Endogenous Double-Stranded RNA

{kind=link}
{kind=link}
{kind=link}
Abstract
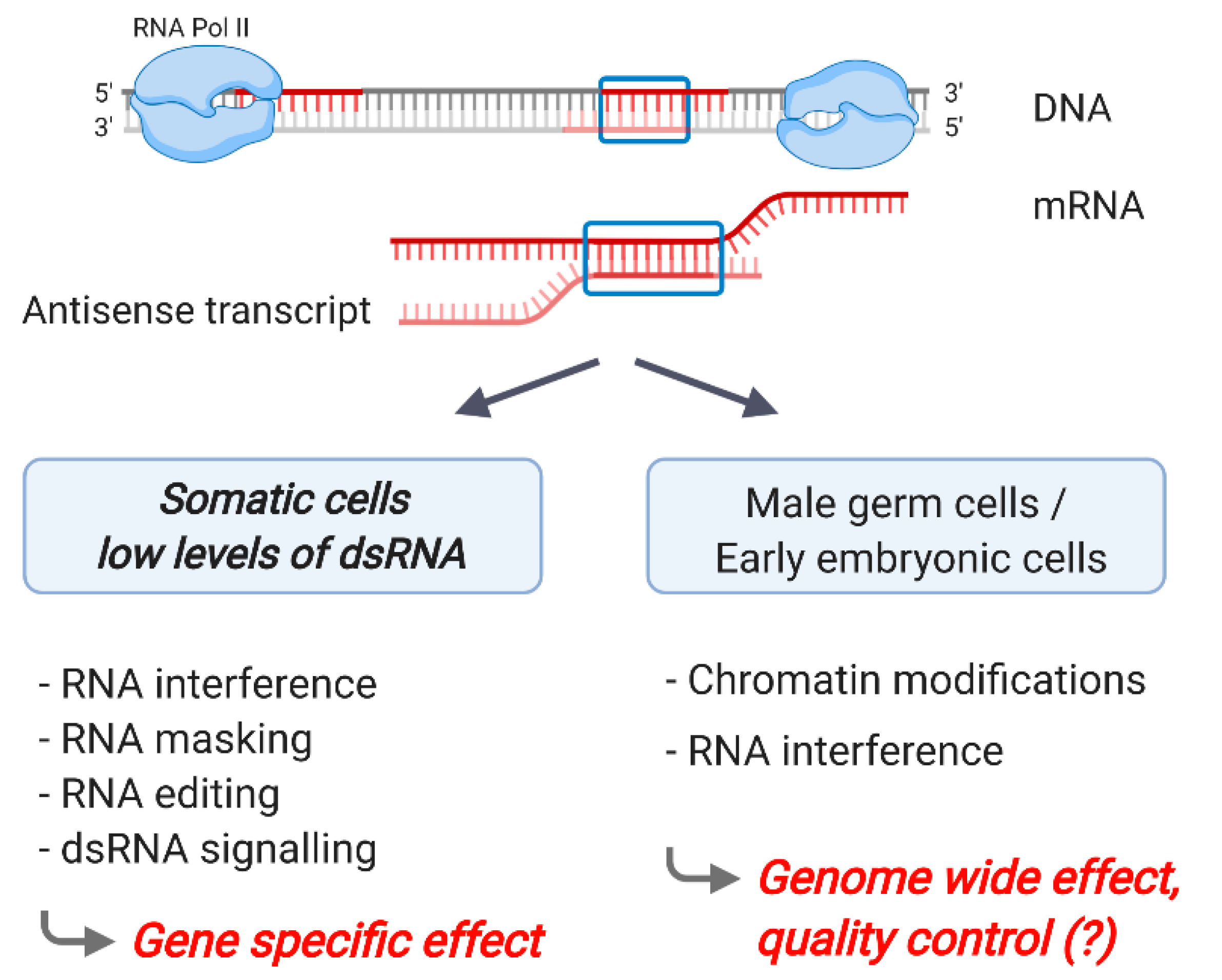
1. Introduction
2. Sources of Endogenous dsRNA
2.1. Mitochondrial Transcripts
2.2. Repetitive DNA Sequences
3. Proteins Binding dsRNA
4. Physiological and Pathophysiological Roles of dsRNA
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- de Koning, A.P.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011, 7, e1002384. [Google Scholar] [CrossRef] [PubMed]
- Haubold, B.; Wiehe, T. How repetitive are genomes? BMC Bioinform. 2006, 7, 541. [Google Scholar] [CrossRef]
- Canapa, A.; Barucca, M.; Biscotti, M.A.; Forconi, M.; Olmo, E. Transposons, Genome Size, and Evolutionary Insights in Animals. Cytogenet. Genome Res. 2015, 147, 217–239. [Google Scholar] [CrossRef] [PubMed]
- Kidwell, M.G.; Lisch, D.R. Transposable elements and host genome evolution. Trends Ecol. Evol. 2000, 15, 95–99. [Google Scholar] [CrossRef]
- Beck, C.R.; Collier, P.; Macfarlane, C.; Malig, M.; Kidd, J.M.; Eichler, E.E.; Badge, R.M.; Moran, J.V. LINE-1 retrotransposition activity in human genomes. Cell 2010, 141, 1159–1170. [Google Scholar] [CrossRef]
- Sanchez-Luque, F.J.; Kempen, M.H.C.; Gerdes, P.; Vargas-Landin, D.B.; Richardson, S.R.; Troskie, R.L.; Jesuadian, J.S.; Cheetham, S.W.; Carreira, P.E.; Salvador-Palomeque, C.; et al. LINE-1 Evasion of Epigenetic Repression in Humans. Mol. Cell 2019, 75, 590–604.e12. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S. So much “junk” DNA in our genome. Brookhaven Symp. Biol. 1972, 23, 366–370. [Google Scholar]
- Clark, M.B.; Amaral, P.P.; Schlesinger, F.J.; Dinger, M.E.; Taft, R.J.; Rinn, J.L.; Ponting, C.P.; Stadler, P.F.; Morris, K.V.; Morillon, A.; et al. The Reality of Pervasive Transcription. PLoS Biol. 2011, 9, e1000625. [Google Scholar] [CrossRef]
- Lu, J.Y.; Shao, W.; Chang, L.; Yin, Y.; Li, T.; Zhang, H.; Hong, Y.; Percharde, M.; Guo, L.; Wu, Z.; et al. Genomic Repeats Categorize Genes with Distinct Functions for Orchestrated Regulation. Cell Rep. 2020, 30, 3296–3311.e5. [Google Scholar] [CrossRef]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef]
- Dinger, M.E.; Amaral, P.P.; Mercer, T.R.; Mattick, J.S. Pervasive transcription of the eukaryotic genome: Functional indices and conceptual implications. Brief. Funct. Genom. Proteom. 2009, 8, 407–423. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Belgard, T.G. Transcribed dark matter: Meaning or myth? Hum. Mol. Genet. 2010, 19, R162–R168. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, A.F.; Koonin, E.V. Functional Long Non-coding RNAs Evolve from Junk Transcripts. Cell 2020, 183, 1151–1161. [Google Scholar] [CrossRef]
- Ganesh, S.; Svoboda, P. Retrotransposon-associated long non-coding RNAs in mice and men. Pflug. Arch 2016, 468, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, A.; Kronenberg, Z.; Lynch, V.J.; Zhuo, X.; Ramsay, L.; Bourque, G.; Yandell, M.; Feschotte, C. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet. 2013, 9, e1003470. [Google Scholar] [CrossRef] [PubMed]
- Achour, C.; Aguilo, F. Long non-coding RNA and Polycomb: An intricate partnership in cancer biology. Front. Biosci. (Landmark Ed.) 2018, 23, 2106–2132. [Google Scholar]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef]
- Fabbri, M.; Girnita, L.; Varani, G.; Calin, G.A. Decrypting noncoding RNA interactions, structures, and functional networks. Genome Res. 2019, 29, 1377–1388. [Google Scholar] [CrossRef]
- Dhir, A.; Dhir, S.; Borowski, L.S.; Jimenez, L.; Teitell, M.; Rotig, A.; Crow, Y.J.; Rice, G.I.; Duffy, D.; Tamby, C.; et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 2018, 560, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Hur, S. Double-Stranded RNA Sensors and Modulators in Innate Immunity. Annu. Rev. Immunol. 2019, 37, 349–375. [Google Scholar] [CrossRef]
- Barak, M.; Porath, H.T.; Finkelstein, G.; Knisbacher, B.A.; Buchumenski, I.; Roth, S.H.; Levanon, E.Y.; Eisenberg, E. Purifying selection of long dsRNA is the first line of defense against false activation of innate immunity. Genome Biol. 2020, 21, 26. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Fultheim, R.; Levanon, E.Y. Detection of A-to-I Hyper-edited RNA Sequences. In RNA Editing: Methods and Protocols; Picardi, E., Pesole, G., Eds.; Springer: New York, NY, USA, 2021; pp. 213–227. [Google Scholar] [CrossRef]
