
Advances in Non-Coding RNA Sequencing



Abstract
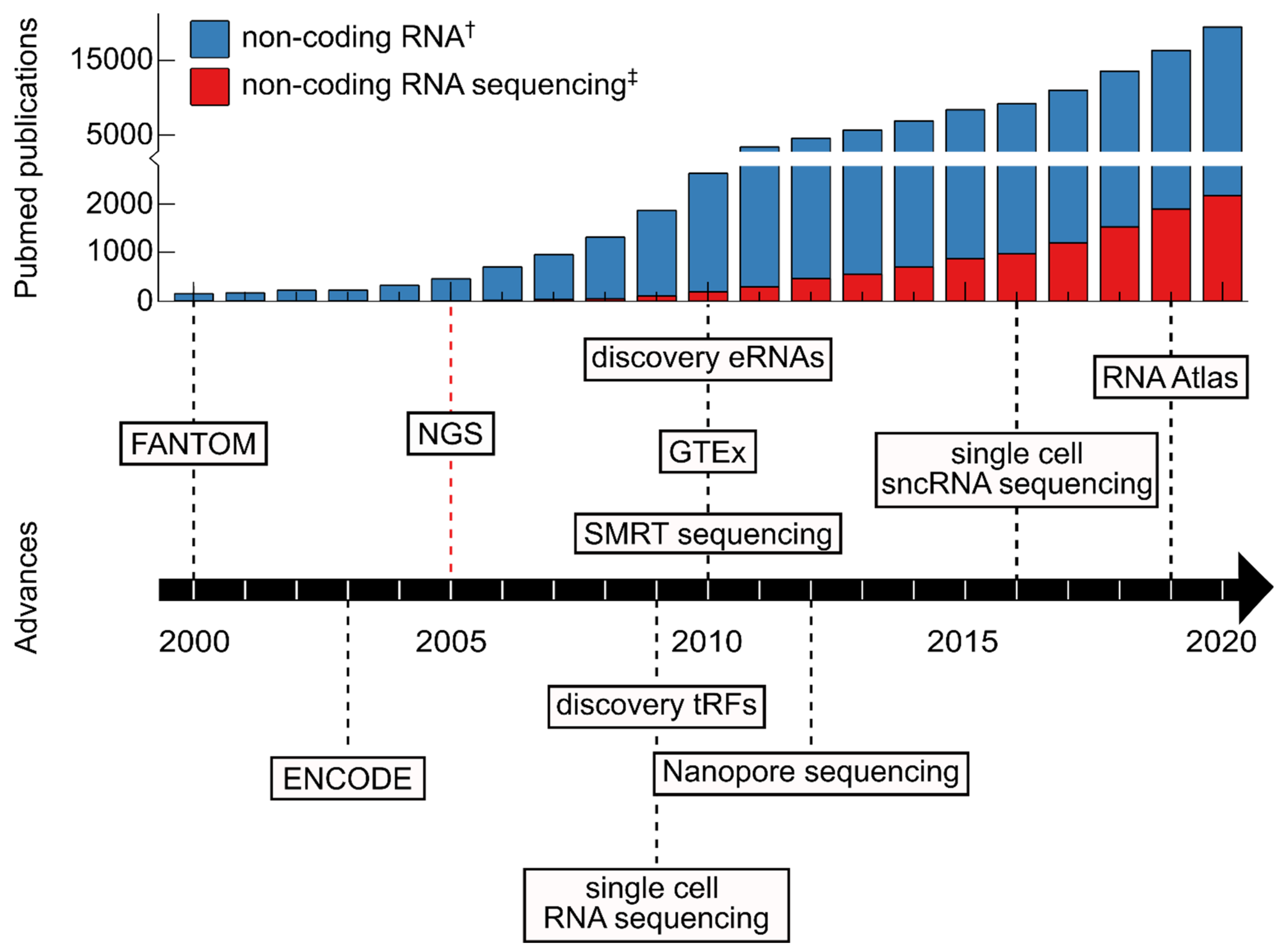
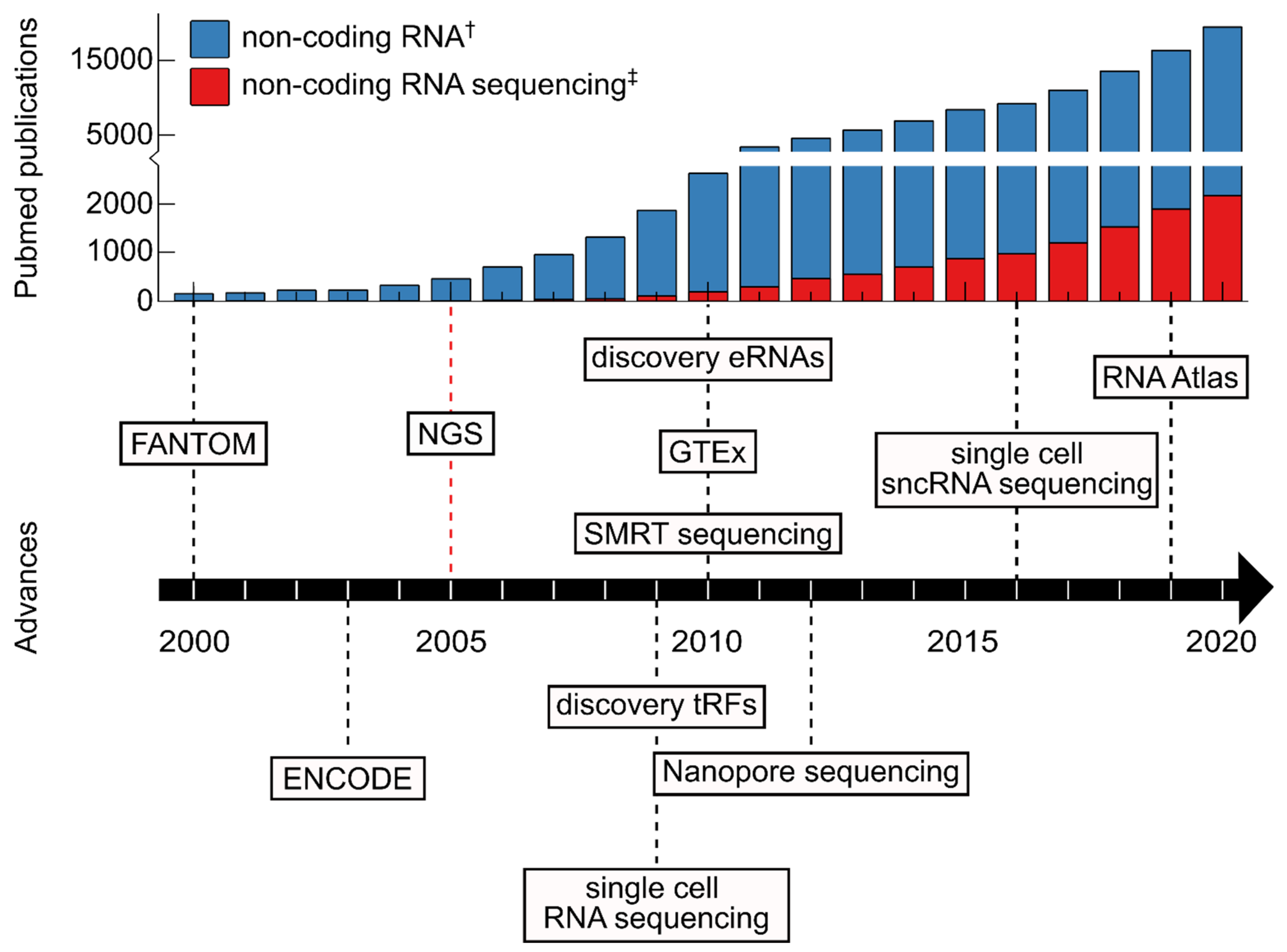
:1. The Dawn of Molecular Biology and Nucleic Acid Sequencing
2. Advancing Our Understanding of ncRNA Biology through RNA Sequencing
2.1. Sequencing Total RNA from Single Cells
2.2. Transcriptional Regulation
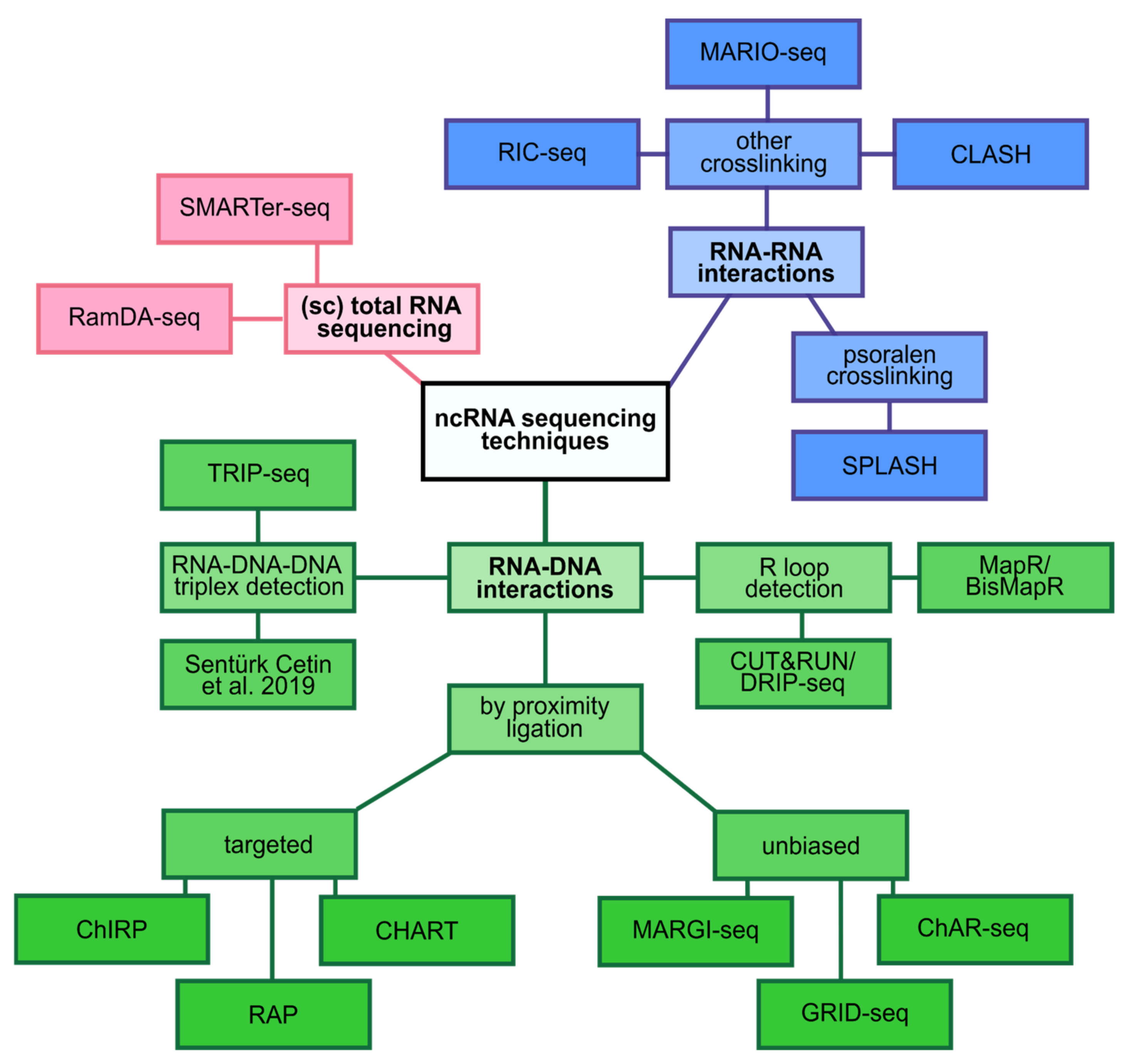
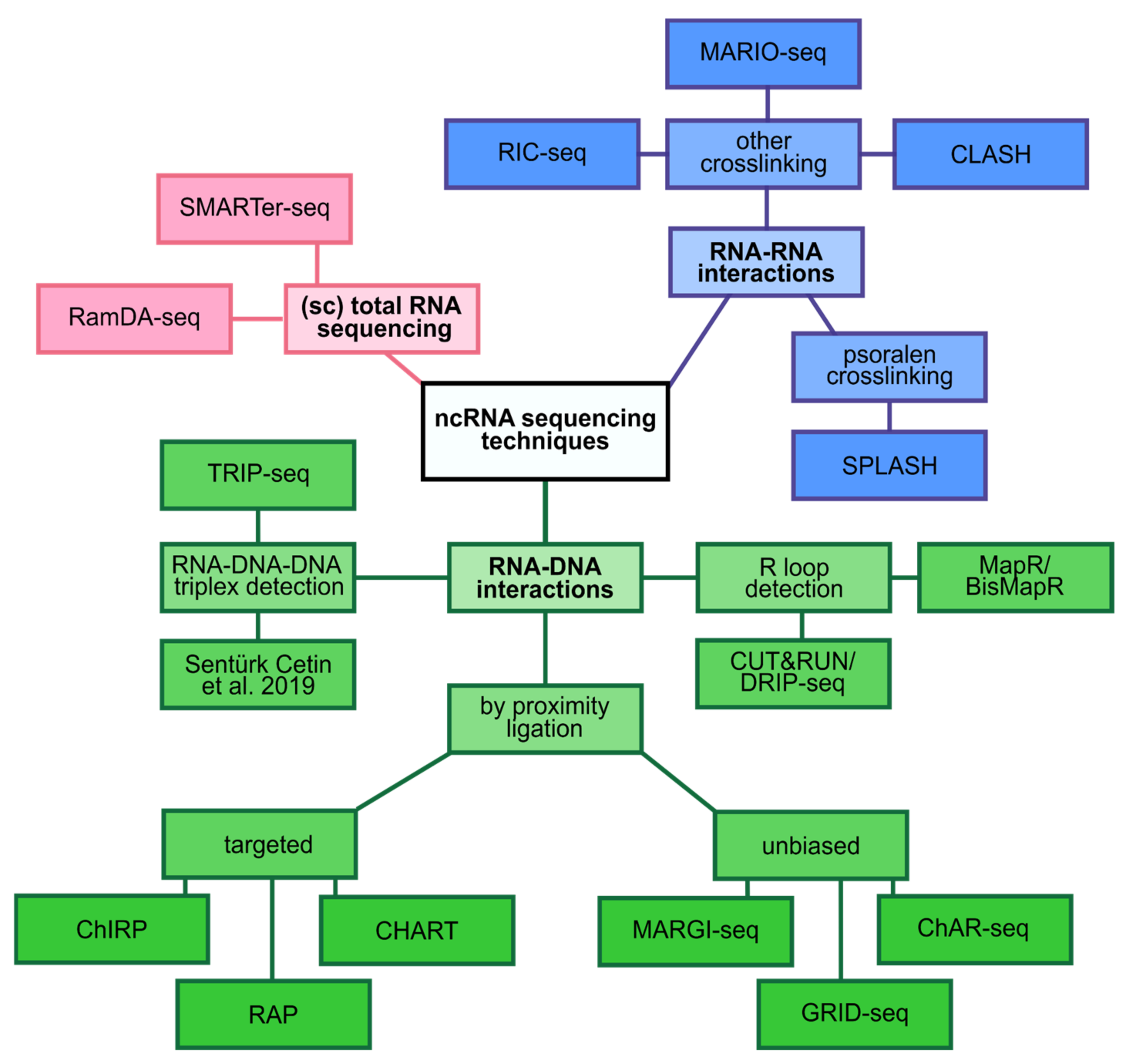
2.3. Regulation on the Chromatin Level
2.4. Post-Transcriptional Regulation
2.5. Scaffolding
3. ncRNA in Human Health and Disease
3.1. Unraveling the ncRNA Landscape in Humans
3.2. ncRNAs Associated with Pathological Mechanisms
3.3. Discovery of Diagnostic ncRNAs
4. Exploring the ncRNA Landscape beyond Humans
4.1. Viruses
4.2. Bacteria and Archaea
4.3. Plants
4.4. Fungi
4.5. Protists
4.6. Invertebrates
4.7. Vertebrates
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cobb, M. 60 Years Ago, Francis Crick Changed the Logic of Biology. PLoS Biol. 2017, 15, e2003243. [Google Scholar] [CrossRef] [Green Version]
- Jacob, F.; Monod, J. Genetic Regulatory Mechanisms in the Synthesis of Proteins. J. Mol. Biol. 1961, 3, 318–356. [Google Scholar] [CrossRef]
- Hoagland, M.B.; Stephenson, M.L.; Scott, J.F.; Hecht, L.I.; Zamecnik, P.C. A soluble ribonucleic acid intermediate in protein synthesis. J. Biol. Chem. 1958, 231, 241–257. [Google Scholar] [CrossRef]
- Palade, G.E. A small particulate component of the cytoplasm. J. Biophys. Biochem. Cytol. 1955, 1, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, R.A.; Penman, S. Small Molecular Weight Monodisperse Nuclear RNA. J. Mol. Biol. 1968, 38, 289–304. [Google Scholar] [CrossRef]
- Comings, D.E. The Structure and Function of Chromatin. In Advances in Human Genetics; Harris, H., Hirschhorn, K., Eds.; Advances in Human Genetics; Springer US: Boston, MA, USA, 1972; pp. 237–431. ISBN 978-1-4757-4429-3. [Google Scholar]
- Ohno, S. So Much “Junk” DNA in Our Genome. Brookhaven Symp. Biol. 1972, 23, 366–370. [Google Scholar]
- Orgel, L.E.; Crick, F.H.C. Selfish DNA: The Ultimate Parasite. Nature 1980, 284, 604–607. [Google Scholar] [CrossRef]
- Coleman, J.; Green, P.J.; Inouye, M. The Use of RNAs Complementary to Specific MRNAs to Regulate the Expression of Individual Bacterial Genes. Cell 1984, 37, 429–436. [Google Scholar] [CrossRef]
- Brannan, C.I.; Dees, E.C.; Ingram, R.S.; Tilghman, S.M. The Product of the H19 Gene May Function as an RNA. Mol. Cell. Biol. 1990, 10, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Brockdorff, N.; Ashworth, A.; Kay, G.F.; McCabe, V.M.; Norris, D.P.; Cooper, P.J.; Swift, S.; Rastan, S. The Product of the Mouse Xist Gene Is a 15 Kb Inactive X-Specific Transcript Containing No Conserved ORF and Located in the Nucleus. Cell 1992, 71, 515–526. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. Elegans Heterochronic Gene Lin-4 Encodes Small RNAs with Antisense Complementarity to Lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA Sequencing with Chain-Terminating Inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanger, F.; Air, G.M.; Barrell, B.G.; Brown, N.L.; Coulson, A.R.; Fiddes, C.A.; Hutchison, C.A.; Slocombe, P.M.; Smith, M. Nucleotide Sequence of Bacteriophage Phi X174 DNA. Nature 1977, 265, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.D.; Adams, M.D.; White, O.; Clayton, R.A.; Kirkness, E.F.; Kerlavage, A.R.; Bult, C.J.; Tomb, J.F.; Dougherty, B.A.; Merrick, J.M. Whole-Genome Random Sequencing and Assembly of Haemophilus Influenzae Rd. Science 1995, 269, 496–512. [Google Scholar] [CrossRef] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A. The Sequence of the Human Genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.; et al. Genome Sequencing in Microfabricated High-Density Picolitre Reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigó, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; et al.; ENCODE Project Consortium Identification and Analysis of Functional Elements in 1% of the Human Genome by the ENCODE Pilot Project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, S.; Kanamori, M.; Hayashizaki, Y. Integrated Analysis of the Genome and the Transcriptome by FANTOM. Brief. Bioinform. 2004, 5, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The Transcriptional Landscape of the Mammalian Genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levene, M.J.; Korlach, J.; Turner, S.W.; Foquet, M.; Craighead, H.G.; Webb, W.W. Zero-Mode Waveguides for Single-Molecule Analysis at High Concentrations. Science 2003, 299, 682–686. [Google Scholar]
- Deamer, D.W.; Branton, D. Characterization of Nucleic Acids by Nanopore Analysis. Acc. Chem. Res. 2002, 35, 817–825. [Google Scholar]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-Read Human Genome Sequencing and Its Applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) Project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Midha, M.K.; Wu, M.; Chiu, K.-P. Long-Read Sequencing in Deciphering Human Genetics to a Greater Depth. Hum. Genet. 2019, 138, 1201–1215. [Google Scholar] [CrossRef]
- Deamer, D.; Akeson, M.; Branton, D. Three Decades of Nanopore Sequencing. Nat. Biotechnol. 2016, 34, 518–524. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. MRNA-Seq Whole-Transcriptome Analysis of a Single Cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Faridani, O.R.; Abdullayev, I.; Hagemann-Jensen, M.; Schell, J.P.; Lanner, F.; Sandberg, R. Single-Cell Sequencing of the Small-RNA Transcriptome. Nat. Biotechnol. 2016, 34, 1264–1266. [Google Scholar] [CrossRef]
- Lorenzi, L.; Chiu, H.-S.; Cobos, F.A.; Gross, S.; Volders, P.-J.; Cannoodt, R.; Nuytens, J.; Vanderheyden, K.; Anckaert, J.; Lefever, S.; et al. The RNA Atlas, a Single Nucleotide Resolution Map of the Human Transcriptome. BioRxiv 2019, 807529. [Google Scholar] [CrossRef]
- Lee, Y.S.; Shibata, Y.; Malhotra, A.; Dutta, A. A Novel Class of Small RNAs: TRNA-Derived RNA Fragments (TRFs). Genes Dev. 2009, 23, 2639–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.-K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread Transcription at Neuronal Activity-Regulated Enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA Sequencing at 40: Past, Present and Future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef]
- Singh, A.; Vivek, A.T.; Kumar, S. AlnC: An Extensive Database of Long Non-Coding RNAs in Angiosperms. PLoS ONE 2021, 16, e0247215. [Google Scholar] [CrossRef]
- Fehlmann, T.; Backes, C.; Pirritano, M.; Laufer, T.; Galata, V.; Kern, F.; Kahraman, M.; Gasparoni, G.; Ludwig, N.; Lenhof, H.-P.; et al. The SncRNA Zoo: A Repository for Circulating Small Noncoding RNAs in Animals. Nucleic Acids Res. 2019, 47, 4431–4441. [Google Scholar] [CrossRef]
- Lyu, Y.; Caudron-Herger, M.; Diederichs, S. Circ2GO: A Database Linking Circular RNAs to Gene Function. Cancers 2020, 12, 2975. [Google Scholar] [CrossRef]
- Wu, W.; Ji, P.; Zhao, F. CircAtlas: An Integrated Resource of One Million Highly Accurate Circular RNAs from 1070 Vertebrate Transcriptomes. Genome Biol. 2020, 21, 101. [Google Scholar] [CrossRef]
- Liu, M.; Wang, Q.; Shen, J.; Yang, B.B.; Ding, X. Circbank: A Comprehensive Database for CircRNA with Standard Nomenclature. RNA Biol. 2019, 16, 899–905. [Google Scholar] [CrossRef]
- Glažar, P.; Papavasileiou, P.; Rajewsky, N. CircBase: A Database for Circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Hu, D.; Zhang, P.; Chen, Q.; Chen, M. CircFunBase: A Database for Functional Circular RNAs. Database 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Dong, R.; Ma, X.-K.; Li, G.-W.; Yang, L. CIRCpedia v2: An Updated Database for Comprehensive Circular RNA Annotation and Expression Comparison. Genom. Proteom. Bioinform. 2018, 16, 226–233. [Google Scholar] [CrossRef]
