meyenberg, M.; Loizou, J.I. Tissue specificity of DNA repair: The CRISPR compass. Trends Genet. 2021, 37, 958–962. [Google Scholar] [CrossRef]
- Simonatto, M.; Latella, L.; Puri, P.L. DNA damage and cellular differentiation: More questions than responses. J. Cell Physiol. 2007, 213, 642–648. [Google Scholar] [CrossRef]
- Ait Saada, A.; Costa, A.B.; Sheng, Z.; Guo, W.; Haber, J.E.; Lobachev, K.S. Structural Parameters of Palindromic Repeats Determine the Specificity of Nuclease Attack of Secondary Structures. Nucleic Acids Res. 2021, 49, 3932–3947. [Google Scholar] [CrossRef] [PubMed]
- Svetec Miklenić, M.; Svetec, I.K. Palindromes in DNA—A Risk for Genome Stability and Implications in Cancer. Int. J. Mol. Sci. 2021, 22, 2840. [Google Scholar] [CrossRef]
- Villarreal, D.D.; Lee, K.; Deem, A.; Shim, E.Y.; Malkova, A.; Lee, S.E. Microhomology Directs Diverse DNA Break Repair Pathways and Chromosomal Translocations. PLoS Genet. 2012, 8, e1003026. [Google Scholar] [CrossRef]
- McVey, M.; Lee, S.E. MMEJ Repair of Double-Strand Breaks (Director’s Cut): Deleted Sequences and Alternative Endings. Trends Genet. 2008, 24, 529–538. [Google Scholar] [CrossRef]
- Dianov, G.L.; Hübscher, U. Mammalian base excision repair: The forgotten archangel. Nucleic Acids Res. 2013, 41, 3483–3490. [Google Scholar] [CrossRef]
- Xia, Y.; Wang, Y.; Wang, Q.; Ghaffar, M.; Wang, Y.; Sheng, W.; Zhang, F. Increased miR-203-3p and reduced miR-21-5p synergistically inhibit proliferation, migration, and invasion in esophageal cancer cells. Anticancer. Drugs 2019, 30, 38–45. [Google Scholar] [CrossRef]
- Báez-Vega, P.M.; Echevarría Vargas, I.M.; Valiyeva, F.; Encarnación-Rosado, J.; Roman, A.; Flores, J.; Marcos-Martínez, M.J.; Vivas-Mejía, P.E. Targeting miR-21-3p inhibits proliferation and invasion of ovarian cancer cells. Oncotarget 2016, 7, 36321–36337. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, C.; Ji, L.; Wang, N.; Liu, Y.; Wang, M.; Ruan, L. The circACC1/miR-29c-3p/FOXP1 network plays a key role in gastric cancer by regulating cell proliferation. Biochem. Biophys. Res. Commun. 2021, 557, 221–227. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, H.; Sun, L.; Zhu, K.; Lang, W. miR-29c-3p Increases Cell Viability and Suppresses Apoptosis by Regulating the TNFAIP1/NF-κB Signaling Pathway via TNFAIP1 in Aβ-Treated Neuroblastoma Cells. Neurochem. Res. 2020, 45, 2375–2384. [Google Scholar] [CrossRef]
- Sun, L.-H.; Tian, D.; Yang, Z.-C.; Li, J.-L. Exosomal MiR-21 Promotes Proliferation, Invasion and Therapy Resistance of Colon Adenocarcinoma Cells through Its Target PDCD4. Sci. Rep. 2020, 10, 8271. [Google Scholar] [CrossRef]
- Huang, H.; Huang, X.; Luo, S.; Zhang, H.; Hu, F.; Chen, R.; Huang, C.; Su, Z. The MicroRNA MiR-29c Alleviates Renal Fibrosis via TPM1-Mediated Suppression of the Wnt/β-Catenin Pathway. Front. Physiol. 2020, 11, 331. [Google Scholar] [CrossRef] [PubMed]