- Molder, T.; Speek, M. [Letter to the Editor] Accelerated RNA-RNA hybridization by concentrated guanidinium thiocyanate solution in single-step RNA isolation. BioTechniques 2016, 61, 61–65. [Google Scholar] [CrossRef]
- Aloni, Y.; Attardi, G. Symmetrical in vivo transcription of mitochondrial DNA in HeLa cells. Proc. Natl. Acad. Sci. USA 1971, 68, 1757–1761. [Google Scholar] [CrossRef]
- Young, P.G.; Attardi, G. Characterization of double-stranded RNA from HeLa cell mitochondria. Biochem. Biophys. Res. Commun. 1975, 65, 1201–1207. [Google Scholar] [CrossRef]
- Krishnan, K.J.; Turnbull, D.M. Mitochondrial DNA and genetic disease. Essays Biochem. 2010, 47, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Borowski, L.S.; Dziembowski, A.; Hejnowicz, M.S.; Stepien, P.P.; Szczesny, R.J. Human mitochondrial RNA decay mediated by PNPase-hSuv3 complex takes place in distinct foci. Nucleic Acids Res. 2013, 41, 1223–1240. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef]
- Pajak, A.; Laine, I.; Clemente, P.; El-Fissi, N.; Schober, F.A.; Maffezzini, C.; Calvo-Garrido, J.; Wibom, R.; Filograna, R.; Dhir, A.; et al. Defects of mitochondrial RNA turnover lead to the accumulation of double-stranded RNA in vivo. PLoS Genet. 2019, 15, e1008240. [Google Scholar] [CrossRef]
- Rius, R.; Van Bergen, N.J.; Compton, A.G.; Riley, L.G.; Kava, M.P.; Balasubramaniam, S.; Amor, D.J.; Fanjul-Fernandez, M.; Cowley, M.J.; Fahey, M.C.; et al. Clinical Spectrum and Functional Consequences Associated with Bi-Allelic Pathogenic PNPT1 Variants. J. Clin. Med. 2019, 8, 2020. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.; Kim, S.; Kim, M.; Kang, M.G.; Kwak, C.; Kang, M.; Kim, B.; Rhee, H.W.; Kim, V.N. PKR Senses Nuclear and Mitochondrial Signals by Interacting with Endogenous Double-Stranded RNAs. Mol. Cell 2018, 71, 1051–1063.e6. [Google Scholar] [CrossRef]
- Arnaiz, E.; Miar, A.; Dias, A.G.; Prasad, N.; Schulze, U.; Waithe, D.; Rehwinkel, J.; Harris, A. Hypoxia regulates endogenous double-stranded RNA production via reduced mitochondrial DNA transcription. bioRxiv 2020. [Google Scholar] [CrossRef]
- Porath, H.T.; Knisbacher, B.A.; Eisenberg, E.; Levanon, E.Y. Massive A-to-I RNA editing is common across the Metazoa and correlates with dsRNA abundance. Genome Biol. 2017, 18, 185. [Google Scholar] [CrossRef]
- Reich, D.P.; Bass, B.L. Mapping the dsRNA World. Cold Spring Harb. Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Quentin, Y. Origin of the Alu family: A family of Alu-like monomers gave birth to the left and the right arms of the Alu elements. Nucleic Acids Res. 1992, 20, 3397–3401. [Google Scholar] [CrossRef] [PubMed]
- Deininger, P. Alu elements: Know the SINEs. Genome Biol. 2011, 12, 236. [Google Scholar] [CrossRef]
- Ullu, E.; Tschudi, C. Alu sequences are processed 7SL RNA genes. Nature 1984, 312, 171–172. [Google Scholar] [CrossRef]
- Batzer, M.A.; Deininger, P.L. Alu repeats and human genomic diversity. Nat. Rev. 2002, 3, 370–379. [Google Scholar] [CrossRef]
- Li, T.-H.; Schmid, C.W. Differential stress induction of individual Alu loci: Implications for transcription and retrotransposition. Gene 2001, 276, 135–141. [Google Scholar] [CrossRef]
- Berger, A.; Strub, K. Multiple Roles of Alu-Related Noncoding RNAs. Prog. Mol. Subcell. Biol. 2011, 51, 119–146. [Google Scholar] [CrossRef]
- Caudron-Herger, M.; Pankert, T.; Seiler, J.; Németh, A.; Voit, R.; Grummt, I.; Rippe, K. Alu element-containing RNAs maintain nucleolar structure and function. EMBO J. 2015, 34, 2758–2774. [Google Scholar] [CrossRef] [PubMed]
- Bazak, L.; Levanon, E.Y.; Eisenberg, E. Genome-wide analysis of Alu editability. Nucleic Acids Res. 2014, 42, 6876–6884. [Google Scholar] [CrossRef]
- Kawahara, Y.; Nishikura, K. Extensive adenosine-to-inosine editing detected in Alu repeats of antisense RNAs reveals scarcity of sense-antisense duplex formation. FEBS Lett. 2006, 580, 2301–2305. [Google Scholar] [CrossRef]
