Seeing Is Believing: Visualizing Circular RNAs


Abstract
1. Introduction
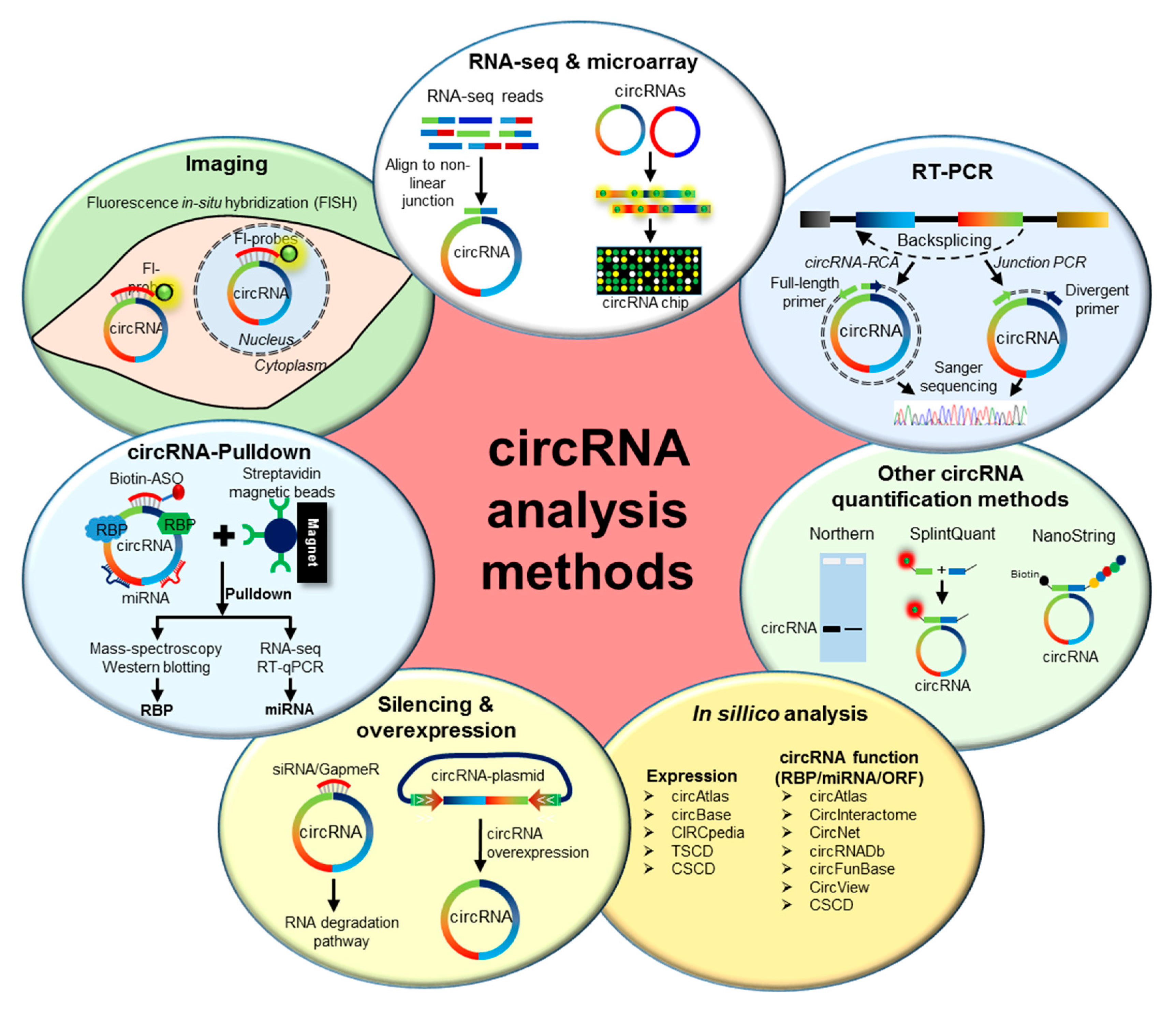
2. Methods to Analyze circRNAs
2.1. Genome-Wide Analysis of circRNA Expression
2.2. Validation of circRNA Expression
2.3. Prediction of circRNA Expression and Function
2.4. Functional Characterization of circRNAs
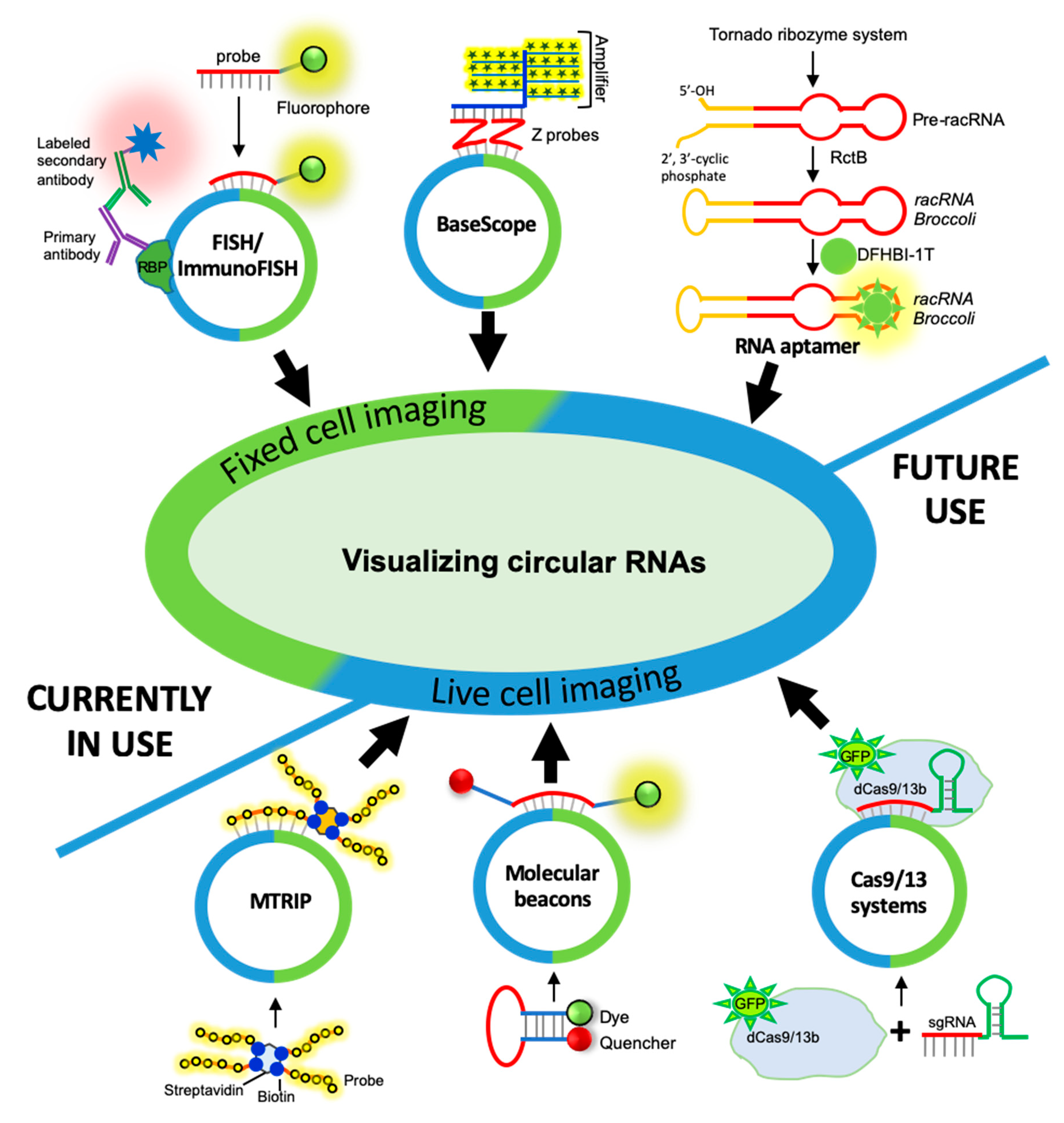
3. CircRNA Detection and Quantification by RNA Imaging Techniques
3.1. Fixed-Cell circRNA Imaging
3.1.1. circRNA Imaging Using smFISH
3.1.2. circRNA Imaging Using BaseScope Assay
3.2. Live-Cell Imaging of circRNAs
3.2.1. Fluorescent RNA Aptamers
3.2.2. Cas-Derived Fluorescent Protein
3.2.3. Molecular Beacons
3.2.4. Multiply Labeled Tetravalent RNA Imaging Probes
4. Limitations and Additional Considerations for circRNA Imaging Techniques
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Palazzo, A.F.; Lee, E.S. Non-coding RNA: What is functional and what is junk? Front. Genet. 2015, 6, 2. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef]
- Salzman, J.; Chen, R.E.; Olsen, M.N.; Wang, P.L.; Brown, P.O. Cell-type specific features of circular RNA expression. PLoS Genet. 2013, 9, e1003777. [Google Scholar] [CrossRef]
- Ji, P.; Wu, W.; Chen, S.; Zheng, Y.; Zhou, L.; Zhang, J.; Cheng, H.; Yan, J.; Zhang, S.; Yang, P.; et al. Expanded Expression Landscape and Prioritization of Circular RNAs in Mammals. Cell Rep. 2019, 26, 3444–3460. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.O.; Dong, R.; Zhang, Y.; Zhang, J.L.; Luo, Z.; Zhang, J.; Chen, L.L.; Yang, L. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 2016, 26, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Guria, A.; Sharma, P.; Natesan, S.; Pandi, G. Circular RNAs-The Road Less Traveled. Front. Mol. Biosci. 2019, 6, 146. [Google Scholar] [CrossRef] [PubMed]
- Noto, J.J.; Schmidt, C.A.; Matera, A.G. Engineering and expressing circular RNAs via tRNA splicing. RNA Biol. 2017, 14, 978–984. [Google Scholar] [CrossRef]
- Gardner, E.J.; Nizami, Z.F.; Talbot, C.C., Jr.; Gall, J.G. Stable intronic sequence RNA (sisRNA), a new class of noncoding RNA from the oocyte nucleus of Xenopus tropicalis. Genes Dev. 2012, 26, 2550–2559. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.O.; Chen, T.; Xiang, J.F.; Yin, Q.F.; Xing, Y.H.; Zhu, S.; Yang, L.; Chen, L.L. Circular intronic long noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef]
- Chen, L.L.; Yang, L. Regulation of circRNA biogenesis. RNA Biol 2015, 12, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Zheng, H.; Wu, Z.; Chen, M.; Huang, Y. Circular RNA-protein interactions: Functions, mechanisms, and identification. Theranostics 2020, 10, 3503–3517. [Google Scholar] [CrossRef] [PubMed]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. circRNA biogenesis competes with pre-mRNA splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, C.X.; Xue, W.; Zhang, Y.; Jiang, S.; Yin, Q.F.; Wei, J.; Yao, R.W.; Yang, L.; Chen, L.L. Coordinated circRNA Biogenesis and Function with NF90/NF110 in Viral Infection. Mol. Cell 2017, 67, 214–227 e217. [Google Scholar] [CrossRef]
- Aktas, T.; Avsar Ilik, I.; Maticzka, D.; Bhardwaj, V.; Pessoa Rodrigues, C.; Mittler, G.; Manke, T.; Backofen, R.; Akhtar, A. DHX9 suppresses RNA processing defects originating from the Alu invasion of the human genome. Nature 2017, 544, 115–119. [Google Scholar] [CrossRef]