- Xie, F.; Liu, S.; Wang, J.; Xuan, J.; Zhang, X.; Qu, L.; Zheng, L.; Yang, J. DeepBase v3.0: Expression Atlas and Interactive Analysis of NcRNAs from Thousands of Deep-Sequencing Data. Nucleic Acids Res. 2021, 49, D877–D883. [Google Scholar] [CrossRef] [PubMed]
- Karagkouni, D.; Paraskevopoulou, M.D.; Tastsoglou, S.; Skoufos, G.; Karavangeli, A.; Pierros, V.; Zacharopoulou, E.; Hatzigeorgiou, A.G. DIANA-LncBase v3: Indexing Experimentally Supported MiRNA Targets on Non-Coding Transcripts. Nucleic Acids Res. 2020, 48, D101–D110. [Google Scholar] [CrossRef] [PubMed]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A Decade-Long Collection of Experimentally Supported MiRNA–Gene Interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-J.; Xie, G.-Y.; Miao, Y.-R.; Xia, M.; Wang, Y.; Lei, Q.; Zhang, Q.; Guo, A.-Y. EVAtlas: A Comprehensive Database for NcRNA Expression in Human Extracellular Vesicles. Nucleic Acids Res. 2021. [Google Scholar] [CrossRef]
- Li, S.; Li, Y.; Chen, B.; Zhao, J.; Yu, S.; Tang, Y.; Zheng, Q.; Li, Y.; Wang, P.; He, X.; et al. ExoRBase: A Database of CircRNA, LncRNA and MRNA in Human Blood Exosomes. Nucleic Acids Res. 2018, 46, D106–D112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- THE GTEX CONSORTIUM The GTEx Consortium Atlas of Genetic Regulatory Effects across Human Tissues. Science 2020, 369, 1318–1330. [CrossRef]
- Daulatabad, S.V.; Srivastava, R.; Janga, S.C. Lantern: An Integrative Repository of Functional Annotations for LncRNAs in the Human Genome. BMC Bioinform. 2021, 22, 279. [Google Scholar] [CrossRef]
- Mas-Ponte, D.; Carlevaro-Fita, J.; Palumbo, E.; Hermoso Pulido, T.; Guigo, R.; Johnson, R. LncATLAS Database for Subcellular Localization of Long Noncoding RNAs. RNA 2017, 23, 1080–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Cao, J.; Liu, L.; Du, Q.; Li, Z.; Zou, D.; Bajic, V.B.; Zhang, Z. LncBook: A Curated Knowledgebase of Human Long Non-Coding RNAs. Nucleic Acids Res. 2019, 47, D128–D134. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Liu, L.; Jiang, S.; Li, Q.; Feng, C.; Du, Q.; Zou, D.; Xiao, J.; Zhang, Z.; Ma, L. LncExpDB: An Expression Database of Human Long Non-Coding RNAs. Nucleic Acids Res. 2021, 49, D962–D968. [Google Scholar] [CrossRef]
- Volders, P.-J.; Anckaert, J.; Verheggen, K.; Nuytens, J.; Martens, L.; Mestdagh, P.; Vandesompele, J. LNCipedia 5: Towards a Reference Set of Human Long Non-Coding RNAs. Nucleic Acids Res. 2019, 47, D135–D139. [Google Scholar] [CrossRef] [Green Version]
- Seifuddin, F.; Singh, K.; Suresh, A.; Judy, J.T.; Chen, Y.-C.; Chaitankar, V.; Tunc, I.; Ruan, X.; Li, P.; Chen, Y.; et al. LncRNAKB, a Knowledgebase of Tissue-Specific Functional Annotation and Trait Association of Long Noncoding RNA. Sci. Data 2020, 7, 326. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, J.; Gao, Y.; Li, Y.; Feng, C.; Song, C.; Ning, Z.; Zhou, X.; Zhao, J.; Feng, M.; et al. LncSEA: A Platform for Long Non-Coding RNA Related Sets and Enrichment Analysis. Nucleic Acids Res. 2021, 49, D969–D980. [Google Scholar] [CrossRef]
- Pliatsika, V.; Loher, P.; Magee, R.; Telonis, A.G.; Londin, E.; Shigematsu, M.; Kirino, Y.; Rigoutsos, I. MINTbase v2.0: A Comprehensive Database for TRNA-Derived Fragments That Includes Nuclear and Mitochondrial Fragments from All The Cancer Genome Atlas Projects. Nucleic Acids Res. 2018, 46, D152–D159. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From MicroRNA Sequences to Function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Marceca, G.P.; Distefano, R.; Tomasello, L.; Lagana, A.; Russo, F.; Calore, F.; Romano, G.; Bagnoli, M.; Gasparini, P.; Ferro, A.; et al. MiREDiBase, a Manually Curated Database of Validated and Putative Editing Events in MicroRNAs. Sci. Data 2021, 8, 199. [Google Scholar] [CrossRef] [PubMed]
- Fromm, B.; Domanska, D.; Høye, E.; Ovchinnikov, V.; Kang, W.; Aparicio-Puerta, E.; Johansen, M.; Flatmark, K.; Mathelier, A.; Hovig, E.; et al. MirGeneDB 2.0: The Metazoan MicroRNA Complement. Nucleic Acids Res. 2020, 48, D132–D141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.-Y.; Lin, Y.-C.-D.; Li, J.; Huang, K.-Y.; Shrestha, S.; Hong, H.-C.; Tang, Y.; Chen, Y.-G.; Jin, C.-N.; Yu, Y.; et al. MiRTarBase 2020: Updates to the Experimentally Validated MicroRNA–Target Interaction Database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sticht, C.; Torre, C.D.L.; Parveen, A.; Gretz, N. MiRWalk: An Online Resource for Prediction of MicroRNA Binding Sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, H.; Fang, S.; Kang, Y.; wu, W.; Hao, Y.; Li, Z.; Bu, D.; Sun, N.; Zhang, M.Q.; et al. NONCODE 2016: An Informative and Valuable Data Source of Long Non-Coding RNAs. Nucleic Acids Res. 2016, 44, D203–D208. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Xie, M.; Wang, Y.; Yang, L.; Xie, Z.; Wang, H. RiboCIRC: A Comprehensive Database of Translatable CircRNAs. Genome Biol. 2021, 22, 79. [Google Scholar] [CrossRef]
- Kalvari, I.; Nawrocki, E.P.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z.; et al. Rfam 14: Expanded Coverage of Metagenomic, Viral and MicroRNA Families. Nucleic Acids Res. 2021, 49, D192–D200. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, L.; Chiu, H.-S.; Avila Cobos, F.; Gross, S.; Volders, P.-J.; Cannoodt, R.; Nuytens, J.; Vanderheyden, K.; Anckaert, J.; Lefever, S.; et al. The RNA Atlas Expands the Catalog of Human Non-Coding RNAs. Nat. Biotechnol. 2021, 1–13. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, P.; Lu, Y.; Li, Y.; Zheng, Y.; Kan, Y.; Chen, R.; He, S. PiRBase: A Comprehensive Database of PiRNA Sequences. Nucleic Acids Res. 2019, 47, D175–D180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenkranz, D.; Zischler, H.; Gebert, D. PiRNAclusterDB 2.0: Update and Expansion of the PiRNA Cluster Database. Nucleic Acids Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.-S.; Brown, J.S.; Chen, T.-T.; Chu, Y.-H.; Huang, W.-C.; Tu, S.; Lee, H.-C. PiRTarBase: A Database of PiRNA Targeting Sites and Their Roles in Gene Regulation. Nucleic Acids Res. 2019, 47, D181–D187. [Google Scholar] [CrossRef]
- Chu, Q.; Zhang, X.; Zhu, X.; Liu, C.; Mao, L.; Ye, C.; Zhu, Q.-H.; Fan, L. PlantcircBase: A Database for Plant Circular RNAs. Mol. Plant 2017, 10, 1126–1128. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Singh, A.; Zahra, S.; Kumar, S. PtRFdb: A Database for Plant Transfer RNA-Derived Fragments. Database J. Biol. Databases Curation 2018, 2018, bay063. [Google Scholar] [CrossRef] [PubMed]
- Bouchard-Bourelle, P.; Desjardins-Henri, C.; Mathurin-St-Pierre, D.; Deschamps-Francoeur, G.; Fafard-Couture, É.; Garant, J.-M.; Elela, S.A.; Scott, M.S. SnoDB: An Interactive Database of Human SnoRNA Sequences, Abundance and Interactions. Nucleic Acids Res. 2020, 48, D220–D225. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, X.; Zhang, S.; Liang, S.; Luan, W.; Ma, X. TarDB: An Online Database for Plant MiRNA Targets and MiRNA-Triggered Phased SiRNAs. BMC Genomics 2021, 22, 348. [Google Scholar] [CrossRef]
- Huang, W.; Ling, Y.; Zhang, S.; Xia, Q.; Cao, R.; Fan, X.; Fang, Z.; Wang, Z.; Zhang, G. TransCirc: An Interactive Database for Translatable Circular RNAs Based on Multi-Omics Evidence. Nucleic Acids Res. 2021, 49, D236–D242. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Feng, J.; Lei, L.; Hu, J.; Xia, L.; Wang, J.; Xiang, Y.; Liu, L.; Zhong, S.; Han, L.; et al. Comprehensive Characterization of Tissue-Specific Circular RNAs in the Human and Mouse Genomes. Brief. Bioinform. 2017, 18, 984–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Hoon, M.; Shin, J.W.; Carninci, P. Paradigm Shifts in Genomics through the FANTOM Projects. Mamm. Genome 2015, 26, 391–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, J.; Shinagawa, A.; Shibata, K.; Yoshino, M.; Itoh, M.; Ishii, Y.; Arakawa, T.; Hara, A.; Fukunishi, Y.; Konno, H. Functional Annotation of a Full-Length Mouse CDNA Collection. Nature 2001, 409, 685–689. [Google Scholar] [PubMed]