- Ahmad, S.; Mu, X.; Yang, F.; Greenwald, E.; Park, J.W.; Jacob, E.; Zhang, C.Z.; Hur, S. Breaching Self-Tolerance to Alu Duplex RNA Underlies MDA5-Mediated Inflammation. Cell 2018, 172, 797–810.e13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liang, J.Q.; Zheng, S. Expressional activation and functional roles of human endogenous retroviruses in cancers. Rev. Med Virol. 2019, 29, e2025. [Google Scholar] [CrossRef]
- Hurst, T.P.; Magiorkinis, G. Activation of the innate immune response by endogenous retroviruses. J. Gen. Virol 2015, 96, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Di Cristofano, A.; Strazzullo, M.; Longo, L.; La Mantia, G. Characterization and genomic mapping of the ZNF80 locus: Expression of this zinc-finger gene is driven by a solitary LTR of ERV9 endogenous retroviral family. Nucleic Acids Res. 1995, 23, 2823–2830. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Domansky, A.N.; Kopantzev, E.P.; Snezhkov, E.V.; Lebedev, Y.B.; Leib-Mosch, C.; Sverdlov, E.D. Solitary HERV-K LTRs possess bi-directional promoter activity and contain a negative regulatory element in the U5 region. FEBS Lett. 2000, 472, 191–195. [Google Scholar] [CrossRef]
- Strick, R.; Strissel, P.L.; Baylin, S.B.; Chiappinelli, K.B. Unraveling the molecular pathways of DNA-methylation inhibitors: Human endogenous retroviruses induce the innate immune response in tumors. Oncoimmunology 2016, 5, e1122160. [Google Scholar] [CrossRef]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef]
- Richardson, S.R.; Doucet, A.J.; Kopera, H.C.; Moldovan, J.B.; Garcia-Perez, J.L.; Moran, J.V. The Influence of LINE-1 and SINE Retrotransposons on Mammalian Genomes. Microbiol. Spectr. 2015, 3, 1165–1208. [Google Scholar] [CrossRef]
- Martin, S.L. The ORF1 protein encoded by LINE-1: Structure and function during L1 retrotransposition. J. Biomed. Biotechnol. 2006, 2006, 45621. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, G.J.; Billon, V. L1 retrotransposition in the soma: A field jumping ahead. Mob. DNA 2018, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Ku, Y.; Ku, J.; Kim, Y. Evidence of Aberrant Immune Response by Endogenous Double-Stranded RNAs: Attack from Within. Bioessays 2019, 41, e1900023. [Google Scholar] [CrossRef] [PubMed]
- Bahn, J.H.; Ahn, J.; Lin, X.; Zhang, Q.; Lee, J.-H.; Civelek, M.; Xiao, X. Genomic analysis of ADAR1 binding and its involvement in multiple RNA processing pathways. Nat. Commun. 2015, 6, 6355. [Google Scholar] [CrossRef] [PubMed]
- Orecchini, E.; Doria, M.; Antonioni, A.; Galardi, S.; Ciafrè, S.A.; Frassinelli, L.; Mancone, C.; Montaldo, C.; Tripodi, M.; Michienzi, A. ADAR1 restricts LINE-1 retrotransposition. Nucleic Acids Res. 2017, 45, 155–168. [Google Scholar] [CrossRef]
- Balbin, O.A.; Malik, R.; Dhanasekaran, S.M.; Prensner, J.R.; Cao, X.; Wu, Y.M.; Robinson, D.; Wang, R.; Chen, G.; Beer, D.G.; et al. The landscape of antisense gene expression in human cancers. Genome Res. 2015, 25, 1068–1079. [Google Scholar] [CrossRef]
- Katayama, S.; Tomaru, Y.; Kasukawa, T.; Waki, K.; Nakanishi, M.; Nakamura, M.; Nishida, H.; Yap, C.C.; Suzuki, M.; Kawai, J.; et al. Antisense transcription in the mammalian transcriptome. Science 2005, 309, 1564–1566. [Google Scholar]
- Sleutels, F.; Zwart, R.; Barlow, D.P. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 2002, 415, 810–813. [Google Scholar] [CrossRef]
- Thakur, N.; Tiwari, V.K.; Thomassin, H.; Pandey, R.R.; Kanduri, M.; Gondor, A.; Grange, T.; Ohlsson, R.; Kanduri, C. An antisense RNA regulates the bidirectional silencing property of the Kcnq1 imprinting control region. Mol. Cell. Biol. 2004, 24, 7855–7862. [Google Scholar] [CrossRef] [PubMed]
- Tufarelli, C.; Frischauf, A.M.; Hardison, R.; Flint, J.; Higgs, D.R. Characterization of a widely expressed gene (LUC7-LIKE; LUC7L) defining the centromeric boundary of the human alpha-globin domain. Genomics 2001, 71, 307–314. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wong, I.H.; Lo, Y.M.; Yeo, W.; Lau, W.Y.; Johnson, P.J. Frequent p15 promoter methylation in tumor and peripheral blood from hepatocellular carcinoma patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 3516–3521. [Google Scholar]