- Conn, S.J.; Pillman, K.A.; Toubia, J.; Conn, V.M.; Salmanidis, M.; Phillips, C.A.; Roslan, S.; Schreiber, A.W.; Gregory, P.A.; Goodall, G.J. The RNA binding protein quaking regulates formation of circRNAs. Cell 2015, 160, 1125–1134. [Google Scholar] [CrossRef]
- Vromman, M.; Vandesompele, J.; Volders, P.J. Closing the circle: Current state and perspectives of circular RNA databases. Brief. Bioinform. 2020. [Google Scholar] [CrossRef]
- Panda, A.C.; Grammatikakis, I.; Munk, R.; Gorospe, M.; Abdelmohsen, K. Emerging roles and context of circular RNAs. Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef]
- Chao, C.W.; Chan, D.C.; Kuo, A.; Leder, P. The mouse formin (Fmn) gene: Abundant circular RNA transcripts and gene-targeted deletion analysis. Mol. Med. 1998, 4, 614–628. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.C. Circular RNAs Act as miRNA Sponges. Adv. Exp. Med. Biol. 2018, 1087, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Wiklund, E.D.; Bramsen, J.B.; Villadsen, S.B.; Statham, A.L.; Clark, S.J.; Kjems, J. miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J. 2011, 30, 4414–4422. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.H.; Lee, S.K.; Sood, A.K. Circular RNAs in Cancer. Mol. Ther. Nucleic Acids 2019, 16, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Das, A.; Panda, A.C. Emerging role of long noncoding RNAs and circular RNAs in pancreatic β cells. Non-coding RNA Investig. 2018, 2. [Google Scholar] [CrossRef]
- Das, A.; Das, A.; Das, D.; Abdelmohsen, K.; Panda, A.C. Circular RNAs in myogenesis. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194372. [Google Scholar] [CrossRef]
- Yang, D.; Yang, K.; Yang, M. Circular RNA in Aging and Age-Related Diseases. Adv. Exp. Med. Biol. 2018, 1086, 17–35. [Google Scholar] [CrossRef]
- Haque, S.; Harries, L.W. Circular RNAs (circRNAs) in Health and Disease. Genes 2017, 8, 353. [Google Scholar] [CrossRef]
- Szabo, L.; Salzman, J. Detecting circular RNAs: Bioinformatic and experimental challenges. Nat. Rev. Genet. 2016, 17, 679–692. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef]
- Pandey, P.R.; Rout, P.K.; Das, A.; Gorospe, M.; Panda, A.C. RPAD (RNase R treatment, polyadenylation, and poly(A)+ RNA depletion) method to isolate highly pure circular RNA. Methods 2019, 155, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Teng, S.; Xu, J.; Su, G.; Zhang, Y.; Zhao, J.; Zhang, S.; Wang, H.; Qin, W.; Lu, Z.J.; et al. Microarray is an efficient tool for circRNA profiling. Brief. Bioinform. 2019, 20, 1420–1433. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.C.; Gorospe, M. Detection and Analysis of Circular RNAs by RT-PCR. Bio Protoc. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Rout, P.K.; Gorospe, M.; Panda, A.C. Rolling Circle cDNA Synthesis Uncovers Circular RNA Splice Variants. Int. J. Mol. Sci. 2019, 20, 3988. [Google Scholar] [CrossRef] [PubMed]
- Conn, V.; Conn, S.J. SplintQuant: A method for accurately quantifying circular RNA transcript abundance without reverse transcription bias. RNA 2019, 25, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Dahl, M.; Daugaard, I.; Andersen, M.S.; Hansen, T.B.; Gronbaek, K.; Kjems, J.; Kristensen, L.S. Enzyme-free digital counting of endogenous circular RNA molecules in B-cell malignancies. Lab. Investig. 2018, 98, 1657–1669. [Google Scholar] [CrossRef]
- Xia, S.; Feng, J.; Lei, L.; Hu, J.; Xia, L.; Wang, J.; Xiang, Y.; Liu, L.; Zhong, S.; Han, L.; et al. Comprehensive characterization of tissue-specific circular RNAs in the human and mouse genomes. Brief. Bioinform. 2017, 18, 984–992. [Google Scholar] [CrossRef]