- Shiraki, T.; Kondo, S.; Katayama, S.; Waki, K.; Kasukawa, T.; Kawaji, H.; Kodzius, R.; Watahiki, A.; Nakamura, M.; Arakawa, T. Cap Analysis Gene Expression for High-Throughput Analysis of Transcriptional Starting Point and Identification of Promoter Usage. Proc. Natl. Acad. Sci. USA 2003, 100, 15776–15781. [Google Scholar] [CrossRef] [Green Version]
- Kodzius, R.; Kojima, M.; Nishiyori, H.; Nakamura, M.; Fukuda, S.; Tagami, M.; Sasaki, D.; Imamura, K.; Kai, C.; Harbers, M. CAGE: Cap Analysis of Gene Expression. Nat. Methods 2006, 3, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F. Landscape of Transcription in Human Cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Palazzo, A.F.; Lee, E.S. Non-Coding RNA: What Is Functional and What Is Junk? Front. Genet. 2015, 6, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camp, J.G.; Platt, R.; Treutlein, B. Mapping Human Cell Phenotypes to Genotypes with Single-Cell Genomics. Science 2019, 365, 1401–1405. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Klaus, J.; Kanton, S.; Kyrousi, C.; Ayo-Martin, A.C.; Di Giaimo, R.; Riesenberg, S.; O’Neill, A.C.; Camp, J.G.; Tocco, C.; Santel, M. Altered Neuronal Migratory Trajectories in Human Cerebral Organoids Derived from Individuals with Neuronal Heterotopia. Nat. Med. 2019, 25, 561–568. [Google Scholar] [CrossRef]
- Marioni, J.C.; Arendt, D. How Single-Cell Genomics Is Changing Evolutionary and Developmental Biology. Annu. Rev. Cell Dev. Biol. 2017, 33, 537–553. [Google Scholar] [CrossRef]
- Hayashi, T.; Ozaki, H.; Sasagawa, Y.; Umeda, M.; Danno, H.; Nikaido, I. Single-Cell Full-Length Total RNA Sequencing Uncovers Dynamics of Recursive Splicing and Enhancer RNAs. Nat. Commun. 2018, 9, 619. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, D.; Glowatz, H.; Schlumpberger, M. Ribosomal RNA Depletion for Efficient Use of RNA-Seq Capacity. Curr. Protoc. Mol. Biol. 2013, 103, 4–19. [Google Scholar] [CrossRef]
- Verboom, K.; Everaert, C.; Bolduc, N.; Livak, K.J.; Yigit, N.; Rombaut, D.; Anckaert, J.; Lee, S.; Venø, M.T.; Kjems, J.; et al. SMARTer Single Cell Total RNA Sequencing. Nucleic Acids Res. 2019, 47, e93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, C.; Qu, K.; Zhong, F.; Artandi, S.E.; Chang, H.Y. Genomic Maps of LincRNA Occupancy Reveal Principles of RNA-Chromatin Interactions. Mol. Cell 2011, 44, 667–678. [Google Scholar] [CrossRef] [Green Version]
- Engreitz, J.M.; Pandya-Jones, A.; McDonel, P.; Shishkin, A.; Sirokman, K.; Surka, C.; Kadri, S.; Xing, J.; Goren, A.; Lander, E.S.; et al. The Xist LncRNA Exploits Three-Dimensional Genome Architecture to Spread across the X-Chromosome. Science 2013, 341, 1237973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, M.D.; Wang, C.I.; Kharchenko, P.V.; West, J.A.; Chapman, B.A.; Alekseyenko, A.A.; Borowsky, M.L.; Kuroda, M.I.; Kingston, R.E. The Genomic Binding Sites of a Noncoding RNA. Proc. Natl. Acad. Sci. USA 2011, 108, 20497–20502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridhar, B.; Rivas-Astroza, M.; Nguyen, T.C.; Chen, W.; Yan, Z.; Cao, X.; Hebert, L.; Zhong, S. Systematic Mapping of RNA-Chromatin Interactions in Vivo. Curr. Biol. CB 2017, 27, 602–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhou, B.; Chen, L.; Gou, L.-T.; Li, H.; Fu, X.-D. GRID-Seq Reveals the Global RNA–Chromatin Interactome. Nat. Biotechnol. 2017, 35, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.C.; Jukam, D.; Teran, N.A.; Risca, V.I.; Smith, O.K.; Johnson, W.L.; Skotheim, J.M.; Greenleaf, W.J.; Straight, A.F. Chromatin-Associated RNA Sequencing (ChAR-Seq) Maps Genome-Wide RNA-to-DNA Contacts. eLife 2018, 7, e27024. [Google Scholar] [CrossRef]
- Maldonado, R.; Schwartz, U.; Silberhorn, E.; Längst, G. Nucleosomes Stabilize SsRNA-DsDNA Triple Helices in Human Cells. Mol. Cell 2019, 73, 1243–1254.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sentürk Cetin, N.; Kuo, C.-C.; Ribarska, T.; Li, R.; Costa, I.G.; Grummt, I. Isolation and Genome-Wide Characterization of Cellular DNA:RNA Triplex Structures. Nucleic Acids Res. 2019, 47, 2306–2321. [Google Scholar] [CrossRef]
- Skene, P.J.; Henikoff, S. An Efficient Targeted Nuclease Strategy for High-Resolution Mapping of DNA Binding Sites. Elife 2017, 6, e21856. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.A.; Aristizabal, M.J.; Lu, P.Y.; Luo, Z.; Hamza, A.; Kobor, M.S.; Stirling, P.C.; Hieter, P. Genome-Wide Profiling of Yeast DNA: RNA Hybrid Prone Sites with DRIP-Chip. PLoS Genet. 2014, 10, e1004288. [Google Scholar] [CrossRef] [Green Version]
- Yan, Q.; Shields, E.J.; Bonasio, R.; Sarma, K. Mapping Native R-Loops Genome-Wide Using a Targeted Nuclease Approach. Cell Rep. 2019, 29, 1369–1380.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wulfridge, P.; Sarma, K. A Nuclease- and Bisulfite-Based Strategy Captures Strand-Specific R-Loops Genome-Wide. eLife 2021, 10, e65146. [Google Scholar] [CrossRef]
- Cai, Z.; Cao, C.; Ji, L.; Ye, R.; Wang, D.; Xia, C.; Wang, S.; Du, Z.; Hu, N.; Yu, X.; et al. RIC-Seq for Global in Situ Profiling of RNA–RNA Spatial Interactions. Nature 2020, 582, 432–437. [Google Scholar] [CrossRef]
- Nguyen, T.C.; Cao, X.; Yu, P.; Xiao, S.; Lu, J.; Biase, F.H.; Sridhar, B.; Huang, N.; Zhang, K.; Zhong, S. Mapping RNA–RNA Interactome and RNA Structure in Vivo by MARIO. Nat. Commun. 2016, 7, 12023. [Google Scholar] [CrossRef] [Green Version]
- Helwak, A.; Tollervey, D. Mapping the MiRNA Interactome by Cross-Linking Ligation and Sequencing of Hybrids (CLASH). Nat. Protoc. 2014, 9, 711–728. [Google Scholar] [CrossRef] [Green Version]
- Aw, J.G.A.; Shen, Y.; Wilm, A.; Sun, M.; Lim, X.N.; Boon, K.-L.; Tapsin, S.; Chan, Y.-S.; Tan, C.-P.; Sim, A.Y.L.; et al. In Vivo Mapping of Eukaryotic RNA Interactomes Reveals Principles of Higher-Order Organization and Regulation. Mol. Cell 2016, 62, 603–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noviello, T.M.R.; Ceccarelli, F.; Ceccarelli, M.; Cerulo, L. Deep Learning Predicts Short Non-Coding RNA Functions from Only Raw Sequence Data. PLoS Comput. Biol. 2020, 16, e1008415. [Google Scholar] [CrossRef] [PubMed]
- Chantsalnyam, T.; Lim, D.Y.; Tayara, H.; Chong, K.T. NcRDeep: Non-Coding RNA Classification with Convolutional Neural Network. Comput. Biol. Chem. 2020, 88, 107364. [Google Scholar] [CrossRef]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-Coding RNA Networks in Cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Loda, A.; Heard, E. Xist RNA in Action: Past, Present, and Future. PLoS Genet. 2019, 15, e1008333. [Google Scholar] [CrossRef] [Green Version]
- Hoki, Y.; Kimura, N.; Kanbayashi, M.; Amakawa, Y.; Ohhata, T.; Sasaki, H.; Sado, T. A Proximal Conserved Repeat in the Xist Gene Is Essential as a Genomic Element for X-Inactivation in Mouse. Development 2009, 136, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Bousard, A.; Raposo, A.C.; Żylicz, J.J.; Picard, C.; Pires, V.B.; Qi, Y.; Gil, C.; Syx, L.; Chang, H.Y.; Heard, E. The Role of Xist-mediated Polycomb Recruitment in the Initiation of X-chromosome Inactivation. EMBO Rep. 2019, 20, e48019. [Google Scholar] [CrossRef]