- Yu, W.; Gius, D.; Onyango, P.; Muldoon-Jacobs, K.; Karp, J.; Feinberg, A.P.; Cui, H. Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature 2008, 451, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Sun, M.; Kent, W.J.; Huang, X.; Xie, H.; Wang, W.; Zhou, G.; Shi, R.Z.; Rowley, J.D. Over 20% of human transcripts might form sense-antisense pairs. Nucleic Acids Res. 2004, 32, 4812–4820. [Google Scholar] [CrossRef]
- Kiyosawa, H.; Yamanaka, I.; Osato, N.; Kondo, S.; Hayashizaki, Y. Antisense transcripts with FANTOM2 clone set and their implications for gene regulation. Genome Res. 2003, 13, 1324–1334. [Google Scholar] [CrossRef]
- Wight, M.; Werner, A. The functions of natural antisense transcripts. Essays Biochem. 2013, 54, 91–101. [Google Scholar] [CrossRef]
- Faghihi, M.A.; Wahlestedt, C. Regulatory roles of natural antisense transcripts. Nat. Rev. Mol. Cell Biol. 2009, 10, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Zinad, H.S.; Natasya, I.; Werner, A. Natural Antisense Transcripts at the Interface between Host Genome and Mobile Genetic Elements. Front. Microbiol. 2017, 8, 2292. [Google Scholar] [CrossRef]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; St Laurent, G., 3rd; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat. Med. 2008, 14, 723–730. [Google Scholar] [CrossRef]
- Faghihi, M.A.; Zhang, M.; Huang, J.; Modarresi, F.; Van der Brug, M.P.; Nalls, M.A.; Cookson, M.R.; St-Laurent, G., 3rd; Wahlestedt, C. Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome Biol. 2010, 11, R56. [Google Scholar] [CrossRef]
- Piatek, M.J.; Henderson, V.; Zynad, H.S.; Werner, A. Natural antisense transcription from a comparative perspective. Genomics 2016. [Google Scholar] [CrossRef]
- Jadaliha, M.; Gholamalamdari, O.; Tang, W.; Zhang, Y.; Petracovici, A.; Hao, Q.; Tariq, A.; Kim, T.G.; Holton, S.E.; Singh, D.K.; et al. A natural antisense lncRNA controls breast cancer progression by promoting tumor suppressor gene mRNA stability. PLoS Genet. 2018, 14, e1007802. [Google Scholar] [CrossRef]
- Deng, S.J.; Chen, H.Y.; Zeng, Z.; Deng, S.; Zhu, S.; Ye, Z.; He, C.; Liu, M.L.; Huang, K.; Zhong, J.X.; et al. Nutrient Stress-Dysregulated Antisense lncRNA GLS-AS Impairs GLS-Mediated Metabolism and Represses Pancreatic Cancer Progression. Cancer Res. 2019, 79, 1398–1412. [Google Scholar] [CrossRef] [PubMed]
- Werner, A.; Cockell, S.; Falconer, J.; Carlile, M.; Alnumeir, S.; Robinson, J. Contribution of natural antisense transcription to an endogenous siRNA signature in human cells. BMC Genom. 2014, 15, 19. [Google Scholar] [CrossRef]
- Piatek, M.J.; Henderson, V.; Fearn, A.; Chaudhry, B.; Werner, A. Ectopically expressed Slc34a2a sense-antisense transcripts cause a cerebellar phenotype in zebrafish embryos depending on RNA complementarity and Dicer. PLoS ONE 2017, 12, e0178219. [Google Scholar] [CrossRef]
- Dahary, D.; Elroy-Stein, O.; Sorek, R. Naturally occurring antisense: Transcriptional leakage or real overlap? Genome Res. 2005, 15, 364–368. [Google Scholar] [CrossRef][Green Version]
- Pillay, S.; Takahashi, H.; Carninci, P.; Kanhere, A. Antisense ncRNAs during early vertebrate development are divided in groups with distinct features. bioRxiv 2020. [Google Scholar] [CrossRef]
- Goll, M.G.; Halpern, M.E. DNA methylation in zebrafish. Prog. Mol. Biol. Transl. Sci. 2011, 101, 193–218. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.D.; Santos, F.; Green, K.; Dean, W.; Reik, W. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 2005, 14, R47–R58. [Google Scholar] [CrossRef]
- Li, N.; Wang, T.; Han, D. Structural, cellular and molecular aspects of immune privilege in the testis. Front. Immunol. 2012, 3, 152. [Google Scholar] [CrossRef] [PubMed]
- Nejepinska, J.; Malik, R.; Filkowski, J.; Flemr, M.; Filipowicz, W.; Svoboda, P. dsRNA expression in the mouse elicits RNAi in oocytes and low adenosine deamination in somatic cells. Nucleic Acids Res. 2012, 40, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Hennig, G.W.; Wu, Q.; Jose, C.; Zheng, H.; Yan, W. Male germ cells express abundant endogenous siRNAs. Proc. Natl. Acad. Sci. USA 2011, 108, 13159–13164. [Google Scholar] [CrossRef]