- Xia, S.; Feng, J.; Chen, K.; Ma, Y.; Gong, J.; Cai, F.; Jin, Y.; Gao, Y.; Xia, L.; Chang, H.; et al. CSCD: A database for cancer-specific circular RNAs. Nucleic Acids Res. 2018, 46, D925–D929. [Google Scholar] [CrossRef]
- Dong, R.; Ma, X.K.; Li, G.W.; Yang, L. CIRCpedia v2: An Updated Database for Comprehensive Circular RNA Annotation and Expression Comparison. Genomics Proteomics Bioinformatics 2018, 16, 226–233. [Google Scholar] [CrossRef]
- Glazar, P.; Papavasileiou, P.; Rajewsky, N. circBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef]
- Wu, W.; Ji, P.; Zhao, F. CircAtlas: An integrated resource of one million highly accurate circular RNAs from 1070 vertebrate transcriptomes. Genome Biol. 2020, 21, 101. [Google Scholar] [CrossRef] [PubMed]
- Dudekula, D.B.; Panda, A.C.; Grammatikakis, I.; De, S.; Abdelmohsen, K.; Gorospe, M. CircInteractome: A web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol. 2016, 13, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Li, J.R.; Sun, C.H.; Andrews, E.; Chao, R.F.; Lin, F.M.; Weng, S.L.; Hsu, S.D.; Huang, C.C.; Cheng, C.; et al. CircNet: A database of circular RNAs derived from transcriptome sequencing data. Nucleic Acids Res. 2016, 44, D209–D215. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Hu, D.; Zhang, P.; Chen, Q.; Chen, M. CircFunBase: A database for functional circular RNAs. Database 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, Q.; Shen, J.; Yang, B.B.; Ding, X. Circbank: A comprehensive database for circRNA with standard nomenclature. RNA Biol. 2019, 16, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Han, P.; Zhou, T.; Guo, X.; Song, X.; Li, Y. circRNADb: A comprehensive database for human circular RNAs with protein-coding annotations. Sci. Rep. 2016, 6, 34985. [Google Scholar] [CrossRef]
- Liu, D.; Conn, V.; Goodall, G.J.; Conn, S.J. A Highly Efficient Strategy for Overexpressing circRNAs. Methods Mol. Biol. 2018, 1724, 97–105. [Google Scholar] [CrossRef]
- Ho-Xuan, H.; Glazar, P.; Latini, C.; Heizler, K.; Haase, J.; Hett, R.; Anders, M.; Weichmann, F.; Bruckmann, A.; Van den Berg, D.; et al. Comprehensive analysis of translation from overexpressed circular RNAs reveals pervasive translation from linear transcripts. Nucleic Acids Res. 2020, 48, 10368–10382. [Google Scholar] [CrossRef]
- Panda, A.C.; Grammatikakis, I.; Kim, K.M.; De, S.; Martindale, J.L.; Munk, R.; Yang, X.; Abdelmohsen, K.; Gorospe, M. Identification of senescence-associated circular RNAs (SAC-RNAs) reveals senescence suppressor CircPVT1. Nucleic Acids Res. 2017, 45, 4021–4035. [Google Scholar] [CrossRef]
- Pandey, P.R.; Yang, J.H.; Tsitsipatis, D.; Panda, A.C.; Noh, J.H.; Kim, K.M.; Munk, R.; Nicholson, T.; Hanniford, D.; Argibay, D.; et al. circSamd4 represses myogenic transcriptional activity of PUR proteins. Nucleic Acids Res. 2020, 48, 3789–3805. [Google Scholar] [CrossRef] [PubMed]
- Kocks, C.; Boltengagen, A.; Piwecka, M.; Rybak-Wolf, A.; Rajewsky, N. Single-Molecule Fluorescence In Situ Hybridization (FISH) of Circular RNA CDR1as. Methods Mol. Biol. 2018, 1724, 77–96. [Google Scholar] [CrossRef] [PubMed]