- Penny, G.D.; Kay, G.F.; Sheardown, S.A.; Rastan, S.; Brockdorff, N. Requirement for Xist in X Chromosome Inactivation. Nature 1996, 379, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.M.; Kadonaga, J.T. Going the Distance: A Current View of Enhancer Action. Science 1998, 281, 60–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin Accessibility and the Regulatory Epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Machyna, M.; Simon, M.D. Catching RNAs on Chromatin Using Hybridization Capture Methods. Brief. Funct. Genomics 2018, 17, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Syed, J.; Sugiyama, H. RNA-DNA Triplex Formation by Long Noncoding RNAs. Cell Chem. Biol. 2016, 23, 1325–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.-C.; Hänzelmann, S.; Sentürk Cetin, N.; Frank, S.; Zajzon, B.; Derks, J.-P.; Akhade, V.S.; Ahuja, G.; Kanduri, C.; Grummt, I. Detection of RNA–DNA Binding Sites in Long Noncoding RNAs. Nucleic Acids Res. 2019, 47, e32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niehrs, C.; Luke, B. Regulatory R-Loops as Facilitators of Gene Expression and Genome Stability. Nat. Rev. Mol. Cell Biol. 2020, 21, 167–178. [Google Scholar] [CrossRef]
- El Hage, A.; Webb, S.; Kerr, A.; Tollervey, D. Genome-Wide Distribution of RNA-DNA Hybrids Identifies RNase H Targets in TRNA Genes, Retrotransposons and Mitochondria. PLoS Genet. 2014, 10, e1004716. [Google Scholar] [CrossRef]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of Post-Transcriptional Regulation by MicroRNAs: Are the Answers in Sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef]
- Alles, J.; Fehlmann, T.; Fischer, U.; Backes, C.; Galata, V.; Minet, M.; Hart, M.; Abu-Halima, M.; Grässer, F.A.; Lenhof, H.-P.; et al. An Estimate of the Total Number of True Human MiRNAs. Nucleic Acids Res. 2019, 47, 3353–3364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Z.; Du, R.; Edwards, A.; Flemington, E.K.; Zhang, K. The Sequence Structures of Human MicroRNA Molecules and Their Implications. PLoS ONE 2013, 8, e54215. [Google Scholar] [CrossRef] [PubMed]
- Huntzinger, E.; Izaurralde, E. Gene Silencing by MicroRNAs: Contributions of Translational Repression and MRNA Decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef]
- Benesova, S.; Kubista, M.; Valihrach, L. Small RNA-Sequencing: Approaches and Considerations for MiRNA Analysis. Diagnostics 2021, 11, 964. [Google Scholar] [CrossRef]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the Human MiRNA Interactome by CLASH Reveals Frequent Noncanonical Binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichholf, B.; Herzog, V.A.; Fasching, N.; Manzenreither, R.A.; Sowemimo, I.; Ameres, S.L. Time-Resolved Small RNA Sequencing Unravels the Molecular Principles of MicroRNA Homeostasis. Mol. Cell 2019, 75, 756–768.e7. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular Mechanisms of Long Noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E. Functional Demarcation of Active and Silent Chromatin Domains in Human HOX Loci by Noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Jiang, T.; Zhou, W.; Li, J.; Li, X.; Wang, Q.; Jin, X.; Yin, J.; Chen, L.; Zhang, Y.; et al. Pan-Cancer Characterization of Immune-Related LncRNAs Identifies Potential Oncogenic Biomarkers. Nat. Commun. 2020, 11, 1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giral, H.; Landmesser, U.; Kratzer, A. Into the Wild: GWAS Exploration of Non-Coding RNAs. Front. Cardiovasc. Med. 2018, 5, 181. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs Are the Predominant Transcript Isoform from Hundreds of Human Genes in Diverse Cell Types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, L.; Morey, R.; Palpant, N.J.; Wang, P.L.; Afari, N.; Jiang, C.; Parast, M.M.; Murry, C.E.; Laurent, L.C.; Salzman, J. Statistically Based Splicing Detection Reveals Neural Enrichment and Tissue-Specific Induction of Circular RNA during Human Fetal Development. Genome Biol. 2015, 16, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Falaleeva, M.; Stamm, S. Processing of SnoRNAs as a New Source of Regulatory Non-Coding RNAs SnoRNA Fragments Form a New Class of Functional RNAs. BioEssays News Rev. Mol. Cell. Dev. Biol. 2013, 35, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Sun, Y.; Yang, X.; Wu, Z.; Guo, K.; Niu, X.; Wang, Q.; Ruan, J.; Bu, W.; Gao, S. Two Featured Series of RRNA-Derived RNA Fragments (RRFs) Constitute a Novel Class of Small RNAs. PLoS ONE 2017, 12, e0176458. [Google Scholar] [CrossRef] [Green Version]
- Zeng, T.; Hua, Y.; Sun, C.; Zhang, Y.; Yang, F.; Yang, M.; Yang, Y.; Li, J.; Huang, X.; Wu, H.; et al. Relationship between TRNA-Derived Fragments and Human Cancers. Int. J. Cancer 2020, 147, 3007–3018. [Google Scholar] [CrossRef]
- Shen, L.; Hong, X.; Zhou, W.; Zhang, Y. Expression Profiles of TRNA-Derived Fragments and Their Potential Roles in Ovarian Endometriosis. Epigenomics 2020, 12, 183–197. [Google Scholar] [CrossRef]
- Wu, W.; Lee, I.; Spratt, H.; Fang, X.; Bao, X. TRNA-Derived Fragments in Alzheimer’s Disease: Implications for New Disease Biomarkers and Neuropathological Mechanisms. J. Alzheimers Dis. JAD 2021, 79, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Wu, W.; Chen, S.; Zheng, Y.; Zhou, L.; Zhang, J.; Cheng, H.; Yan, J.; Zhang, S.; Yang, P.; et al. Expanded Expression Landscape and Prioritization of Circular RNAs in Mammals. Cell Rep. 2019, 26, 3444–3460.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maass, P.G.; Glažar, P.; Memczak, S.; Dittmar, G.; Hollfinger, I.; Schreyer, L.; Sauer, A.V.; Toka, O.; Aiuti, A.; Luft, F.C.; et al. A Map of Human Circular RNAs in Clinically Relevant Tissues. J. Mol. Med. 2017, 95, 1179–1189. [Google Scholar] [CrossRef]
- Jiang, C.; Li, Y.; Zhao, Z.; Lu, J.; Chen, H.; Ding, N.; Wang, G.; Xu, J.; Li, X. Identifying and Functionally Characterizing Tissue-Specific and Ubiquitously Expressed Human LncRNAs. Oncotarget 2016, 7, 7120–7133. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.J.; Nowakowski, T.J.; Pollen, A.A.; Lui, J.H.; Horlbeck, M.A.; Attenello, F.J.; He, D.; Weissman, J.S.; Kriegstein, A.R.; Diaz, A.A.; et al. Single-Cell Analysis of Long Non-Coding RNAs in the Developing Human Neocortex. Genome Biol. 2016, 17, 67. [Google Scholar] [CrossRef] [Green Version]
- Gong, S.; Gaccioli, F.; Dopierala, J.; Sovio, U.; Cook, E.; Volders, P.-J.; Martens, L.; Kirk, P.D.W.; Richardson, S.; Smith, G.C.S.; et al. The RNA Landscape of the Human Placenta in Health and Disease. Nat. Commun. 2021, 12, 2639. [Google Scholar] [CrossRef]
- Ruan, H.; Xiang, Y.; Ko, J.; Li, S.; Jing, Y.; Zhu, X.; Ye, Y.; Zhang, Z.; Mills, T.; Feng, J.; et al. Comprehensive Characterization of Circular RNAs in ~ 1000 Human Cancer Cell Lines. Genome Med. 2019, 11, 55. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Wang, K.; Wu, F.; Wang, W.; Zhang, K.; Hu, H.; Liu, Y.; Jiang, T. CircRNA Disease: A Manually Curated Database of Experimentally Supported CircRNA-Disease Associations. Cell Death Dis. 2018, 9, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Chen, W.; Dong, X.; Wang, J.; Mei, X.; Deng, J.; Yang, S.; Zhuo, C.; Huang, X.; Shao, L.; et al. CSCD2: An Integrated Interactional Database of Cancer-Specific Circular RNAs. Nucleic Acids Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wu, L.; Wang, A.; Tang, W.; Zhao, Y.; Zhao, H.; Teschendorff, A.E. DbDEMC 2.0: Updated Database of Differentially Expressed MiRNAs in Human Cancers. Nucleic Acids Res. 2017, 45, D812–D818. [Google Scholar] [CrossRef]
- Gao, Y.; Shang, S.; Guo, S.; Li, X.; Zhou, H.; Liu, H.; Sun, Y.; Wang, J.; Wang, P.; Zhi, H.; et al. Lnc2Cancer 3.0: An Updated Resource for Experimentally Supported LncRNA/CircRNA Cancer Associations and Web Tools Based on RNA-Seq and ScRNA-Seq Data. Nucleic Acids Res. 2021, 49, D1251–D1258. [Google Scholar] [CrossRef]
- Bao, Z.; Yang, Z.; Huang, Z.; Zhou, Y.; Cui, Q.; Dong, D. LncRNADisease 2.0: An Updated Database of Long Non-Coding RNA-Associated Diseases. Nucleic Acids Res. 2019, 47, D1034–D1037. [Google Scholar] [CrossRef]