- Watanabe, T.; Totoki, Y.; Toyoda, A.; Kaneda, M.; Kuramochi-Miyagawa, S.; Obata, Y.; Chiba, H.; Kohara, Y.; Kono, T.; Nakano, T.; et al. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature 2008, 453, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Masliah, G.; Barraud, P.; Allain, F.H. RNA recognition by double-stranded RNA binding domains: A matter of shape and sequence. Cell. Mol. Life Sci. 2013, 70, 1875–1895. [Google Scholar] [CrossRef] [PubMed]
- Stefl, R.; Oberstrass, F.C.; Hood, J.L.; Jourdan, M.; Zimmermann, M.; Skrisovska, L.; Maris, C.; Peng, L.; Hofr, C.; Emeson, R.B.; et al. The solution structure of the ADAR2 dsRBM-RNA complex reveals a sequence-specific readout of the minor groove. Cell 2010, 143, 225–237. [Google Scholar] [CrossRef]
- Yadav, D.K.; Zigáčková, D.; Zlobina, M.; Klumpler, T.; Beaumont, C.; Kubíčková, M.; Vaňáčová, Š.; Lukavsky, P.J. Staufen1 reads out structure and sequence features in ARF1 dsRNA for target recognition. Nucleic Acids Res. 2020, 48, 2091–2106. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T.; Akira, S. Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity. Immunol. Rev. 2011, 243, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Matz, K.M.; Guzman, R.M.; Goodman, A.G. The Role of Nucleic Acid Sensing in Controlling Microbial and Autoimmune Disorders. Int. Rev. Cell Mol. Biol. 2019, 345, 35–136. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, X.; Wang, G.; Zheng, H. The laboratory of genetics and physiology 2: Emerging insights into the controversial functions of this RIG-I-like receptor. BioMed Res. Int. 2014, 2014, 960190. [Google Scholar] [CrossRef]
- Cui, J.; Chen, Y.; Wang, H.Y.; Wang, R.F. Mechanisms and pathways of innate immune activation and regulation in health and cancer. Hum. Vaccines Immunother. 2014, 10, 3270–3285. [Google Scholar] [CrossRef]
- Sohn, J.; Hur, S. Filament assemblies in foreign nucleic acid sensors. Curr. Opin. Struct. Biol. 2016, 37, 134–144. [Google Scholar] [CrossRef]
- Kowalinski, E.; Lunardi, T.; McCarthy, A.A.; Louber, J.; Brunel, J.; Grigorov, B.; Gerlier, D.; Cusack, S. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 2011, 147, 423–435. [Google Scholar] [CrossRef]
- Schlee, M.; Roth, A.; Hornung, V.; Hagmann, C.A.; Wimmenauer, V.; Barchet, W.; Coch, C.; Janke, M.; Mihailovic, A.; Wardle, G.; et al. Recognition of 5’ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 2009, 31, 25–34. [Google Scholar] [CrossRef]
- Peisley, A.; Jo, M.H.; Lin, C.; Wu, B.; Orme-Johnson, M.; Walz, T.; Hohng, S.; Hur, S. Kinetic mechanism for viral dsRNA length discrimination by MDA5 filaments. Proc. Natl. Acad. Sci. USA 2012, 109, E3340–E3349. [Google Scholar] [CrossRef] [PubMed]
- Reikine, S.; Nguyen, J.B.; Modis, Y. Pattern Recognition and Signaling Mechanisms of RIG-I and MDA5. Front. Immunol. 2014, 5, 342. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Ding, T.; Zuo, Z.; Xu, Z.; Deng, J.; Wei, Z. Regulation of MAVS Expression and Signaling Function in the Antiviral Innate Immune Response. Front. Immunol. 2020, 11, 1030. [Google Scholar] [CrossRef] [PubMed]
- Chukwurah, E.; Farabaugh, K.T.; Guan, B.J.; Ramakrishnan, P.; Hatzoglou, M. A tale of two proteins: PACT and PKR and their roles in inflammation. FEBS J. 2021. [Google Scholar] [CrossRef]
- Toth, A.M.; Zhang, P.; Das, S.; George, C.X.; Samuel, C.E. Interferon action and the double-stranded RNA-dependent enzymes ADAR1 adenosine deaminase and PKR protein kinase. Prog. Nucleic Acid Res. Mol. Biol. 2006, 81, 369–434. [Google Scholar] [CrossRef]
- Bevilacqua, P.C.; Cech, T.R. Minor-groove recognition of double-stranded RNA by the double-stranded RNA-binding domain from the RNA-activated protein kinase PKR. Biochemistry 1996, 35, 9983–9994. [Google Scholar] [CrossRef] [PubMed]
- Manche, L.; Green, S.R.; Schmedt, C.; Mathews, M.B. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol. Cell. Biol. 1992, 12, 5238–5248. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.L. Activation of PKR: An open and shut case? Trends Biochem. Sci. 2007, 32, 57–62. [Google Scholar] [CrossRef] [PubMed]