- Zirkel, A.; Papantonis, A. Detecting Circular RNAs by RNA Fluorescence In Situ Hybridization. Methods Mol. Biol. 2018, 1724, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.S.; Ebbesen, K.K.; Sokol, M.; Jakobsen, T.; Korsgaard, U.; Eriksen, A.C.; Hansen, T.B.; Kjems, J.; Hager, H. Spatial expression analyses of the putative oncogene ciRS-7 in cancer reshape the microRNA sponge theory. Nat. Commun. 2020, 11, 4551. [Google Scholar] [CrossRef] [PubMed]
- Gall, J.G.; Pardue, M.L. Formation and detection of RNA-DNA hybrid molecules in cytological preparations. Proc. Natl. Acad. Sci. USA 1969, 63, 378–383. [Google Scholar] [CrossRef]
- Rudkin, G.T.; Stollar, B.D. High resolution detection of DNA-RNA hybrids in situ by indirect immunofluorescence. Nature 1977, 265, 472–473. [Google Scholar] [CrossRef]
- Haimovich, G.; Gerst, J.E. Single-molecule Fluorescence in situ Hybridization (smFISH) for RNA Detection in Adherent Animal Cells. Bioprotocol 2018, 8. [Google Scholar] [CrossRef]
- Han, B.; Zhang, Y.; Zhang, Y.; Bai, Y.; Chen, X.; Huang, R.; Wu, F.; Leng, S.; Chao, J.; Zhang, J.H.; et al. Novel insight into circular RNA HECTD1 in astrocyte activation via autophagy by targeting MIR142-TIPARP: Implications for cerebral ischemic stroke. Autophagy 2018, 14, 1164–1184. [Google Scholar] [CrossRef]
- Jin, M.; Shi, C.; Yang, C.; Liu, J.; Huang, G. Upregulated circRNA ARHGAP10 Predicts an Unfavorable Prognosis in NSCLC through Regulation of the miR-150-5p/GLUT-1 Axis. Mol. Ther. Nucleic Acids 2019, 18, 219–231. [Google Scholar] [CrossRef]
- Zhan, W.; Liao, X.; Chen, Z.; Li, L.; Tian, T.; Yu, L.; Wang, W.; Hu, Q. Circular RNA hsa_circRNA_103809 promoted hepatocellular carcinoma development by regulating miR-377-3p/FGFR1/ERK axis. J. Cell. Physiol. 2020, 235, 1733–1745. [Google Scholar] [CrossRef]
- Li, B.; Jin, M.; Cao, F.; Li, J.; Wu, J.; Xu, L.; Liu, X.; Shi, Y.; Chen, W. Hsa_circ_0017639 expression promotes gastric cancer proliferation and metastasis by sponging miR-224-5p and upregulating USP3. Gene 2020, 750, 144753. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, H.; Li, Y.; Mao, R.; Yang, H.; Zhang, Y.; Zhang, Y.; Guo, P.; Zhan, D.; Zhang, T. Circular RNA SAMD4A controls adipogenesis in obesity through the miR-138-5p/EZH2 axis. Theranostics 2020, 10, 4705–4719. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Z.; Jiang, H.; Li, Q.; Wang, R.; Pan, H.; Niu, Y.; Liu, F.; Gu, H.; Fan, X.; et al. Circular RNA circPVT1 Promotes Proliferation and Invasion Through Sponging miR-125b and Activating E2F2 Signaling in Non-Small Cell Lung Cancer. Cell. Physiol. Biochem. 2018, 51, 2324–2340. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cao, X.; Dong, D.; Shen, X.; Cheng, J.; Jiang, R.; Yang, Z.; Peng, S.; Huang, Y.; Lan, X.; et al. Circular RNA TTN Acts As a miR-432 Sponge to Facilitate Proliferation and Differentiation of Myoblasts via the IGF2/PI3K/AKT Signaling Pathway. Mol. Ther. Nucleic Acids 2019, 18, 966–980. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Hao, X.; Song, W.; Niu, K.; Yang, X.; Zhang, X.; Shang, R.; Wang, Q.; Li, H.; Liu, Z. Circular RNA circRHOT1 is upregulated and promotes cell proliferation and invasion in pancreatic cancer. Epigenomics 2019, 11, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Piwecka, M.; Glazar, P.; Hernandez-Miranda, L.R.; Memczak, S.; Wolf, S.A.; Rybak-Wolf, A.; Filipchyk, A.; Klironomos, F.; Cerda Jara, C.A.; Fenske, P.; et al. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 2017, 357. [Google Scholar] [CrossRef]
- Liu, G.; Huang, K.; Jie, Z.; Wu, Y.; Chen, J.; Chen, Z.; Fang, X.; Shen, S. CircFAT1 sponges miR-375 to promote the expression of Yes-associated protein 1 in osteosarcoma cells. Mol. Cancer 2018, 17, 170. [Google Scholar] [CrossRef]