- Miao, Y.-R.; Liu, W.; Zhang, Q.; Guo, A.-Y. LncRNASNP2: An Updated Database of Functional SNPs and Mutations in Human and Mouse LncRNAs. Nucleic Acids Res. 2018, 46, D276–D280. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Shi, J.; Zhang, Y.; Xie, A.; Yu, L.; Zhang, C.; Lei, J.; Xu, H.; Leng, Z.; Li, T.; et al. LncTarD: A Manually-Curated Database of Experimentally-Supported Functional LncRNA–Target Regulations in Human Diseases. Nucleic Acids Res. 2020, 48, D118–D126. [Google Scholar] [CrossRef]
- Vo, J.N.; Cieslik, M.; Zhang, Y.; Shukla, S.; Xiao, L.; Zhang, Y.; Wu, Y.-M.; Dhanasekaran, S.M.; Engelke, C.G.; Cao, X.; et al. The Landscape of Circular RNA in Cancer. Cell 2019, 176, 869–881.e13. [Google Scholar] [CrossRef] [Green Version]
- Ning, L.; Cui, T.; Zheng, B.; Wang, N.; Luo, J.; Yang, B.; Du, M.; Cheng, J.; Dou, Y.; Wang, D. MNDR v3.0: Mammal NcRNA–Disease Repository with Increased Coverage and Annotation. Nucleic Acids Res. 2021, 49, D160–D164. [Google Scholar] [CrossRef]
- Li, J.; Xue, Y.; Amin, M.T.; Yang, Y.; Yang, J.; Zhang, W.; Yang, W.; Niu, X.; Zhang, H.-Y.; Gong, J. NcRNA-EQTL: A Database to Systematically Evaluate the Effects of SNPs on Non-Coding RNA Expression across Cancer Types. Nucleic Acids Res. 2020, 48, D956–D963. [Google Scholar] [CrossRef]
- Zhang, W.; Yao, G.; Wang, J.; Yang, M.; Wang, J.; Zhang, H.; Li, W. NcRPheno: A Comprehensive Database Platform for Identification and Validation of Disease Related Noncoding RNAs. RNA Biol. 2020, 17, 943–955. [Google Scholar] [CrossRef]
- Wang, J.; Cao, Y.; Zhang, H.; Wang, T.; Tian, Q.; Lu, X.; Lu, X.; Kong, X.; Liu, Z.; Wang, N.; et al. NSDNA: A Manually Curated Database of Experimentally Supported NcRNAs Associated with Nervous System Diseases. Nucleic Acids Res. 2017, 45, D902–D907. [Google Scholar] [CrossRef] [PubMed]
- Avican, K.; Aldahdooh, J.; Togninalli, M.; Mahmud, A.K.M.F.; Tang, J.; Borgwardt, K.M.; Rhen, M.; Fällman, M. RNA Atlas of Human Bacterial Pathogens Uncovers Stress Dynamics Linked to Infection. Nat. Commun. 2021, 12, 3282. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Han, L.; Roebuck, P.; Diao, L.; Liu, L.; Yuan, Y.; Weinstein, J.N.; Liang, H. TANRIC: An Interactive Open Platform to Explore the Function of LncRNAs in Cancer. Cancer Res. 2015, 75, 3728–3737. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, C.; Miao, Z.; Bi, X.; Wu, D.; Jin, N.; Wang, L.; Wu, H.; Qian, K.; Li, C.; et al. ViRBase: A Resource for Virus–Host NcRNA-Associated Interactions. Nucleic Acids Res. 2015, 43, D578–D582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Fan, Y.; Zhang, Z.; Lu, C.; Zhu, Z.; Jiang, T.; Shan, T.; Peng, Y. VirusCircBase: A Database of Virus Circular RNAs. Brief. Bioinform. 2021, 22, 2182–2190. [Google Scholar] [CrossRef]
- Toden, S.; Zumwalt, T.J.; Goel, A. Non-Coding RNAs and Potential Therapeutic Targeting in Cancer. Biochim. Biophys. Acta BBA Rev. Cancer 2021, 1875, 188491. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.-Y.; Ju, C.J.-T.; Hao, J.; Chen, M.; Wang, W. JEDI: Circular RNA Prediction Based on Junction Encoders and Deep Interaction among Splice Sites. Bioinformatics 2021, 37, i289–i298. [Google Scholar] [CrossRef]
- Chaabane, M.; Williams, R.M.; Stephens, A.T.; Park, J.W. CircDeep: Deep Learning Approach for Circular RNA Classification from Other Long Non-Coding RNA. Bioinformatics 2020, 36, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Chaabane, M.; Andreeva, K.; Hwang, J.Y.; Kook, T.L.; Park, J.W.; Cooper, N.G.F. SeekCRIT: Detecting and Characterizing Differentially Expressed Circular RNAs Using High-Throughput Sequencing Data. PLoS Comput. Biol. 2020, 16, e1008338. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A CeRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.-D.; Shi, Y.; Liu, J.-B.; Yang, X.-L.; Xin, R.; Wang, H.-M.; Wang, P.-Y.; Jia, C.-Y.; Zhang, W.-J.; Ma, Y.-S.; et al. Construction of a Myc-Associated CeRNA Network Reveals a Prognostic Signature in Hepatocellular Carcinoma. Mol. Ther. Nucleic Acids 2021, 24, 1033–1050. [Google Scholar] [CrossRef]
- Wang, Q.-C.; Wang, Z.-Y.; Xu, Q.; Chen, X.-L.; Shi, R.-Z. LncRNA Expression Profiles and Associated CeRNA Network Analyses in Epicardial Adipose Tissue of Patients with Coronary Artery Disease. Sci. Rep. 2021, 11, 1567. [Google Scholar] [CrossRef]
- Li, J.; Qian, Y.; Zhang, C.; Wang, W.; Qiao, Y.; Song, H.; Li, L.; Guo, J.; Lu, D.; Deng, X. LncRNA LINC00473 Is Involved in the Progression of Invasive Pituitary Adenoma by Upregulating KMT5A via CeRNA-Mediated MiR-502-3p Evasion. Cell Death Dis. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Yang, S.L.; DeFalco, L.; Anderson, D.E.; Zhang, Y.; Aw, J.G.A.; Lim, S.Y.; Lim, X.N.; Tan, K.Y.; Zhang, T.; Chawla, T.; et al. Comprehensive Mapping of SARS-CoV-2 Interactions in Vivo Reveals Functional Virus-Host Interactions. Nat. Commun. 2021, 12, 5113. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Moshitch-Moshkovitz, S.; Han, D.; Kol, N.; Amariglio, N.; Rechavi, G.; Dominissini, D.; He, C. Nm-Seq Maps 2′-O-Methylation Sites in Human MRNA with Base Precision. Nat. Methods 2017, 14, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Kern, F.; Fehlmann, T.; Violich, I.; Alsop, E.; Hutchins, E.; Kahraman, M.; Grammes, N.L.; Guimarães, P.; Backes, C.; Poston, K.L.; et al. Deep Sequencing of SncRNAs Reveals Hallmarks and Regulatory Modules of the Transcriptome during Parkinson’s Disease Progression. Nat. Aging 2021, 1, 309–322. [Google Scholar] [CrossRef]
- Beal, M.F. Mitochondria, Oxidative Damage, and Inflammation in Parkinson’s Disease. Ann.-N. Y. Acad. Sci. 2003, 991, 120–131. [Google Scholar] [CrossRef]
- Basu, M.; Wang, K.; Ruppin, E.; Hannenhalli, S. Predicting Tissue-Specific Gene Expression from Whole Blood Transcriptome. Sci. Adv. 2021, 7, eabd6991. [Google Scholar] [CrossRef]
- Hücker, S.M.; Fehlmann, T.; Werno, C.; Weidele, K.; Lüke, F.; Schlenska-Lange, A.; Klein, C.A.; Keller, A.; Kirsch, S. Single-Cell MicroRNA Sequencing Method Comparison and Application to Cell Lines and Circulating Lung Tumor Cells. Nat. Commun. 2021, 12, 4316. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, J.; Sen, S. MicroRNA as Biomarkers and Diagnostics. J. Cell. Physiol. 2016, 231, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Ganini, C.; Candi, E.; Melino, G. The Role of Noncoding RNAs in Epithelial Cancer. Cell Death Discov. 2020, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Sun, L.; Wen, S.; Deng, D.; Wan, F.; He, X.; Tian, L.; Liang, L.; Wei, C.; Gao, K.; et al. RNA Sequencing of Plasma Exosomes Revealed Novel Functional Long Noncoding RNAs in Hepatocellular Carcinoma. Cancer Sci. 2020, 111, 3338–3349. [Google Scholar] [CrossRef]
- Khan, I.A.; Rashid, S.; Singh, N.; Rashid, S.; Singh, V.; Gunjan, D.; Das, P.; Dash, N.R.; Pandey, R.M.; Chauhan, S.S.; et al. Panel of Serum MiRNAs as Potential Non-Invasive Biomarkers for Pancreatic Ductal Adenocarcinoma. Sci. Rep. 2021, 11, 2824. [Google Scholar] [CrossRef]
- Hulstaert, E.; Morlion, A.; Cobos, F.A.; Verniers, K.; Nuytens, J.; Eynde, E.V.; Yigit, N.; Anckaert, J.; Geerts, A.; Hindryckx, P.; et al. Charting Extracellular Transcriptomes in The Human Biofluid RNA Atlas. Cell Rep. 2020, 33. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, X.; Jing, S.; Zhang, H.; Zhang, Y. Non-Coding RNAs and Retroviruses. Retrovirology 2018, 15, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern-Ginossar, N.; Elefant, N.; Zimmermann, A.; Wolf, D.G.; Saleh, N.; Biton, M.; Horwitz, E.; Prokocimer, Z.; Prichard, M.; Hahn, G.; et al. Host Immune System Gene Targeting by a Viral MiRNA. Science 2007, 317, 376–381. [Google Scholar] [CrossRef] [Green Version]
- Tycowski, K.T.; Guo, Y.E.; Lee, N.; Moss, W.N.; Vallery, T.K.; Xie, M.; Steitz, J.A. Viral Noncoding RNAs: More Surprises. Genes Dev. 2015, 29, 567–584. [Google Scholar] [CrossRef] [Green Version]