- García, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of protein kinase PKR in cell biology: From antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef] [PubMed]
- Gal-Ben-Ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A Kinase to Remember. Front. Mol. Neurosci. 2018, 11, 480. [Google Scholar] [CrossRef]
- von Roretz, C.; Gallouzi, I.E. Protein kinase RNA/FADD/caspase-8 pathway mediates the proapoptotic activity of the RNA-binding protein human antigen R (HuR). J. Biol. Chem. 2010, 285, 16806–16813. [Google Scholar] [CrossRef]
- Patterson, J.B.; Samuel, C.E. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: Evidence for two forms of the deaminase. Mol. Cell. Biol. 1995, 15, 5376–5388. [Google Scholar] [CrossRef]
- Walkley, C.R.; Li, J.B. Rewriting the transcriptome: Adenosine-to-inosine RNA editing by ADARs. Genome Biol. 2017, 18, 205. [Google Scholar] [CrossRef] [PubMed]
- Mannion, N.; Arieti, F.; Gallo, A.; Keegan, L.P.; O’Connell, M.A. New Insights into the Biological Role of Mammalian ADARs; the RNA Editing Proteins. Biomolecules 2015, 5, 2338–2362. [Google Scholar] [CrossRef]
- George, C.X.; Samuel, C.E. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc. Natl. Acad. Sci. USA 1999, 96, 4621–4626. [Google Scholar] [CrossRef]
- Hundley, H.A.; Bass, B.L. ADAR editing in double-stranded UTRs and other noncoding RNA sequences. Trends Biochem. Sci. 2010, 35, 377–383. [Google Scholar] [CrossRef]
- Tan, M.H.; Li, Q.; Shanmugam, R.; Piskol, R.; Kohler, J.; Young, A.N.; Liu, K.I.; Zhang, R.; Ramaswami, G.; Ariyoshi, K.; et al. Dynamic landscape and regulation of RNA editing in mammals. Nature 2017, 550, 249–254. [Google Scholar] [CrossRef]
- Hood, J.L.; Emeson, R.B. Editing of neurotransmitter receptor and ion channel RNAs in the nervous system. Curr. Top. Microbiol. Immunol. 2012, 353, 61–90. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, M.; Maas, S.; Single, F.N.; Hartner, J.; Rozov, A.; Burnashev, N.; Feldmeyer, D.; Sprengel, R.; Seeburg, P.H. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 2000, 406, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Stulić, M.; Jantsch, M.F. Spatio-temporal profiling of Filamin A RNA-editing reveals ADAR preferences and high editing levels outside neuronal tissues. RNA Biol. 2013, 10, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- Oakes, E.; Anderson, A.; Cohen-Gadol, A.; Hundley, H.A. Adenosine Deaminase That Acts on RNA 3 (ADAR3) Binding to Glutamate Receptor Subunit B Pre-mRNA Inhibits RNA Editing in Glioblastoma. J. Biol. Chem. 2017, 292, 4326–4335. [Google Scholar] [CrossRef]
- Pfaller, C.K.; Li, Z.; George, C.X.; Samuel, C.E. Protein kinase PKR and RNA adenosine deaminase ADAR1: New roles for old players as modulators of the interferon response. Curr. Opin. Immunol. 2011, 23, 573–582. [Google Scholar] [CrossRef]
- Yang, S.; Deng, P.; Zhu, Z.; Zhu, J.; Wang, G.; Zhang, L.; Chen, A.F.; Wang, T.; Sarkar, S.N.; Billiar, T.R.; et al. Adenosine deaminase acting on RNA 1 limits RIG-I RNA detection and suppresses IFN production responding to viral and endogenous RNAs. J. Immunol. 2014, 193, 3436–3445. [Google Scholar] [CrossRef]
- Wang, Q.; Carmichael, G.G. Effects of length and location on the cellular response to double-stranded RNA. Microbiol. Mol. Biol. Rev. 2004, 68, 432–452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Carmichael, G.G. The fate of dsRNA in the nucleus: A p54(nrb)-containing complex mediates the nuclear retention of promiscuously A-to-I edited RNAs. Cell 2001, 106, 465–475. [Google Scholar] [CrossRef]
- Jeffrey, I.W.; Kadereit, S.; Meurs, E.F.; Metzger, T.; Bachmann, M.; Schwemmle, M.; Hovanessian, A.G.; Clemens, M.J. Nuclear localization of the interferon-inducible protein kinase PKR in human cells and transfected mouse cells. Exp. Cell Res. 1995, 218, 17–27. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, J.H.; Park, J.E.; Cho, J.; Yi, H.; Kim, V.N. PKR is activated by cellular dsRNAs during mitosis and acts as a mitotic regulator. Genes Dev. 2014, 28, 1310–1322. [Google Scholar] [CrossRef]