- Wu, Y.; Xie, Z.; Chen, J.; Chen, J.; Ni, W.; Ma, Y.; Huang, K.; Wang, G.; Wang, J.; Ma, J.; et al. Circular RNA circTADA2A promotes osteosarcoma progression and metastasis by sponging miR-203a-3p and regulating CREB3 expression. Mol. Cancer 2019, 18, 73. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Li, P.; Yang, X.; Yuan, W.; Yang, C.; Zhang, X.; Han, J.; Wang, J.; Deng, X.; Yang, H.; Li, P.; et al. CircRNA-Cdr1as Exerts Anti-Oncogenic Functions in Bladder Cancer by Sponging MicroRNA-135a. Cell. Physiol. Biochem. 2018, 46, 1606–1616. [Google Scholar] [CrossRef]
- Han, D.; Wang, Y.; Wang, Y.; Dai, X.; Zhou, T.; Chen, J.; Tao, B.; Zhang, J.; Cao, F. The Tumor-Suppressive Human Circular RNA CircITCH Sponges miR-330-5p to Ameliorate Doxorubicin-Induced Cardiotoxicity Through Upregulating SIRT6, Survivin, and SERCA2a. Circ. Res. 2020, 127, e108–e125. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Zhang, Y.; Han, B.; Yang, L.; Chen, X.; Huang, R.; Wu, F.; Chao, J.; Liu, P.; Hu, G.; et al. Circular RNA DLGAP4 Ameliorates Ischemic Stroke Outcomes by Targeting miR-143 to Regulate Endothelial-Mesenchymal Transition Associated with Blood-Brain Barrier Integrity. J. Neurosci. 2018, 38, 32–50. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Mou, T.; He, J.; Chen, D.; Lv, D.; Liu, H.; Yu, J.; Wang, S.; Li, G. Circular RNA circRHOBTB3 acts as a sponge for miR-654-3p inhibiting gastric cancer growth. J. Exp. Clin. Cancer Res. 2020, 39, 1. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Rong, Z.; Zhang, J.; Zhu, Z.; Yu, Z.; Li, T.; Fu, Z.; Qiu, Z.; Huang, C. Circular RNA circCCDC9 acts as a miR-6792-3p sponge to suppress the progression of gastric cancer through regulating CAV1 expression. Mol. Cancer 2020, 19, 86. [Google Scholar] [CrossRef]
- Liu, T.; Lu, Q.; Liu, J.; Xie, S.; Feng, B.; Zhu, W.; Liu, M.; Liu, Y.; Zhou, X.; Sun, W.; et al. Circular RNA FAM114A2 suppresses progression of bladder cancer via regulating NP63 by sponging miR-762. Cell Death. Dis. 2020, 11, 47. [Google Scholar] [CrossRef]
- Jiang, Q.; Liu, C.; Li, C.P.; Xu, S.S.; Yao, M.D.; Ge, H.M.; Sun, Y.N.; Li, X.M.; Zhang, S.J.; Shan, K.; et al. Circular RNA-ZNF532 regulates diabetes-induced retinal pericyte degeneration and vascular dysfunction. J. Clin. Investig. 2020, 130, 3833–3847. [Google Scholar] [CrossRef]
- Huang, X.; He, M.; Huang, S.; Lin, R.; Zhan, M.; Yang, D.; Shen, H.; Xu, S.; Cheng, W.; Yu, J.; et al. Circular RNA circERBB2 promotes gallbladder cancer progression by regulating PA2G4-dependent rDNA transcription. Mol. Cancer 2019, 18, 166. [Google Scholar] [CrossRef]
- Wang, L.; Long, H.; Zheng, Q.; Bo, X.; Xiao, X.; Li, B. Circular RNA circRHOT1 promotes hepatocellular carcinoma progression by initiation of NR2F6 expression. Mol. Cancer 2019, 18, 119. [Google Scholar] [CrossRef]
- Xie, M.; Yu, T.; Jing, X.; Ma, L.; Fan, Y.; Yang, F.; Ma, P.; Jiang, H.; Wu, X.; Shu, Y.; et al. Exosomal circSHKBP1 promotes gastric cancer progression via regulating the miR-582-3p/HUR/VEGF axis and suppressing HSP90 degradation. Mol. Cancer 2020, 19, 112. [Google Scholar] [CrossRef]