- Denli, A.M.; Tops, B.B.; Plasterk, R.H.; Ketting, R.F.; Hannon, G.J. Processing of primary microRNAs by the Microprocessor complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485. [Google Scholar] [CrossRef]
- Han, J.; Lee, Y.; Yeom, K.H.; Kim, Y.K.; Jin, H.; Kim, V.N. The Drosha-DGCR8 complex in primary microRNA processing. Genes. Dev. 2004, 18, 3016–3027. [Google Scholar] [CrossRef]
- Okada, C.; Yamashita, E.; Lee, S.J.; Shibata, S.; Katahira, J.; Nakagawa, A.; Yoneda, Y.; Tsukihara, T. A high-resolution structure of the pre-microRNA nuclear export machinery. Science 2009, 326, 1275–1279. [Google Scholar] [CrossRef]
- Jones, A.N.; Walbrun, A.; Falleroni, F.; Rief, M.; Sattler, M. Conformational Effects of a Cancer-Linked Mutation in Pri-miR-30c RNA. J. Mol. Biol. 2022, 434, 167705. [Google Scholar] [CrossRef]
- Iivonen, A.P.; Känsäkoski, J.; Vaaralahti, K.; Raivio, T. Screening for mutations in selected miRNA genes in hypogonadotropic hypogonadism patients. Endocr. Connect. 2019, 8, 506–509. [Google Scholar] [CrossRef]
- Amiel, J.; de Pontual, L.; Henrion-Caude, A. miRNA, development and disease. Adv. Genet. 2012, 80, 1–36. [Google Scholar] [CrossRef]
- Karagyaur, M.N.; Rubtsov, Y.P.; Vasiliev, P.A.; Tkachuk, V.A. Practical Recommendations for Improving Efficiency and Accuracy of the CRISPR/Cas9 Genome Editing System. Biochemistry 2018, 83, 629–642. [Google Scholar] [CrossRef]
- Cradick, T.J.; Qiu, P.; Lee, C.M.; Fine, E.J.; Bao, G. COSMID: A Web-Based Tool for Identifying and Validating CRISPR/Cas Off-Target Sites. Mol. Ther. Nucleic Acids 2014, 3, e214. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Longo, P.A.; Kavran, J.M.; Kim, M.-S.; Leahy, D.J. Transient Mammalian Cell Transfection with Polyethylenimine (PEI). In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 529, pp. 227–240. ISBN 978-0-12-418687-3. [Google Scholar]
- Tyurin-Kuzmin, P.A.; Karagyaur, M.N.; Kulebyakin, K.Y.; Dyikanov, D.T.; Chechekhin, V.I.; Ivanova, A.M.; Skryabina, M.N.; Arbatskiy, M.S.; Sysoeva, V.Y.; Kalinina, N.I.; et al. Functional Heterogeneity of Protein Kinase A Activation in Multipotent Stromal Cells. Int. J. Mol. Sci. 2020, 21, 4442. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy Quantitative Assessment of Genome Editing by Sequence Trace Decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Bell, J.; Hendrix, D.A. Predicting Drosha and Dicer Cleavage Sites with DeepMirCut. Front. Mol. Biosci. 2022, 8, 799056. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5′->3′) | Tm, °C | Cut-Out DNA Fragment Amplicon Length, bp |
|---|---|---|---|
| miR-21-gRNA-site1-f | CACCGATGGAGATGTCACGATGGT | - | 498 |
| miR-21-gRNA-site1-r | AAACACCATCGTGACATCTCCATC | ||
| miR-21-gRNA-site2-f | CACCGCTAAGTCAGCCTCTAGTCG | ||