- Bennett, R.L.; Pan, Y.; Christian, J.; Hui, T.; May, W.S., Jr. The RAX/PACT-PKR stress response pathway promotes p53 sumoylation and activation, leading to G1 arrest. Cell Cycle 2012, 11, 407–417. [Google Scholar] [CrossRef]
- Cuddihy, A.R.; Wong, A.H.; Tam, N.W.; Li, S.; Koromilas, A.E. The double-stranded RNA activated protein kinase PKR physically associates with the tumor suppressor p53 protein and phosphorylates human p53 on serine 392 in vitro. Oncogene 1999, 18, 2690–2702. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.S.; Li, A.; Ratliff, T.S.; Melsom, M.; Garza, L.A. After Skin Wounding, Noncoding dsRNA Coordinates Prostaglandins and Wnts to Promote Regeneration. J. Investig. Dermatol. 2017, 137, 1562–1568. [Google Scholar] [CrossRef]
- Kim, D.; Chen, R.; Sheu, M.; Kim, N.; Kim, S.; Islam, N.; Wier, E.M.; Wang, G.; Li, A.; Park, A.; et al. Noncoding dsRNA induces retinoic acid synthesis to stimulate hair follicle regeneration via TLR3. Nat. Commun. 2019, 10, 2811. [Google Scholar] [CrossRef]
- Nelson, A.M.; Reddy, S.K.; Ratliff, T.S.; Hossain, M.Z.; Katseff, A.S.; Zhu, A.S.; Chang, E.; Resnik, S.R.; Page, C.; Kim, D.; et al. dsRNA Released by Tissue Damage Activates TLR3 to Drive Skin Regeneration. Cell Stem Cell 2015, 17, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Stojanovich, L.; Marisavljevich, D. Stress as a trigger of autoimmune disease. Autoimmun. Rev. 2008, 7, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Schur, P.H.; Stollar, B.D.; Steinberg, A.D.; Talal, N. Incidence of antibodies to double-stranded RNA in systemic lupus erythematosus and related diseases. Arthritis Rheum. 1971, 14, 342–347. [Google Scholar] [CrossRef]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef]
- Crow, M.K.; Kirou, K.A.; Wohlgemuth, J. Microarray analysis of interferon-regulated genes in SLE. Autoimmunity 2003, 36, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Fenando, A.; Firn, K.; Louissa, S.; Hussain, A. Case of anti-MDA-5 positive dermatomyositis with rapidly progressive interstitial lung disease. BMJ Case Rep. 2020, 13, e235493. [Google Scholar] [CrossRef]
- Cufi, P.; Dragin, N.; Weiss, J.M.; Martinez-Martinez, P.; De Baets, M.H.; Roussin, R.; Fadel, E.; Berrih-Aknin, S.; Le Panse, R. Implication of double-stranded RNA signaling in the etiology of autoimmune myasthenia gravis. Ann. Neurol. 2013, 73, 281–293. [Google Scholar] [CrossRef]
- Rácz, E.; Prens, E.P.; Kant, M.; Florencia, E.; Jaspers, N.G.; Laman, J.D.; de Ridder, D.; van der Fits, L. Narrowband ultraviolet B inhibits innate cytosolic double-stranded RNA receptors in psoriatic skin and keratinocytes. Br. J. Dermatol. 2011, 164, 838–847. [Google Scholar] [CrossRef]
- Afshar, M.; Martinez, A.D.; Gallo, R.L.; Hata, T.R. Induction and exacerbation of psoriasis with Interferon-alpha therapy for hepatitis C: A review and analysis of 36 cases. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.M.; Davis, C.; Olerud, J. Concurrent antiphospholipid syndrome and cutaneous [corrected] sarcoidosis due to interferon alfa and ribavirin treatment for hepatitis C. J. Drugs Dermatol. 2009, 8, 870–872. [Google Scholar]
- Roth, S.H.; Danan-Gotthold, M.; Ben-Izhak, M.; Rechavi, G.; Cohen, C.J.; Louzoun, Y.; Levanon, E.Y. Increased RNA Editing May Provide a Source for Autoantigens in Systemic Lupus Erythematosus. Cell Rep. 2018, 23, 50–57. [Google Scholar] [CrossRef]
- Vlachogiannis, N.I.; Gatsiou, A.; Silvestris, D.A.; Stamatelopoulos, K.; Tektonidou, M.G.; Gallo, A.; Sfikakis, P.P.; Stellos, K. Increased adenosine-to-inosine RNA editing in rheumatoid arthritis. J. Autoimmun. 2020, 106, 102329. [Google Scholar] [CrossRef]
- Paz-Yaacov, N.; Bazak, L.; Buchumenski, I.; Porath, H.T.; Danan-Gotthold, M.; Knisbacher, B.A.; Eisenberg, E.; Levanon, E.Y. Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in Tumors. Cell Rep. 2015, 13, 267–276. [Google Scholar] [CrossRef]
- Peng, X.; Xu, X.; Wang, Y.; Hawke, D.H.; Yu, S.; Han, L.; Zhou, Z.; Mojumdar, K.; Jeong, K.J.; Labrie, M.; et al. A-to-I RNA Editing Contributes to Proteomic Diversity in Cancer. Cancer Cell 2018, 33, 817–828.e7. [Google Scholar] [CrossRef]