- Luo, J.; Li, Y.; Zheng, W.; Xie, N.; Shi, Y.; Long, Z.; Xie, L.; Fazli, L.; Zhang, D.; Gleave, M.; et al. Characterization of a Prostate- and Prostate Cancer-Specific Circular RNA Encoded by the Androgen Receptor Gene. Mol. Ther. Nucleic Acids 2019, 18, 916–926. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, Q.; Qiu, Q.; Hou, L.; Wu, M.; Li, J.; Li, X.; Lu, B.; Cheng, X.; Liu, P.; et al. CircPLEKHM3 acts as a tumor suppressor through regulation of the miR-9/BRCA1/DNAJB6/KLF4/AKT1 axis in ovarian cancer. Mol. Cancer 2019, 18, 144. [Google Scholar] [CrossRef] [PubMed]
- Suenkel, C.; Cavalli, D.; Massalini, S.; Calegari, F.; Rajewsky, N. A Highly Conserved Circular RNA Is Required to Keep Neural Cells in a Progenitor State in the Mammalian Brain. Cell Rep. 2020, 30, 2170–2179 e2175. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Chen, D.; Wang, Z.; Ma, J.; Zhou, J.; Chen, N.; Lv, L.; Zheng, Y.; Hu, X.; Zhang, Y.; et al. Annotation and functional clustering of circRNA expression in rhesus macaque brain during aging. Cell Discov. 2018, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Talhouarne, G.J.S.; Gall, J.G. Lariat intronic RNAs in the cytoplasm of vertebrate cells. Proc. Natl. Acad. Sci. USA 2018, 115, 7970–7977. [Google Scholar] [CrossRef]
- Moldovan, L.I.; Hansen, T.B.; Veno, M.T.; Okholm, T.L.H.; Andersen, T.L.; Hager, H.; Iversen, L.; Kjems, J.; Johansen, C.; Kristensen, L.S. High-throughput RNA sequencing from paired lesional- and non-lesional skin reveals major alterations in the psoriasis circRNAome. BMC Med. Genomics 2019, 12, 174. [Google Scholar] [CrossRef]
- Abere, B.; Li, J.; Zhou, H.; Toptan, T.; Moore, P.S.; Chang, Y. Kaposi’s Sarcoma-Associated Herpesvirus-Encoded circRNAs Are Expressed in Infected Tumor Tissues and Are Incorporated into Virions. mBio 2020, 11. [Google Scholar] [CrossRef]
- Ungerleider, N.; Concha, M.; Lin, Z.; Roberts, C.; Wang, X.; Cao, S.; Baddoo, M.; Moss, W.N.; Yu, Y.; Seddon, M.; et al. The Epstein Barr virus circRNAome. PLoS Pathog. 2018, 14. [Google Scholar] [CrossRef]
- Schmidt, C.A.; Noto, J.J.; Filonov, G.S.; Matera, A.G. A Method for Expressing and Imaging Abundant, Stable, Circular RNAs In Vivo Using tRNA Splicing. Methods Enzymol. 2016, 572, 215–236. [Google Scholar] [CrossRef]
- Litke, J.L.; Jaffrey, S.R. Highly efficient expression of circular RNA aptamers in cells using autocatalytic transcripts. Nat. Biotechnol. 2019, 37, 667–675. [Google Scholar] [CrossRef]
- Wang, F.; Flanagan, J.; Su, N.; Wang, L.C.; Bui, S.; Nielson, A.; Wu, X.; Vo, H.T.; Ma, X.J.; Luo, Y. RNAscope: A novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 2012, 14, 22–29. [Google Scholar] [CrossRef]
- Gross-Thebing, T.; Paksa, A.; Raz, E. Simultaneous high-resolution detection of multiple transcripts combined with localization of proteins in whole-mount embryos. BMC Biol. 2014, 12, 55. [Google Scholar] [CrossRef] [PubMed]
- Braselmann, E.; Rathbun, C.; Richards, E.M.; Palmer, A.E. Illuminating RNA Biology: Tools for Imaging RNA in Live Mammalian Cells. Cell Chem. Biol. 2020, 27, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Urbanek, M.O.; Galka-Marciniak, P.; Olejniczak, M.; Krzyzosiak, W.J. RNA imaging in living cells - methods and applications. RNA Biol. 2014, 11, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Querido, E.; Chartrand, P. Using fluorescent proteins to study mRNA trafficking in living cells. Methods Cell Biol. 2008, 85, 273–292. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, L.; Zou, X.; Duan, S.; Li, Z.; Deng, Z.; Luo, J.; Lee, S.Y.; Chen, S. Advances in CRISPR-Cas systems for RNA targeting, tracking and editing. Biotechnol. Adv. 2019, 37, 708–729. [Google Scholar] [CrossRef]