- Gorbea, C.; Mosbruger, T.; Cazalla, D. A Viral Sm-Class RNA Base-Pairs with MRNAs and Recruits MicroRNAs to Inhibit Apoptosis. Nature 2017, 550, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.N.; Ronk, A.J.; Widen, S.G.; Wood, T.G.; Basler, C.F.; Bukreyev, A. Ebola Virus Produces Discrete Small Noncoding RNAs Independently of the Host MicroRNA Pathway Which Lack RNA Interference Activity in Bat and Human Cells. J. Virol. 2020, 94, e01441-19. [Google Scholar] [CrossRef]
- Dunn, L.E.M.; Ivens, A.; Netherton, C.L.; Chapman, D.A.G.; Beard, P.M. Identification of a Functional Small Noncoding RNA of African Swine Fever Virus. J. Virol. 2020, 94, e01515-20. [Google Scholar] [CrossRef] [PubMed]
- Mason-D’Croz, D.; Bogard, J.R.; Herrero, M.; Robinson, S.; Sulser, T.B.; Wiebe, K.; Willenbockel, D.; Godfray, H.C.J. Modelling the Global Economic Consequences of a Major African Swine Fever Outbreak in China. Nat. Food 2020, 1, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Shakya, M.; Lo, C.-C.; Chain, P.S.G. Advances and Challenges in Metatranscriptomic Analysis. Front. Genet. 2019, 10, 904. [Google Scholar] [CrossRef] [Green Version]
- Bodor, A.; Bounedjoum, N.; Vincze, G.E.; Erdeiné Kis, Á.; Laczi, K.; Bende, G.; Szilágyi, Á.; Kovács, T.; Perei, K.; Rákhely, G. Challenges of Unculturable Bacteria: Environmental Perspectives. Rev. Environ. Sci. Biotechnol. 2020, 19, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Browne, H.P.; Forster, S.C.; Anonye, B.O.; Kumar, N.; Neville, B.A.; Stares, M.D.; Goulding, D.; Lawley, T.D. Culturing of ‘Unculturable’ Human Microbiota Reveals Novel Taxa and Extensive Sporulation. Nature 2016, 533, 543–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelsinger, D.R.; Uritskiy, G.; Reddy, R.; Munn, A.; Farney, K.; DiRuggiero, J. Regulatory Noncoding Small RNAs Are Diverse and Abundant in an Extremophilic Microbial Community. mSystems 2020, 5, e0058419. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.-W.; Um, J.-H.; Cho, J.-H.; Lee, H.-J. Tiny RNAs and Their Voyage via Extracellular Vesicles: Secretion of Bacterial Small RNA and Eukaryotic MicroRNA. Exp. Biol. Med. 2017, 242, 1475–1481. [Google Scholar] [CrossRef]
- Delihas, N. Discovery and Characterization of the First Non-Coding RNA That Regulates Gene Expression, MicF RNA: A Historical Perspective. World J. Biol. Chem. 2015, 6, 272–280. [Google Scholar] [CrossRef]
- Mizuno, T.; Chou, M.Y.; Inouye, M. A Unique Mechanism Regulating Gene Expression: Translational Inhibition by a Complementary RNA Transcript (MicRNA). Proc. Natl. Acad. Sci. USA 1984, 81, 1966–1970. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.H.; Kim, J.-H. Cis-Encoded Non-Coding Antisense RNAs in Streptococci and Other Low GC Gram (+) Bacterial Pathogens. Front. Genet. 2015, 6, 110. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Fang, Y.; Chen, L.; Wang, J.; Chen, X. Role of Non-Coding RNAs in Plant Immunity. Plant Commun. 2021, 2, 100180. [Google Scholar] [CrossRef] [PubMed]
- Bhogireddy, S.; Mangrauthia, S.K.; Kumar, R.; Pandey, A.K.; Singh, S.; Jain, A.; Budak, H.; Varshney, R.K.; Kudapa, H. Regulatory Non-Coding RNAs: A New Frontier in Regulation of Plant Biology. Funct. Integr. Genomics 2021, 21, 313–330. [Google Scholar] [CrossRef] [PubMed]
- Jannesar, M.; Seyedi, S.M.; Moazzam Jazi, M.; Niknam, V.; Ebrahimzadeh, H.; Botanga, C. A Genome-Wide Identification, Characterization and Functional Analysis of Salt-Related Long Non-Coding RNAs in Non-Model Plant Pistacia Vera L. Using Transcriptome High Throughput Sequencing. Sci. Rep. 2020, 10, 5585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cui, J.; Dai, C.; Liu, T.; Cheng, D.; Luo, C. Whole-Transcriptome RNA Sequencing Reveals the Global Molecular Responses and CeRNA Regulatory Network of MRNAs, LncRNAs, MiRNAs and CircRNAs in Response to Salt Stress in Sugar Beet (Beta Vulgaris). Int. J. Mol. Sci. 2020, 22, 289. [Google Scholar] [CrossRef]
- Chen, Y.; Cheng, C.; Feng, X.; Lai, R.; Gao, M.; Chen, W.; Wu, R. Integrated Analysis of LncRNA and MRNA Transcriptomes Reveals the Potential Regulatory Role of LncRNA in Kiwifruit Ripening and Softening. Sci. Rep. 2021, 11, 1671. [Google Scholar] [CrossRef]
- Zhao, X.; Li, J.; Lian, B.; Gu, H.; Li, Y.; Qi, Y. Global Identification of Arabidopsis LncRNAs Reveals the Regulation of MAF4 by a Natural Antisense RNA. Nat. Commun. 2018, 9, 5056. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Su, T.; Li, P.; Xin, X.; Cao, Y.; Wang, W.; Zhao, X.; Zhang, D.; Yu, Y.; Li, D.; et al. Identification of Long Noncoding RNAs Involved in Resistance to Downy Mildew in Chinese Cabbage. Hortic. Res. 2021, 8, 44. [Google Scholar] [CrossRef]
- Wang, L.-L.; Jin, J.-J.; Li, L.-H.; Qu, S.-H. Long Non-Coding RNAs Responsive to Blast Fungus Infection in Rice. Rice N. Y. N 2020, 13, 77. [Google Scholar] [CrossRef]
- Verstraeten, B.; Atighi, M.R.; Ruiz-Ferrer, V.; Escobar, C.; Meyer, T.; Kyndt, T. Non-Coding RNAs in the Interaction between Rice and Meloidogyne Graminicola. BMC Genomics 2021, 22, 560. [Google Scholar] [CrossRef]
- Song, X.; Hu, J.; Wu, T.; Yang, Q.; Feng, X.; Lin, H.; Feng, S.; Cui, C.; Yu, Y.; Zhou, R.; et al. Comparative Analysis of Long Noncoding RNAs in Angiosperms and Characterization of Long Noncoding RNAs in Response to Heat Stress in Chinese Cabbage. Hortic. Res. 2021, 8, 48. [Google Scholar] [CrossRef]
- Ariel, F.; Lucero, L.; Christ, A.; Mammarella, M.F.; Jegu, T.; Veluchamy, A.; Mariappan, K.; Latrasse, D.; Blein, T.; Liu, C.; et al. R-Loop Mediated Trans Action of the APOLO Long Noncoding RNA. Mol. Cell 2020, 77, 1055–1065.e4. [Google Scholar] [CrossRef] [PubMed]
- Kirov, I.; Dudnikov, M.; Merkulov, P.; Shingaliev, A.; Omarov, M.; Kolganova, E.; Sigaeva, A.; Karlov, G.; Soloviev, A. Nanopore RNA Sequencing Revealed Long Non-Coding and LTR Retrotransposon-Related RNAs Expressed at Early Stages of Triticale SEED Development. Plants 2020, 9. [Google Scholar] [CrossRef]
- Rai, M.I.; Alam, M.; Lightfoot, D.A.; Gurha, P.; Afzal, A.J. Classification and Experimental Identification of Plant Long Non-Coding RNAs. Genomics 2019, 111, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Budak, H.; Kaya, S.B.; Cagirici, H.B. Long Non-Coding RNA in Plants in the Era of Reference Sequences. Front. Plant Sci. 2020, 11, 276. [Google Scholar] [CrossRef] [Green Version]
- Deng, P.; Liu, S.; Nie, X.; Weining, S.; Wu, L. Conservation Analysis of Long Non-Coding RNAs in Plants. Sci. China Life Sci. 2018, 61, 190–198. [Google Scholar] [CrossRef]
- Meng, J.; Kang, Q.; Chang, Z.; Luan, Y. PlncRNA-HDeep: Plant Long Noncoding RNA Prediction Using Hybrid Deep Learning Based on Two Encoding Styles. BMC Bioinform. 2021, 22, 242. [Google Scholar] [CrossRef]
- Cagirici, H.B.; Galvez, S.; Sen, T.Z.; Budak, H. LncMachine: A Machine Learning Algorithm for Long Noncoding RNA Annotation in Plants. Funct. Integr. Genom. 2021, 21, 195–204. [Google Scholar] [CrossRef]
- Ye, C.-Y.; Chen, L.; Liu, C.; Zhu, Q.-H.; Fan, L. Widespread Noncoding Circular RNAs in Plants. New Phytol. 2015, 208, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Tian, X.; Zhang, J.; Sun, P.; Li, G. PCirc: Random Forest-Based Plant CircRNA Identification Software. BMC Bioinform. 2021, 22, 10. [Google Scholar] [CrossRef]
- Zhang, P.; Liu, Y.; Chen, H.; Meng, X.; Xue, J.; Chen, K.; Chen, M. CircPlant: An Integrated Tool for CircRNA Detection and Functional Prediction in Plants. Genom. Proteom. Bioinform. 2020, 18, 352–358. [Google Scholar] [CrossRef]
- Balarezo-Cisneros, L.N.; Parker, S.; Fraczek, M.G.; Timouma, S.; Wang, P.; O’Keefe, R.T.; Millar, C.B.; Delneri, D. Functional and Transcriptional Profiling of Non-Coding RNAs in Yeast Reveal Context-Dependent Phenotypes and in Trans Effects on the Protein Regulatory Network. PLOS Genet. 2021, 17, e1008761. [Google Scholar] [CrossRef]