- Han, L.; Diao, L.; Yu, S.; Xu, X.; Li, J.; Zhang, R.; Yang, Y.; Werner, H.M.J.; Eterovic, A.K.; Yuan, Y.; et al. The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer Cell 2015, 28, 515–528. [Google Scholar] [CrossRef]
- Sakurai, M.; Shiromoto, Y.; Ota, H.; Song, C.; Kossenkov, A.V.; Wickramasinghe, J.; Showe, L.C.; Skordalakes, E.; Tang, H.Y.; Speicher, D.W.; et al. ADAR1 controls apoptosis of stressed cells by inhibiting Staufen1-mediated mRNA decay. Nat. Struct. Mol. Biol. 2017, 24, 534–543. [Google Scholar] [CrossRef]
- Xu, L.D.; Öhman, M. ADAR1 Editing and its Role in Cancer. Genes 2018, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Martinez, H.D.; Jasavala, R.J.; Hinkson, I.; Fitzgerald, L.D.; Trimmer, J.S.; Kung, H.J.; Wright, M.E. RNA editing of androgen receptor gene transcripts in prostate cancer cells. J. Biol. Chem. 2008, 283, 29938–29949. [Google Scholar] [CrossRef]
- Chen, L.; Li, Y.; Lin, C.H.; Chan, T.H.; Chow, R.K.; Song, Y.; Liu, M.; Yuan, Y.F.; Fu, L.; Kong, K.L.; et al. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat. Med. 2013, 19, 209–216. [Google Scholar] [CrossRef]
- Valencia, J.C.; Egbukichi, N.; Erwin-Cohen, R.A. Autoimmunity and Cancer, the Paradox Comorbidities Challenging Therapy in the Context of Preexisting Autoimmunity. J. Interferon Cytokine Res. 2019, 39, 72–84. [Google Scholar] [CrossRef]
- Staege, M.S.; Emmer, A. Editorial: Endogenous Viral Elements-Links Between Autoimmunity and Cancer? Front. Microbiol. 2018, 9, 3171. [Google Scholar] [CrossRef]
- Brütting, C.; Emmer, A.; Kornhuber, M.E.; Staege, M.S. Cooccurrences of Putative Endogenous Retrovirus-Associated Diseases. BioMed Res. Int. 2017, 2017, 7973165. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Hu, S.; Li, Y.; Jiang, T.T.; Jin, H.; Feng, L. Targeting nuclear acid-mediated immunity in cancer immune checkpoint inhibitor therapies. Signal Transduct. Target. Ther. 2020, 5, 270. [Google Scholar] [CrossRef]
- Jin, B.; Cheng, L.F.; Wu, K.; Yu, X.H.; Yeo, A.E. Application of dsRNA in cancer immunotherapy: Current status and future trends. Anti-Cancer Agents Med. Chem. 2014, 14, 241–255. [Google Scholar] [CrossRef]
- Tsai, H.C.; Li, H.; Van Neste, L.; Cai, Y.; Robert, C.; Rassool, F.V.; Shin, J.J.; Harbom, K.M.; Beaty, R.; Pappou, E.; et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 2012, 21, 430–446. [Google Scholar] [CrossRef]
- Li, H.; Chiappinelli, K.B.; Guzzetta, A.A.; Easwaran, H.; Yen, R.W.; Vatapalli, R.; Topper, M.J.; Luo, J.; Connolly, R.M.; Azad, N.S.; et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget 2014, 5, 587–598. [Google Scholar] [CrossRef]
- Banik, D.; Moufarrij, S.; Villagra, A. Immunoepigenetics Combination Therapies: An Overview of the Role of HDACs in Cancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 2241. [Google Scholar] [CrossRef]
- Covre, A.; Coral, S.; Nicolay, H.; Parisi, G.; Fazio, C.; Colizzi, F.; Fratta, E.; Di Giacomo, A.M.; Sigalotti, L.; Natali, P.G.; et al. Antitumor activity of epigenetic immunomodulation combined with CTLA-4 blockade in syngeneic mouse models. Oncoimmunology 2015, 4, e1019978. [Google Scholar] [CrossRef]
- Ammi, R.; De Waele, J.; Willemen, Y.; Van Brussel, I.; Schrijvers, D.M.; Lion, E.; Smits, E.L. Poly(I:C) as cancer vaccine adjuvant: Knocking on the door of medical breakthroughs. Pharmacol. Ther. 2015, 146, 120–131. [Google Scholar] [CrossRef]
- Cheever, M.A. Twelve immunotherapy drugs that could cure cancers. Immunol. Rev. 2008, 222, 357–368. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadeq, S.; Al-Hashimi, S.; Cusack, C.M.; Werner, A. Endogenous Double-Stranded RNA. Non-Coding RNA 2021, 7, 15. https://doi.org/10.3390/ncrna7010015
Sadeq S, Al-Hashimi S, Cusack CM, Werner A. Endogenous Double-Stranded RNA. Non-Coding RNA. 2021; 7(1):15. https://doi.org/10.3390/ncrna7010015
Chicago/Turabian StyleSadeq, Shaymaa, Surar Al-Hashimi, Carmen M. Cusack, and Andreas Werner. 2021. "Endogenous Double-Stranded RNA" Non-Coding RNA 7, no. 1: 15. https://doi.org/10.3390/ncrna7010015
APA StyleSadeq, S., Al-Hashimi, S., Cusack, C. M., & Werner, A. (2021). Endogenous Double-Stranded RNA. Non-Coding RNA, 7(1), 15. https://doi.org/10.3390/ncrna7010015