- Yang, L.Z.; Wang, Y.; Li, S.Q.; Yao, R.W.; Luan, P.F.; Wu, H.; Carmichael, G.G.; Chen, L.L. Dynamic Imaging of RNA in Living Cells by CRISPR-Cas13 Systems. Mol. Cell 2019, 76, 981–997. [Google Scholar] [CrossRef]
- Nelles, D.A.; Fang, M.Y.; O’Connell, M.R.; Xu, J.L.; Markmiller, S.J.; Doudna, J.A.; Yeo, G.W. Programmable RNA Tracking in Live Cells with CRISPR/Cas9. Cell 2016, 165, 488–496. [Google Scholar] [CrossRef]
- Tyagi, S.; Kramer, F.R. Molecular beacons: Probes that fluoresce upon hybridization. Nat. Biotechnol. 1996, 14, 303–308. [Google Scholar] [CrossRef]
- Chen, M.; Ma, Z.; Wu, X.; Mao, S.; Yang, Y.; Tan, J.; Krueger, C.J.; Chen, A.K. A molecular beacon-based approach for live-cell imaging of RNA transcripts with minimal target engineering at the single-molecule level. Sci. Rep. 2017, 7, 1550. [Google Scholar] [CrossRef]
- Monroy-Contreras, R.; Vaca, L. Molecular beacons: Powerful tools for imaging RNA in living cells. J. Nucleic. Acids 2011, 2011, 741723. [Google Scholar] [CrossRef]
- Alonas, E.; Vanover, D.; Blanchard, E.; Zurla, C.; Santangelo, P.J. Imaging viral RNA using multiply labeled tetravalent RNA imaging probes in live cells. Methods 2016, 98, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Santangelo, P.J.; Lifland, A.W.; Curt, P.; Sasaki, Y.; Bassell, G.J.; Lindquist, M.E.; Crowe, J.E., Jr. Single molecule-sensitive probes for imaging RNA in live cells. Nat. Methods 2009, 6, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, T.; Xiao, J. Circular RNAs: Promising Biomarkers for Human Diseases. EBioMedicine 2018, 34, 267–274. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| CircRNA Name | Cell or Tissue | Purpose | Reference | |
|---|---|---|---|---|
| FISH | circHECTD1 | Ischemic brain tissues | Quantification and localization | [58] |
| circARHGAP10 | Human non-small cell lung cancer tissues | Quantification and localization | [59] | |
| hsa_circRNA_103809 | Hepatocellular carcinoma tissues | [60] | ||
| hsa_circ_0017639 | Gastric cancer cells | Localization | [61] | |
| circSAMD4A | Preadipocytes | Localization | [62] | |
| circPVT1 | Human non-small cell lung cancer tissues | Localization | [63] | |
| circTTN | Bovine primary myoblasts | Localization | [64] | |
| circRHOT1 | PANC-1 and Capan-2 pancreatic cancer cells | Localization | [65] | |
| circEIF3J & circPAIP2 | HEK293 cells | Localization | [19] | |
| CDR1as (ciRS-7) | Adult brain, bladder cancer, and HEK293 cells | Localization | [21,66,69,70] | |
| CircFAT1 | HOS and 143B osteosarcoma cells | Localization | [67] | |
| circTADA2A | HOS and 143B osteosarcoma cells | Localization | [68] | |
| circITCH | Cardiomyocytes (hiPSC-CMs) | Localization | [71] | |
| circDLGAP4 | Brain endothelial cells | Quantification and localization | [72] | |
| circRHOBTB3 | HGC27 and AGS cells | Localization | [73] | |
| circCCDC9 | MKN45 and AGS cells | Localization | [74] | |
| circFAM114A2 | UCB cells | Localization | [75] | |
| circZNF532 | Pericytes | Localization | [76] | |
| circERBB2 | GBC-SD cells, SGC-996 cells | Localization | [77] | |
| circRHOT1 | Hepatocellular carcinoma (HCC) | Localization | [78] | |
| BaseScope | circSamd4 | C2C12 myoblasts | Localization | [51] |
| circSHKBP1 | HGC27 cells | Localization | [79] | |
| circAR3 | PCa tumor samples | Localization | [80] | |
| circPLEKHM3 | A2780 and OV90 cells | Localization | [81] | |
