The Role of Long Non-Coding RNAs (lncRNAs) in the Development and Progression of Fibrosis Associated with Nonalcoholic Fatty Liver Disease (NAFLD)



Abstract
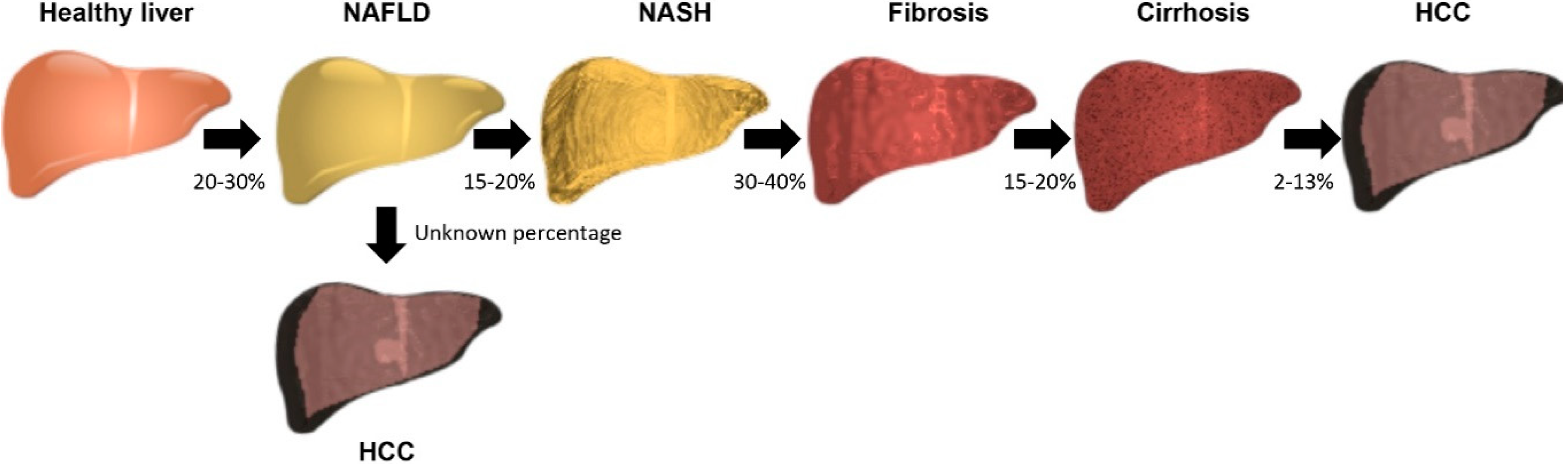
1. Introduction
2. Nonalcoholic Fatty Liver Disease: Prevalence, Clinical Management, and Risk Factors
3. lncRNAs in the Development and Progression of NAFLD-Related Fibrosis
3.1. lncRNAs from In Vivo Studies of NAFLD Fibrosis Animal Models
3.1.1. Maternally Expressed Gene 3 (MEG3)
3.1.2. Alu-Mediated p21 Transcriptional Regulator (APTR)
3.1.3. Metastasis-Associated Lung Adenocarcinoma Transcript 1 (MALAT1)
3.1.4. Plasmacytoma Variant Translocation 1 (PVT1)
3.1.5. Homeobox (HOX) Transcript Antisense RNA (HOTAIR)
3.1.6. LncRNA-Cyclooxygenase 2 (lncRNA-COX2)
3.1.7. Nuclear Enriched Abundant Transcript 1 (NEAT1)
3.1.8. Genome-Wide Identification of lncRNAs in Animal Models of Liver Fibrosis
3.2. Evidence from Studies of Patients with NAFLD Fibrosis
3.3. Circulating lncRNAs in NAFLD Fibrosis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- DiStefano, J.K. The emerging role of long noncoding RNAs in human disease. Methods Mol. Biol. 2018, 1706, 91–110. [Google Scholar] [PubMed]
- Zhao, X.Y.; Lin, J.D. Long noncoding RNAs: A new regulatory code in metabolic control. Trends Biochem. Sci. 2015, 40, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Demir, M.; Lang, S.; Steffen, H.M. Nonalcoholic fatty liver disease—Current status and future directions. J. Dig. Dis. 2015, 16, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Satapathy, S.K.; Sanyal, A.J. Epidemiology and natural history of nonalcoholic fatty liver disease. Semin. Liver Dis. 2015, 35, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Stepanova, M.; Afendy, M.; Fang, Y.; Younossi, Y.; Mir, H.; Srishord, M. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin. Gastroenterol. Hepatol. 2011, 9, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Spengler, E.K.; Loomba, R. Recommendations for diagnosis, referral for liver biopsy, and treatment of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Mayo Clin. Proc. 2015, 90, 1233–1246. [Google Scholar] [CrossRef] [PubMed]
- Dyson, J.K.; Anstee, Q.M.; McPherson, S. Non-alcoholic fatty liver disease: A practical approach to diagnosis and staging. Frontline Gastroenterol. 2014, 5, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Hoyumpa, A.M., Jr.; Greene, H.L.; Dunn, G.D.; Schenker, S. Fatty liver: Biochemical and clinical considerations. Am. J. Dig. Dis. 1975, 20, 1142–1170. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- McPherson, S.; Hardy, T.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 2015, 62, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654. [Google Scholar] [CrossRef] [PubMed]
- White, D.L.; Kanwal, F.; El-Serag, H.B. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin. Gastroenterol. Hepatol. 2012, 10, 1342–1359. [Google Scholar] [CrossRef] [PubMed]
- Cholankeril, G.; Wong, R.J.; Hu, M.; Perumpail, R.B.; Yoo, E.R.; Puri, P.; Younossi, Z.M.; Harrison, S.A.; Ahmed, A. Liver Transplantation for nonalcoholic steatohepatitis in the US: Temporal trends and outcomes. Dig. Dis Sci. 2017, 62, 2915–2922. [Google Scholar] [CrossRef] [PubMed]