- Parker, S.; Fraczek, M.G.; Wu, J.; Shamsah, S.; Manousaki, A.; Dungrattanalert, K.; de Almeida, R.A.; Invernizzi, E.; Burgis, T.; Omara, W.; et al. Large-Scale Profiling of Noncoding RNA Function in Yeast. PLOS Genet. 2018, 14, e1007253. [Google Scholar] [CrossRef] [Green Version]
- Donaldson, M.E.; Ostrowski, L.A.; Goulet, K.M.; Saville, B.J. Transcriptome Analysis of Smut Fungi Reveals Widespread Intergenic Transcription and Conserved Antisense Transcript Expression. BMC Genom. 2017, 18, 340. [Google Scholar] [CrossRef] [PubMed]
- Yassour, M.; Pfiffner, J.; Levin, J.Z.; Adiconis, X.; Gnirke, A.; Nusbaum, C.; Thompson, D.-A.; Friedman, N.; Regev, A. Strand-Specific RNA Sequencing Reveals Extensive Regulated Long Antisense Transcripts That Are Conserved across Yeast Species. Genome Biol. 2010, 11, R87. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Jones, T.A.; Rivas, E. Discovery of 17 Conserved Structural RNAs in Fungi. Nucleic Acids Res. 2021, 49, 6128–6143. [Google Scholar] [CrossRef] [PubMed]
- Parra-Rivero, O.; Pardo-Medina, J.; Gutiérrez, G.; Limón, M.C.; Avalos, J. A Novel LncRNA as a Positive Regulator of Carotenoid Biosynthesis in Fusarium. Sci. Rep. 2020, 10, 678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandmann, G. Carotenoids of Biotechnological Importance. In Biotechnology of Isoprenoids; Schrader, J., Bohlmann, J., Eds.; Advances in Biochemical Engineering/Biotechnology; Springer International Publishing: Cham, Switzerland, 2015; pp. 449–467. ISBN 978-3-319-20107-8. [Google Scholar]
- Li, Y.; Baptista, R.P.; Kissinger, J.C. Noncoding RNAs in Apicomplexan Parasites: An Update. Trends Parasitol. 2020, 36, 835–849. [Google Scholar] [CrossRef]
- Li, Y.; Baptista, R.P.; Sateriale, A.; Striepen, B.; Kissinger, J.C. Analysis of Long Non-Coding RNA in Cryptosporidium Parvum Reveals Significant Stage-Specific Antisense Transcription. Front. Cell. Infect. Microbiol. 2021, 10, 833. [Google Scholar] [CrossRef]
- Bracken-Grissom, H.; Collins, A.G.; Collins, T.; Crandall, K.; Distel, D.; Dunn, C.; Giribet, G.; Haddock, S.; Knowlton, N.; et al.; GIGA Community of Scientists The Global Invertebrate Genomics Alliance (GIGA): Developing Community Resources to Study Diverse Invertebrate Genomes. J. Hered. 2014, 105, 1–18. [Google Scholar] [CrossRef]
- Panwar, B.; Arora, A.; Raghava, G.P. Prediction and Classification of NcRNAs Using Structural Information. BMC Genom. 2014, 15, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, L.; Xi, Y.; Qiao, X.; Huang, C.; Wu, Q.; Yang, N.; Guo, J.; Liu, W.; Fan, W.; Wan, F.; et al. The Landscape of LncRNAs in Cydia Pomonella Provides Insights into Their Signatures and Potential Roles in Transcriptional Regulation. BMC Genom. 2021, 22, 4. [Google Scholar] [CrossRef]
- Shin, G.; Koo, H.J.; Seo, M.; Lee, S.-J.V.; Nam, H.G.; Jung, G.Y. Transfer RNA-Derived Fragments in Aging Caenorhabditis Elegans Originate from Abundant Homologous Gene Copies. Sci. Rep. 2021, 11, 12304. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Lluch, S.; Klein, C.C.; Breschi, A.; Ruiz-Romero, M.; Abad, A.; Palumbo, E.; Bekish, L.; Arnan, C.; Guigó, R. BsAS, an Antisense Long Non-Coding RNA, Essential for Correct Wing Development through Regulation of Blistered/DSRF Isoform Usage. PLOS Genet. 2021, 16, e1009245. [Google Scholar] [CrossRef]
- Azlan, A.; Obeidat, S.M.; Theva Das, K.; Yunus, M.A.; Azzam, G. Genome-Wide Identification of Aedes Albopictus Long Noncoding RNAs and Their Association with Dengue and Zika Virus Infection. PLoS Negl. Trop. Dis. 2021, 15, e0008351. [Google Scholar] [CrossRef]
- Salabi, F.; Jafari, H.; Navidpour, S.; Sadr, A.S. Systematic and Computational Identification of Androctonus Crassicauda Long Non-Coding RNAs. Sci. Rep. 2021, 11, 4720. [Google Scholar] [CrossRef]
- Zhang, W.; Qin, P.; Gong, X.; Huang, L.; Wang, C.; Chen, G.; Chen, J.; Wang, L.; Lv, Z. Identification of CircRNAs in the Liver of Whitespotted Bamboo Shark (Chiloscyllium Plagiosum). Front. Genet. 2020, 11, 1618. [Google Scholar] [CrossRef]
- Ma, X.; Cen, S.; Wang, L.; Zhang, C.; Wu, L.; Tian, X.; Wu, Q.; Li, X.; Wang, X. Genome-Wide Identification and Comparison of Differentially Expressed Profiles of MiRNAs and LncRNAs with Associated CeRNA Networks in the Gonads of Chinese Soft-Shelled Turtle, Pelodiscus Sinensis. BMC Genom. 2020, 21, 443. [Google Scholar] [CrossRef]
- Ren, J.; Li, Q.; Zhang, Q.; Clinton, M.; Sun, C.; Yang, N. Systematic Screening of Long Intergenic Noncoding RNAs Expressed during Chicken Embryogenesis. Poult. Sci. 2021, 100, 101160. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, W.; Zhang, B.; Ling, Y.; Kim, W.K.; Zhang, H. Comprehensive Analysis of Coding and Non-Coding RNA Transcriptomes Related to Hypoxic Adaptation in Tibetan Chickens. J. Anim. Sci. Biotechnol. 2021, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Jehl, F.; Muret, K.; Bernard, M.; Boutin, M.; Lagoutte, L.; Désert, C.; Dehais, P.; Esquerré, D.; Acloque, H.; Giuffra, E.; et al. An Integrative Atlas of Chicken Long Non-Coding Genes and Their Annotations across 25 Tissues. Sci. Rep. 2020, 10, 20457. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jin, W.; Zhai, B.; Chen, Y.; Li, G.; Zhang, Y.; Guo, Y.; Sun, G.; Han, R.; Li, Z.; et al. LncRNAs and Their Regulatory Networks in Breast Muscle Tissue of Chinese Gushi Chickens during Late Postnatal Development. BMC Genom. 2021, 22, 44. [Google Scholar] [CrossRef]
- Li, H.; Cui, P.; Fu, X.; Zhang, L.; Yan, W.; Zhai, Y.; Lei, C.; Wang, H.; Yang, X. Identification and Analysis of Long Non-Coding RNAs and MRNAs in Chicken Macrophages Infected with Avian Infectious Bronchitis Coronavirus. BMC Genom. 2021, 22, 67. [Google Scholar] [CrossRef]
- Mottet, A.; de Haan, C.; Falcucci, A.; Tempio, G.; Opio, C.; Gerber, P. Livestock: On Our Plates or Eating at Our Table? A New Analysis of the Feed/Food Debate. Glob. Food Secur. 2017, 14, 1–8. [Google Scholar] [CrossRef]
- Nolte, W.; Weikard, R.; Brunner, R.M.; Albrecht, E.; Hammon, H.M.; Reverter, A.; Kühn, C. Biological Network Approach for the Identification of Regulatory Long Non-Coding RNAs Associated With Metabolic Efficiency in Cattle. Front. Genet. 2019, 10, 1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Huang, K.; Wang, P.; Feng, T.; Shi, D.; Cui, K.; Luo, C.; Shafique, L.; Qian, Q.; Ruan, J.; et al. Comparison of Long Non-Coding RNA Expression Profiles of Cattle and Buffalo Differing in Muscle Characteristics. Front. Genet. 2020, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; He, Y.; Chen, S.-Y.; Wang, J.; Hu, S.; Lai, S.-J. Genome-Wide Identification and Characterisation of Long Non-Coding RNAs in Two Chinese Cattle Breeds. Ital. J. Anim. Sci. 2020, 19, 383–391. [Google Scholar] [CrossRef]
- Yan, X.-M.; Zhang, Z.; Meng, Y.; Li, H.-B.; Gao, L.; Luo, D.; Jiang, H.; Gao, Y.; Yuan, B.; Zhang, J.-B. Genome-Wide Identification and Analysis of Circular RNAs Differentially Expressed in the Longissimus Dorsi between Kazakh Cattle and Xinjiang Brown Cattle. PeerJ 2020, 8, e8646. [Google Scholar] [CrossRef]
- Amiri Yekta, A.; Dalman, A.; Eftekhari-Yazdi, P.; Sanati, M.H.; Shahverdi, A.H.; Fakheri, R.; Vazirinasab, H.; Daneshzadeh, M.T.; Vojgani, M.; Zomorodipour, A.; et al. Production of Transgenic Goats Expressing Human Coagulation Factor IX in the Mammary Glands after Nuclear Transfer Using Transfected Fetal Fibroblast Cells. Transgenic Res. 2013, 22, 131–142. [Google Scholar] [CrossRef]
- Deng, M.; Wan, Y.; Chen, B.; Dai, X.; Liu, Z.; Yang, Y.; Cai, Y.; Zhang, Y.; Wang, F. Long Non-Coding RNA Lnc_3712 Impedes Nuclear Reprogramming via Repressing Kdm5b. Mol. Ther. Nucleic Acids 2021, 24, 54–66. [Google Scholar] [CrossRef]
- Orlando, L.; Allaby, R.; Skoglund, P.; Der Sarkissian, C.; Stockhammer, P.W.; Ávila-Arcos, M.C.; Fu, Q.; Krause, J.; Willerslev, E.; Stone, A.C.; et al. Ancient DNA Analysis. Nat. Rev. Methods Primer 2021, 1, 1–26. [Google Scholar] [CrossRef]
- Zwir, I.; Del-Val, C.; Hintsanen, M.; Cloninger, K.M.; Romero-Zaliz, R.; Mesa, A.; Arnedo, J.; Salas, R.; Poblete, G.F.; Raitoharju, E.; et al. Evolution of Genetic Networks for Human Creativity. Mol. Psychiatry 2021, 1–23. [Google Scholar] [CrossRef]
- Rahimi, K.; Venø, M.T.; Dupont, D.M.; Kjems, J. Nanopore Sequencing of Brain-Derived Full-Length CircRNAs Reveals CircRNA-Specific Exon Usage, Intron Retention and Microexons. Nat. Commun. 2021, 12, 4825. [Google Scholar] [CrossRef] [PubMed]
- van Nimwegen, K.J.M.; van Soest, R.A.; Veltman, J.A.; Nelen, M.R.; van der Wilt, G.J.; Vissers, L.E.L.M.; Grutters, J.P.C. Is the $1000 Genome as Near as We Think? A Cost Analysis of Next-Generation Sequencing. Clin. Chem. 2016, 62, 1458–1464. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Seki, M. Recent Advances in the Detection of Base Modifications Using the Nanopore Sequencer. J. Hum. Genet. 2020, 65, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Szabo, L.; Salzman, J. Detecting Circular RNAs: Bioinformatic and Experimental Challenges. Nat. Rev. Genet. 2016, 17, 679–692. [Google Scholar] [CrossRef] [Green Version]
- Behrens, A.; Rodschinka, G.; Nedialkova, D.D. High-Resolution Quantitative Profiling of TRNA Abundance and Modification Status in Eukaryotes by Mim-TRNAseq. Mol. Cell 2021, 81, 1802–1815.e7. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.K.; Poodari, V.C.; Jain, M.; Olsen, H.E.; Akeson, M.; Abu-Shumays, R.L. Direct Nanopore Sequencing of Individual Full Length TRNA Strands. ACS Nano 2021, 15, 16642–16653. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.M.; Salinas-Giegé, T.; Hummel, G.; Coots, N.L.; Svendsen, J.M.; Brown, K.C.; Drouard, L.; Sloan, D.B. Combining TRNA Sequencing Methods to Characterize Plant TRNA Expression and Post-Transcriptional Modification. RNA Biol. 2021, 18, 64–78. [Google Scholar] [CrossRef]
- Lambert, M.; Benmoussa, A.; Provost, P. Small Non-Coding RNAs Derived from Eukaryotic Ribosomal RNA. Non-Coding RNA 2019, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Goethem, A.; Yigit, N.; Everaert, C.; Moreno-Smith, M.; Mus, L.M.; Barbieri, E.; Speleman, F.; Mestdagh, P.; Shohet, J.; Van Maerken, T.; et al. Depletion of TRNA-Halves Enables Effective Small RNA Sequencing of Low-Input Murine Serum Samples. Sci. Rep. 2016, 6, 37876. [Google Scholar] [CrossRef]
- Vromman, M.; Vandesompele, J.; Volders, P.-J. Closing the Circle: Current State and Perspectives of Circular RNA Databases. Brief. Bioinform. 2021, 22, 288–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosar, J.P.; Rovira, C.; Cayota, A. Non-Coding RNA Fragments Account for the Majority of Annotated PiRNAs Expressed in Somatic Non-Gonadal Tissues. Commun. Biol. 2018, 1, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosar, J.P.; García-Silva, M.R.; Cayota, A. Circulating SNORD57 Rather than PiR-54265 Is a Promising Biomarker for Colorectal Cancer: Common Pitfalls in the Study of Somatic PiRNAs in Cancer. RNA 2021, 27, 403–410. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Name | Species | Database Content | URL | Refs. |
|---|---|---|---|---|
| AlnC | 682 Angiosperms | lncRNA | http://www.nipgr.ac.in/AlnC | [34] |
| ASRA | 21 Vertebrates | Circulating sncRNA | https://ccb-web.cs.uni-saarland.de/asra/ | [35] |
| circ2Go | Human | circRNA | https://circ2go.dkfz.de/ | [36] |
| CircAtlas 2.0 | 6 Vertebrates | circRNA | http://circatlas.biols.ac.cn/ | [37] |
| circBank | Human | circRNA | http://www.circbank.cn/ | [38] |
| circBase | 3 Vertebrates 2 Invertebrates | circRNA | http://www.circbase.org/ | [39] |
| circFunBase | 7 Plants 8 Vertebrates 1 Invertebrate | circRNA | http://bis.zju.edu.cn/CircFunBase/index.php | [40] |
| CIRCpedia 2.0 | 4 Vertebrates 2 Invertebrates | circRNA | https://www.picb.ac.cn/rnomics/circpedia/ | [41] |
| DeepBase v3.0 | 11 Vertebrates 2 Invertebrates | ncRNA | https://rna.sysu.edu.cn/deepbase3/index.html | [42] |
| DIANA-lncBase v.3 | 2 Vertebrates | miRNA targets on non-coding transcripts | https://diana.e-ce.uth.gr/lncbasev3/home | [43] |
| DIANA-Tarbase v8 | 2 Viruses 7 Plants 2 Invertebrates 7 Vertebrates | miRNA-gene interactions | http://www.microrna.gr/tarbase | [44] |
| EVAtlas | Human | Extracellular vesicle ncRNA | http://bioinfo.life.hust.edu.cn/EVAtlas/ | [45] |
| exoRBase 2.0 | Human | Exosome ncRNA | http://www.exorbase.org/ | [46] |
| GTEx version 8 | Human (tissue) | Transcriptome | https://gtexportal.org/home/ | [47] |
| Lantern | Human | lncRNA | https://sysbio.lab.iupui.edu/lantern/ | [48] |
| LncATLAS | Human | lncRNA subcellular localization | https://lncatlas.crg.eu/ | [49] |
| LncBook | Human | lncRNA | https://ngdc.cncb.ac.cn/lncbook/index | [50] |
| LncExpDB | Human | lncRNA | https://ngdc.cncb.ac.cn/lncexpdb/ | [51] |
| LNCipedia 5 | Human | lncRNA | https://lncipedia.org/ | [52] |
| lncRNAKB | Human | lncRNA | http://psychiatry.som.jhmi.edu/lncrnakb/ | [53] |
| LncSEA | Human | lncRNA | http://bio.liclab.net/LncSEA/index.php | [54] |
| MINTbase 2.0 | Human | tRF | https://cm.jefferson.edu/MINTbase/ | [55] |
| miRBase | 271 organisms | miRNA | https://www.mirbase.org/ | [56] |
| MiREDiBase | 4 Vertebrates | Editing events in miRNA | https://ncrnaome.osumc.edu/miredibase/ | [57] |
| MirGeneDB 2.0 | 22 Vertebrates 23 Invertebrates | miRNA | https://mirgenedb.org/ | [58] |
| mirTarBase 9.0 | 16 Vertebrates 3 Invertebrates 5 Plants 3 Viruses | miRNA-target interactions | https://mirtarbase.cuhk.edu.cn/~miRTarBase/miRTarBase_2022/php/index.php | [59] |
| miRWalk | 6 Vertebrates | miRNA binding sites | http://mirwalk.umm.uni-heidelberg.de/ | [60] |
| NONCODE v6.0 | 23 Plants 1 Fungus 2 Invertebrates 13 Vertebrates | ncRNA | http://www.noncode.org/index.php | [61] |
| riboCIRC | 2 Invertebrates 4 Vertebrates | Translatable circRNA | http://www.ribocirc.com/ | [62] |
| Rfam 14.6 | Various | RNA families | https://rfam.org/ | [63] |
| RNA Atlas | Human (tissue and cell lines) | Transcriptome | http://r2platform.com/rna_atlas | [64] |
| piRBase 3.0 | 28 Invertebrates 16 Vertebrates | piRNA | http://bigdata.ibp.ac.cn/piRBase/ | [65] |
| piRNAclusterDB 2.0 | 23 Invertebrates 28 Vertebrates | piRNA clusters | https://www.smallrnagroup.uni-mainz.de/piRNAclusterDB/ | [66] |
| piRTarBase | 2 Invertebrates | piRNA targeting sites | http://cosbi6.ee.ncku.edu.tw/piRTarBase/ | [67] |
| PlantcircBase 6.0 | 20 Plants | circRNA | http://ibi.zju.edu.cn/plantcircbase/index.php | [68] |
| PtRFdb | 10 Plants | tRF | http://14.139.61.8/PtRFdb/index.php | [69] |
| snoDB | Human | snoRNA | http://scottgroup.med.usherbrooke.ca/snoDB/ | [70] |
| TarDB | 43 Plants | miRNA targets | http://www.biosequencing.cn/TarDB/ | [71] |
| TransCirc | Human | Translatable circRNA | https://www.biosino.org/transcirc/ | [72] |
| TSCD | 2 Vertebrates | circRNA | http://gb.whu.edu.cn/TSCD/ | [73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Micheel, J.; Safrastyan, A.; Wollny, D. Advances in Non-Coding RNA Sequencing. Non-Coding RNA 2021, 7, 70. https://doi.org/10.3390/ncrna7040070
Micheel J, Safrastyan A, Wollny D. Advances in Non-Coding RNA Sequencing. Non-Coding RNA. 2021; 7(4):70. https://doi.org/10.3390/ncrna7040070
Chicago/Turabian StyleMicheel, Julia, Aram Safrastyan, and Damian Wollny. 2021. "Advances in Non-Coding RNA Sequencing" Non-Coding RNA 7, no. 4: 70. https://doi.org/10.3390/ncrna7040070
APA StyleMicheel, J., Safrastyan, A., & Wollny, D. (2021). Advances in Non-Coding RNA Sequencing. Non-Coding RNA, 7(4), 70. https://doi.org/10.3390/ncrna7040070