| circSlc45a4 | E15.5 mouse cortices | Localization | [82] | |
| circCACNA2D1 and circCACNA1E | Rhesus macaque brain | Localization | [83] | |
| sisRNAs | HeLa and mouse 3T3 cells | Localization | [84] | |
| CDR1as (ciRS-7) | Colon cancer and lesional skin | Quantification | [54,85] | |
| circPANs and circK7.3s | Kaposi’s sarcoma-associated herpesvirus (KSHV) infected tumor | Quantification | [86] | |
| circBHLF1 | Epstein Barr virus (EBV) | Localization | [87] | |
| Aptamer | tricRNA: Broccoli tricRNA: Spinach2 | HEK293T cells | Live cell tracking | [88] |
| tricY: Broccoli racRNA: Broccoli | HEK293T, HepG2, HeLa, and COS-7 cells | Live cell tracking | [89] |
| CircRNA Imaging Method | smFISH and ImmunoFISH | BaseScope | RNA Aptamer | CRISPR-Cas System | Molecular Beacon | MTRIP |
|---|---|---|---|---|---|---|
| Mechanism | Single fluorescent-labeled antisense probe targeting the backsplice junction of circRNA-associated protein detected with fluorescent antibodies. | One ZZ probe pair targets the circRNA junction. | A short stretch of RNA sequence introduced to target circRNA binds to fluorochrome for live-cell imaging. | SgRNA-mediated specific detection of target RNA by the fluorescent protein-tagged Cas protein. | Hairpin-shaped molecules with an internally quenched fluorophore whose fluorescence is restored when they bind to a target RNA. | Multiply labeled tetravalent RNA imaging probe that identifies RNA, enhanced signal to background ratio. |
| Advantages | Probes are inexpensive, easy to synthesize, and easily penetrate the cells. Multiplexing with other circRNAs, miRNAs, or target proteins. | Very sensitive, allows detection of single-copy circRNAs. | Thermally stable, robust in binding to dye. Cost-effective and low background. Suitable for live-cell imaging. | Live-cell imaging. Very sensitive and specific for target RNA. | Live-cell imaging. Low signal-to-background fluorescence from unbound dye. Capable of multiplexing. | Live-cell imaging. Higher specificity and high signal intensity. |
| Limitations | Time-consuming and works only in fixed cells. Difficult to visualize low-abundance circRNAs. CircRNA probes may target the parent mRNA due to sequence similarity. | Expensive and not suitable for live-cell imaging. | Limited knowledge on the optimal placing of the aptamer within circRNA. Limited availability of fluorophores in aptamer dye systems. Fluorophores can sometimes be cytotoxic. | Limited resources available for designing specific sgRNA. It cannot multiplex. | Requires extensive technical optimization of probe design and hybridization technique. Introduction into the cell may be challenging. | Expensive and difficult to synthesize. Introduction into the cell may be challenging. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bejugam, P.R.; Das, A.; Panda, A.C. Seeing Is Believing: Visualizing Circular RNAs. Non-Coding RNA 2020, 6, 45. https://doi.org/10.3390/ncrna6040045
Bejugam PR, Das A, Panda AC. Seeing Is Believing: Visualizing Circular RNAs. Non-Coding RNA. 2020; 6(4):45. https://doi.org/10.3390/ncrna6040045
Chicago/Turabian StyleBejugam, Pruthvi Raj, Aniruddha Das, and Amaresh Chandra Panda. 2020. "Seeing Is Believing: Visualizing Circular RNAs" Non-Coding RNA 6, no. 4: 45. https://doi.org/10.3390/ncrna6040045
APA StyleBejugam, P. R., Das, A., & Panda, A. C. (2020). Seeing Is Believing: Visualizing Circular RNAs. Non-Coding RNA, 6(4), 45. https://doi.org/10.3390/ncrna6040045