| miR-21-gRNA-site2-r | AAACCGACTAGAGGCTGACTTAGC | ||
| miR-29c-gRNA-site1-f | CACCGAAATCGGTTATGATGTAGG | - | 518 |
| miR-29c-gRNA-site1-r | AAACCCTACATCATAACCGATTTC | ||
| miR-29c-gRNA-site2-f | CACCGACTCAAGTGGCAAGAGGAG | ||
| miR-29c-gRNA-site2-r | AAACCTCCTCTTGCCACTTGAGTC | ||
| mir-21-test-f | ACTTGTTCATTTTGTTTTGCTTGG | 59.5 | 885 |
| mir-21-test-r | ACGTATCAATTAGACCTTCAACCTA | ||
| mir-29c-test-f | GAACAGCACTACATTTCAGCAAA | 58.0 | 906 |
| mir-29c-test-r | TGGAAGCTGGTTTCACATGGT |
| Name | Sequence (5′->3′) | Tm, °C | Cut-Out DNA Fragment Amplicon Length, bp |
|---|---|---|---|
| miR-21-gRNAn-site1-f | CACCGTCATGGCAACACCAGTCGA | - | 498 |
| miR-21-gRNAn-site1-r | AAACTCGACTGGTGTTGCCATGAC | ||
| miR-21-gRNAn-site2-f | CACCGGATAAGCTACCCGACAAGG | ||
| miR-21-gRNAn-site2-r | AAACCCTTGTCGGGTAGCTTATCC | ||
| miR-29c-gRNAn-site1-f | CACCGGTCTAGCACCATTTGAAAT | - | 518 |
| miR-29c-gRNAn-site1-r | AAACATTTCAAATGGTGCTAGACC | ||
| miR-29c-gRNAn-site2-f | CACCGGGTCAGCCTGTGTAAGAGA | ||
| miR-29c-gRNAn-site2-r | AAACTCTCTTACACAGGCTGACCC | ||
| mir-21-nick-test-f | GGAGAGAATTCTAACCCAGTTTTCTTGCCGT | 59.5 | 885 |
| mir-21-nick-test-r | GGAGAGGTACCTCAAAACCCACAATGCAGCTTAG | ||
| mir-29c-nick-test-f | GGAGAGAATTCCAGGACCCACTTCTTATCATCAC | 58.0 | 906 |
| mir-29c-nick-test-r | GGAGAGGTACCTTGACTCCTAGCAGCCATCAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basalova, N.; Illarionova, M.; Skryabina, M.; Vigovskiy, M.; Tolstoluzhinskaya, A.; Primak, A.; Chechekhina, E.; Chechekhin, V.; Karagyaur, M.; Efimenko, A. Advances and Obstacles in Using CRISPR/Cas9 Technology for Non-Coding RNA Gene Knockout in Human Mesenchymal Stromal Cells. Non-Coding RNA 2023, 9, 49. https://doi.org/10.3390/ncrna9050049
Basalova N, Illarionova M, Skryabina M, Vigovskiy M, Tolstoluzhinskaya A, Primak A, Chechekhina E, Chechekhin V, Karagyaur M, Efimenko A. Advances and Obstacles in Using CRISPR/Cas9 Technology for Non-Coding RNA Gene Knockout in Human Mesenchymal Stromal Cells. Non-Coding RNA. 2023; 9(5):49. https://doi.org/10.3390/ncrna9050049
Chicago/Turabian StyleBasalova, Nataliya, Maria Illarionova, Mariya Skryabina, Maksim Vigovskiy, Anastasia Tolstoluzhinskaya, Alexandra Primak, Elizaveta Chechekhina, Vadim Chechekhin, Maxim Karagyaur, and Anastasia Efimenko. 2023. "Advances and Obstacles in Using CRISPR/Cas9 Technology for Non-Coding RNA Gene Knockout in Human Mesenchymal Stromal Cells" Non-Coding RNA 9, no. 5: 49. https://doi.org/10.3390/ncrna9050049
APA StyleBasalova, N., Illarionova, M., Skryabina, M., Vigovskiy, M., Tolstoluzhinskaya, A., Primak, A., Chechekhina, E., Chechekhin, V., Karagyaur, M., & Efimenko, A. (2023). Advances and Obstacles in Using CRISPR/Cas9 Technology for Non-Coding RNA Gene Knockout in Human Mesenchymal Stromal Cells. Non-Coding RNA, 9(5), 49. https://doi.org/10.3390/ncrna9050049