- Charlton, M.R.; Burns, J.M.; Pedersen, R.A.; Watt, K.D.; Heimbach, J.K.; Dierkhising, R.A. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology 2011, 141, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Pais, R.; Barritt, A.S.T.; Calmus, Y.; Scatton, O.; Runge, T.; Lebray, P.; Poynard, T.; Ratziu, V.; Conti, F. NAFLD and liver transplantation: Current burden and expected challenges. J. Hepatol. 2016, 65, 1245–1257. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M. Review article: Current management of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2008, 28, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P. Genes or environment to determine alcoholic liver disease and non-alcoholic fatty liver disease. Liver Int. 2006, 26, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Koek, G.H.; Liedorp, P.R.; Bast, A. The role of oxidative stress in non-alcoholic steatohepatitis. Clin. Chim. Acta 2011, 412, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Jorge, A.S.; Andrade, J.M.; Paraiso, A.F.; Jorge, G.C.; Silveira, C.M.; de Souza, L.R.; Santos, E.P.; Guimaraes, A.L.; Santos, S.H.; De-Paula, A.M. Body mass index and the visceral adipose tissue expression of IL-6 and TNF-alpha are associated with the morphological severity of non-alcoholic fatty liver disease in individuals with class III obesity. Obes. Res. Clin. Pract. 2016, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Baker, R.D.; Bhatia, T.; Zhu, L.; Baker, S.S. Pathogenesis of nonalcoholic steatohepatitis. Cell. Mol. Life Sci. 2016, 73, 1969–1987. [Google Scholar] [CrossRef] [PubMed]
- Arrese, M.; Cabrera, D.; Kalergis, A.M.; Feldstein, A.E. Innate Immunity and Inflammation in NAFLD/NASH. Dig. Dis. Sci. 2016, 61, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Heymann, F.; Tacke, F. Immunology in the liver—From homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Wong, G.L.; Choi, P.C.; Chan, A.W.; Li, M.K.; Chan, H.Y.; Chim, A.M.; Yu, J.; Sung, J.J.; Chan, H.L. Disease progression of non-alcoholic fatty liver disease: A prospective study with paired liver biopsies at 3 years. Gut 2010, 59, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.T.; Newton, C.C.; Patel, A.V.; Jacobs, E.J.; Gapstur, S.M. Diabetes and cause-specific mortality in a prospective cohort of one million U.S. adults. Diabetes Care 2012, 35, 1835–1844. [Google Scholar] [CrossRef] [PubMed]
- Porepa, L.; Ray, J.G.; Sanchez-Romeu, P.; Booth, G.L. Newly diagnosed diabetes mellitus as a risk factor for serious liver disease. CMAJ 2010, 182, E526–E531. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Hazlehurst, J.M.; Woods, C.; Marjot, T.; Cobbold, J.F.; Tomlinson, J.W. Non-alcoholic fatty liver disease and diabetes. Metabolism 2016, 65, 1096–1108. [Google Scholar] [PubMed]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Ong, J.P.; Pitts, A.; Younossi, Z.M. Increased overall mortality and liver-related mortality in non-alcoholic fatty liver disease. J. Hepatol. 2008, 49, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Berardis, S.; Sokal, E. Pediatric non-alcoholic fatty liver disease: An increasing public health issue. Eur. J. Pediatr. 2014, 173, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Ajmera, V.H.; Gunderson, E.P.; VanWagner, L.B.; Lewis, C.E.; Carr, J.J.; Terrault, N.A. Gestational diabetes mellitus is strongly associated with non-alcoholic fatty liver disease. Am. J. Gastroenterol. 2016, 111, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Brumbaugh, D.E.; Tearse, P.; Cree-Green, M.; Fenton, L.Z.; Brown, M.; Scherzinger, A.; Reynolds, R.; Alston, M.; Hoffman, C.; Pan, Z.; et al. Intrahepatic fat is increased in the neonatal offspring of obese women with gestational diabetes. J. Pediatr. 2013, 162, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Celedon, M.A.; Lavine, J.E.; Salem, R.; Campbell, N.; Schork, N.J.; Shiehmorteza, M.; Yokoo, T.; Chavez, A.; Middleton, M.S.; et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology 2009, 136, 1585–1592. [Google Scholar] [PubMed]
- Struben, V.M.; Hespenheide, E.E.; Caldwell, S.H. Nonalcoholic steatohepatitis and cryptogenic cirrhosis within kindreds. Am. J. Med. 2000, 108, 9–13. [Google Scholar] [CrossRef]
- Willner, I.R.; Waters, B.; Patil, S.R.; Reuben, A.; Morelli, J.; Riely, C.A. Ninety patients with nonalcoholic steatohepatitis: Insulin resistance, familial tendency, and severity of disease. Am. J. Gastroenterol. 2001, 96, 2957–2961. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Chen, C.H.; Lo, M.T.; Schork, N.; Bettencourt, R.; Gonzalez, M.P.; Bhatt, A.; Hooker, J.; Shaffer, K.; Nelson, K.E.; et al. Shared genetic effects between hepatic steatosis and fibrosis: A prospective twin study. Hepatology 2016, 64, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Schork, N.; Chen, C.H.; Bettencourt, R.; Bhatt, A.; Ang, B.; Nguyen, P.; Hernandez, C.; Richards, L.; Salotti, J.; et al. Heritability of hepatic fibrosis and steatosis based on a prospective twin study. Gastroenterology 2015, 149, 1784–1793. [Google Scholar] [CrossRef] [PubMed]
- Tarnoki, A.D.; Tarnoki, D.L.; Bata, P.; Littvay, L.; Osztovits, J.; Jermendy, G.; Karlinger, K.; Lannert, A.; Preda, I.; Kiss, R.G.; et al. Heritability of non-alcoholic fatty liver disease and association with abnormal vascular parameters: A twin study. Liver Int. 2012, 32, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Palmer, N.D.; Musani, S.K.; Yerges-Armstrong, L.M.; Feitosa, M.F.; Bielak, L.F.; Hernaez, R.; Kahali, B.; Carr, J.J.; Harris, T.B.; Jhun, M.A.; et al. Characterization of European ancestry nonalcoholic fatty liver disease-associated variants in individuals of African and Hispanic descent. Hepatology 2013, 58, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Speliotes, E.K.; Yerges-Armstrong, L.M.; Wu, J.; Hernaez, R.; Kim, L.J.; Palmer, C.D.; Gudnason, V.; Eiriksdottir, G.; Garcia, M.E.; Launer, L.J.; et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011, 7, e1001324. [Google Scholar] [CrossRef] [PubMed]
- Wagenknecht, L.E.; Scherzinger, A.L.; Stamm, E.R.; Hanley, A.J.; Norris, J.M.; Chen, Y.D.; Bryer-Ash, M.; Haffner, S.M.; Rotter, J.I. Correlates and heritability of nonalcoholic fatty liver disease in a minority cohort. Obesity 2009, 17, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Rich, N.E.; Oji, S.; Mufti, A.R.; Browning, J.D.; Parikh, N.D.; Odewole, M.; Mayo, H.; Singal, A.G. Racial and ethnic disparities in nonalcoholic fatty liver disease prevalence, severity, and outcomes in the United States: A systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 2018, 16, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Bambha, K.; Belt, P.; Abraham, M.; Wilson, L.A.; Pabst, M.; Ferrell, L.; Unalp-Arida, A.; Bass, N.; Nonalcoholic Steatohepatitis Clinical Research Network Research Group. Ethnicity and nonalcoholic fatty liver disease. Hepatology 2012, 55, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Kumar, K.S.; Saboorian, M.H.; Thiele, D.L. Ethnic differences in the prevalence of cryptogenic cirrhosis. Am. J. Gastroenterol. 2004, 99, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Patel, Y.A.; Gifford, E.J.; Glass, L.M.; McNeil, R.; