Targeted Genomic Screen Reveals Focal Long Non-Coding RNA Copy Number Alterations in Cancer Cell Lines

 ,
,  ,
,
Abstract
1. Introduction
2. Results
2.1. A Targeted Platform to Detect Focal Copy Number Changes in lncRNA Genes
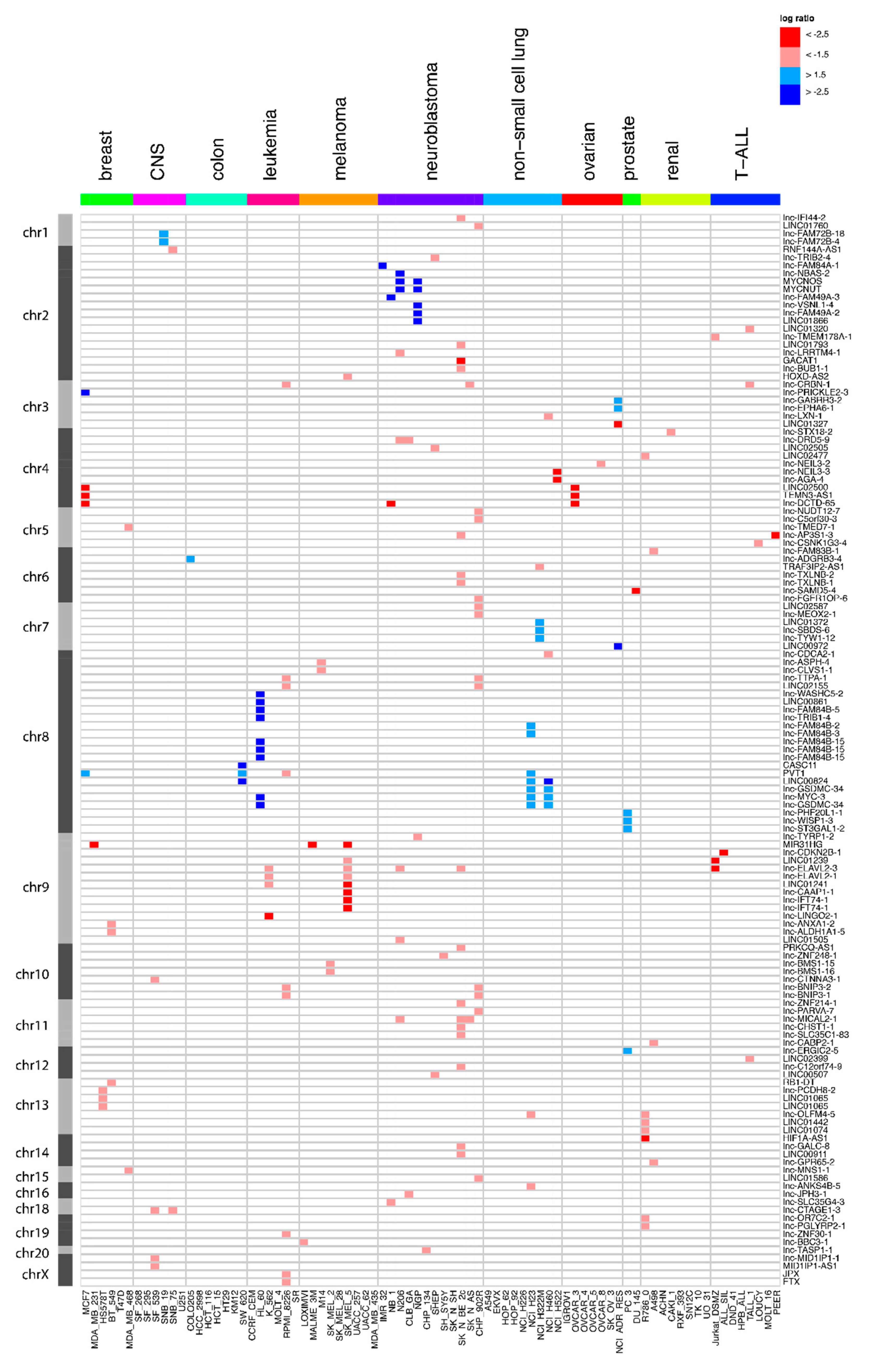
2.2. Frequent Focal lncRNA Copy Number Alterations in Cancer Cell Lines
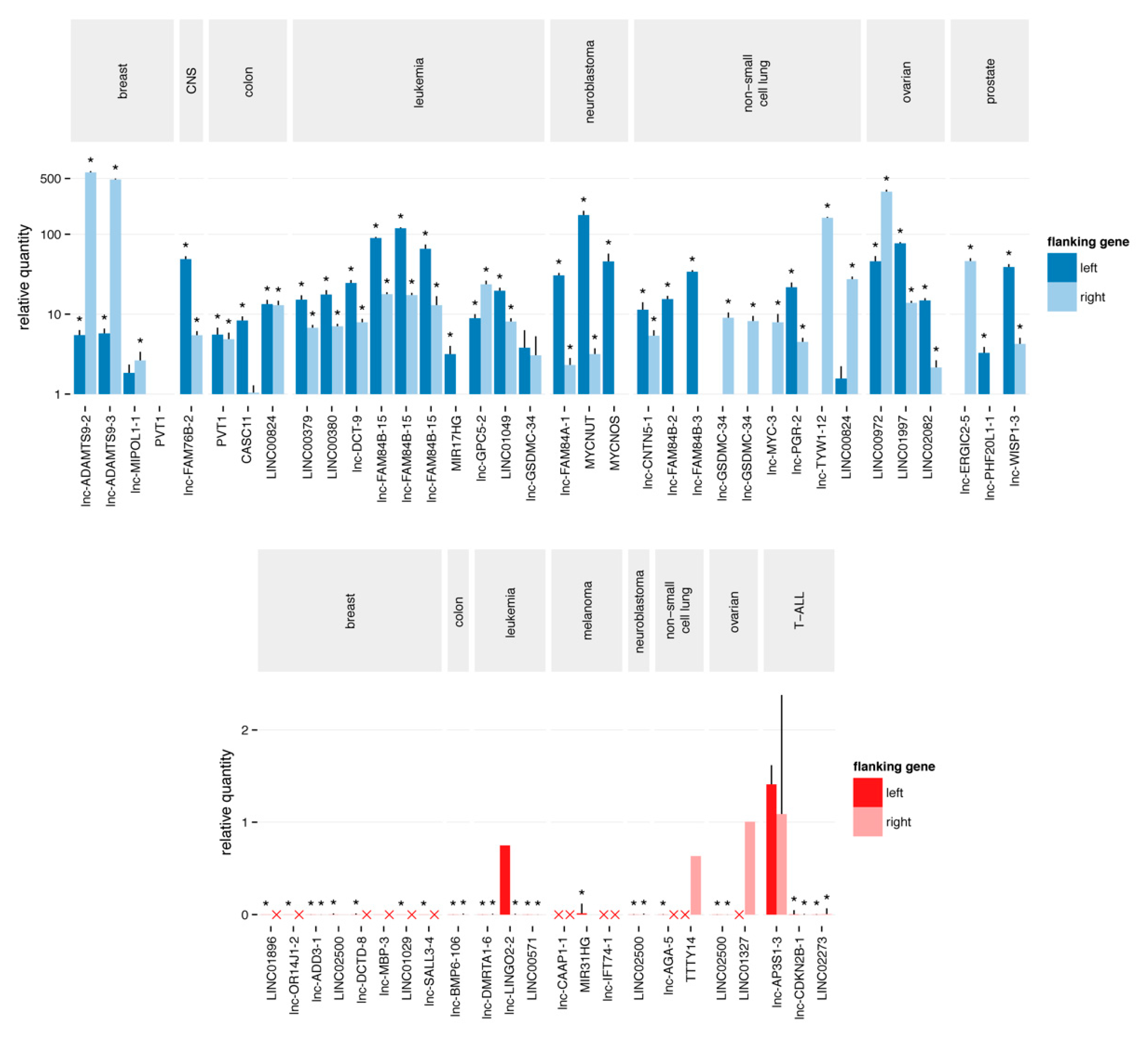
2.3. Quantitative Polymerase Chain Reaction Confirms the Majority of Focal Aberrations
2.4. Most Novel lncRNA Aberrations do not Correspond to Common Somatic Variants
3. Discussion
4. Materials and Methods
4.1. A lncRNA Exon Database
4.2. Array Comparative Genome Hybridization Platform Design
4.3. Cancer Cell Line DNA and RNA
4.4. Array Comparative Genome Hybridization
4.5. Segment Analysis and Filtering
4.6. Quantitive PCR Validation
4.7. Text Mining
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Friend, S.H.; Bernards, R.; Rogelj, S.; Weinberg, R.A.; Rapaport, J.M.; Albert, D.M.; Dryja, T.P. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986, 323, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Nau, M.M.; Brooks, B.J.; Carney, D.N.; Gazdar, A.F.; Battey, J.F.; Sausville, E.A.; Minna, J.D. Human small-cell lung cancers show amplification and expression of the N-myc gene. Proc. Natl. Acad. Sci. USA 1986, 83, 1092–1096. [Google Scholar] [CrossRef] [PubMed]
- Little, C.D.; Nau, M.M.; Carney, D.N.; Gazdar, A.F.; Minna, J.D. Amplification and expression of the c-myc oncogene in human lung cancer cell lines. Nature 1983, 306, 194–196. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, C.; Paez, J.G.; Chin, K.; Janne, P.A.; Chen, T.-H.; Girard, L.; Minna, J.; Christiani, D.; Leo, C.; et al. An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res. 2004, 64, 3060–3071. [Google Scholar] [CrossRef] [PubMed]
- Network, T.C.G.A. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Weir, B.A.; Woo, M.S.; Getz, G.; Perner, S.; Ding, L.; Beroukhim, R.; Lin, W.M.; Province, M.A.; Kraja, A.; Johnson, L.A.; et al. Characterizing the cancer genome in lung adenocarcinoma. Nature 2007, 450, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Volders, P.-J.; Verheggen, K.; Menschaert, G.; Vandepoele, K.; Martens, L.; Vandesompele, J.; Mestdagh, P. An update on LNCipedia: A database for annotated human lncRNA sequences. Nucleic Acids Res. 2015, 43, D174–D180. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T. Epigenetic regulation by long noncoding RNAs. Science 2012, 338, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Kogo, R.; Shimamura, T.; Mimori, K.; Kawahara, K.; Imoto, S.; Sudo, T.; Tanaka, F.; Shibata, K.; Suzuki, A.; Komune, S.; et al. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011, 71, 6320–6326. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Shkumatava, A.; Jan, C.; Sive, H.; Bartel, D.P. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 2011, 147, 1537–1550. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Diederichs, S. The Hallmarks of Cancer: A long non-coding RNA point of view. Rnabiology 2012, 9, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Hämmerle, M.; Eißmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.S.; Arun, G.; Stentrup, M.; Groß, M.; et al. The non-coding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2012, 73, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Kuo, W.L.; Stilwell, J.L.; Takano, H.; Lapuk, A.V.; Fridlyand, J.; Mao, J.H.; Yu, M.; Miller, M.A.; Santos, J.L.; et al. Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clin. Cancer Res. 2007, 13, 5745–5755. [Google Scholar] [CrossRef] [PubMed]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Fei, T.; Verhaak, R.G.W.; Su, Z.; Zhang, Y.; Brown, M.; Chen, Y.; Liu, X.S. Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nat. Struct. Mol. Biol. 2013, 20, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Feng, Y.; Zhang, D.; Zhao, S.D.; Hu, Z.; Greshock, J.; Zhang, Y.; Yang, L.; Zhong, X.; Wang, L.P.; et al. A functional genomic approach identifies FAL1 as an oncogenic long noncoding RNA that associates with BMI1 and represses p21 expression in cancer. Cancer Cell 2014, 26, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Ni, N.; Song, H.; Wang, X.; Xu, X.; Jiang, Y.; Sun, J. Up-regulation of long noncoding RNA FALEC predicts poor prognosis and promotes melanoma cell proliferation through epigenetically silencing p21. Biomed. Pharmacother. 2017, 96, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Akrami, R.; Jacobsen, A.; Hoell, J.; Schultz, N.; Sander, C.; Larsson, E. Comprehensive analysis of long non-coding RNAs in ovarian cancer reveals global patterns and targeted DNA Amplification. PLoS ONE 2013, 8, e80306. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Li, D.; Gong, M.; Wang, J.; Huang, X.; Wu, T.; Wang, C. Expression and clinical significance of the long non-coding RNA PVT1 in human gastric cancer. OncoTargets Ther. 2014, 7, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.-Y.; Moriarity, B.S.; Gong, W.; Akiyama, R.; Tiwari, A.; Kawakami, H.; Ronning, P.; Reuland, B.; Guenther, K.; Beadnell, T.C.; et al. PVT1 dependence in cancer with MYC copy-number increase. Nature 2014, 512, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Lanzós, A.; Carlevaro-Fita, J.; Mularoni, L.; Reverter, F.; Palumbo, E.; Guigó, R.; Johnson, R. Discovery of cancer driver long noncoding RNAs across 1112 tumour genomes: New candidates and distinguishing features. Sci. Rep. 2017, 7, 41544. [Google Scholar] [CrossRef] [PubMed]
- Volders, P.-J.; Helsens, K.; Wang, X.; Menten, B.; Martens, L.; Gevaert, K.; Vandesompele, J.; Mestdagh, P. LNCipedia: A database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Res. 2013, 41, D246–D251. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, T.; Barker, D.; Birney, E.; Cameron, G.; Chen, Y.; Clark, L.; Cox, T.; Cuff, J.; Curwen, V.; Down, T.; et al. The Ensembl genome database project. Nucleic Acids Res. 2002, 30, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Sante, T.; Vergult, S.; Volders, P.-J.; Kloosterman, W.P.; Trooskens, G.; de Preter, K.; Dheedene, A.; Speleman, F.; De Meyer, T.; Menten, B. ViVar: A comprehensive platform for the analysis and visualization of structural genomic variation. PLoS ONE 2014, 9, e113800. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.R.; Ziman, R.; Yuen, R.K.C.; Feuk, L.; Scherer, S.W. The database of genomic variants: A curated collection of structural variation in the human genome. Nucleic Acids Res. 2014, 42, D986–D992. [Google Scholar] [CrossRef] [PubMed]
- Rozen, S.; Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000, 132, 365–386. [Google Scholar] [PubMed]
- Tusnády, G.E.; Simon, I.; Váradi, A.; Arányi, T. BiSearch: Primer-design and search tool for PCR on bisulfite-treated genomes. Nucleic Acids Res. 2005, 33, e9. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cancer Type | # | Cell Lines | Origin |
|---|---|---|---|
| Breast | 6 | MCF7, MDA-MB-231, HS578T, BT-549, T47D, MDA-MB-468 | NCI |
| CNS | 6 | SF-268, SF-295, SF-539, SNB-19, SNB-75, U251 | NCI |
| Colon | 7 | COLO205, HCC-2998, HCT-116, HCT-15, HT29, KM12, SW-620 | NCI |
| Leukemia | 6 | CCRF-CEM *, HL-60, K-562, MOLT-4 *, RPMI-8226, SR | NCI |
| Melanoma | 9 | LOXIMVI, MALME-3M, M14, SK-MEL-2, SK-MEL-28, SK-MEL-5, UACC-257, UACC-62, MDA-MB-435 | NCI |
| Non-small cell lung | 9 | A549, EKVX, HOP-62, HOP-92, NCI-H226, NCI-H23, NCI-H322M, NCI-H460, NCI-H522 | NCI |
| Ovarian | 7 | IGROV1, OVCAR-3, OVCAR-4, OVCAR-5, OVCAR-8, SK-OV-3, NCI-ADR-RES | NCI |
| Prostate | 2 | PC-3, DU-145 | NCI |
| Renal | 8 | 786-0, A498, ACHN, CAKI-1, RXF-393, SN12C, TK-10, UO-31 | NCI |
| T-cell acute lymphoblastic leukemia | 8 | Jurkat-DSMZ, ALL-SIL, DND-41, HPB-ALL, TALL-1, LOUCY, MOLT-16, PEER | DSMZ |
| Neuroblastoma | 12 | CLB-GA, IMR-32, NB-1, NGP, N206, SHEP, SH-SY5Y, SK-N-SH, SK-N-BE-2c, CHP-134, SK-N-AS, CHP-902R | CMGG |
| 11 types | 80 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volders, P.-J.; Lefever, S.; Baute, S.; Nuytens, J.; Vanderheyden, K.; Menten, B.; Mestdagh, P.; Vandesompele, J. Targeted Genomic Screen Reveals Focal Long Non-Coding RNA Copy Number Alterations in Cancer Cell Lines. Non-Coding RNA 2018, 4, 21. https://doi.org/10.3390/ncrna4030021
Volders P-J, Lefever S, Baute S, Nuytens J, Vanderheyden K, Menten B, Mestdagh P, Vandesompele J. Targeted Genomic Screen Reveals Focal Long Non-Coding RNA Copy Number Alterations in Cancer Cell Lines. Non-Coding RNA. 2018; 4(3):21. https://doi.org/10.3390/ncrna4030021
Chicago/Turabian StyleVolders, Pieter-Jan, Steve Lefever, Shalina Baute, Justine Nuytens, Katrien Vanderheyden, Björn Menten, Pieter Mestdagh, and Jo Vandesompele. 2018. "Targeted Genomic Screen Reveals Focal Long Non-Coding RNA Copy Number Alterations in Cancer Cell Lines" Non-Coding RNA 4, no. 3: 21. https://doi.org/10.3390/ncrna4030021
APA StyleVolders, P.-J., Lefever, S., Baute, S., Nuytens, J., Vanderheyden, K., Menten, B., Mestdagh, P., & Vandesompele, J. (2018). Targeted Genomic Screen Reveals Focal Long Non-Coding RNA Copy Number Alterations in Cancer Cell Lines. Non-Coding RNA, 4(3), 21. https://doi.org/10.3390/ncrna4030021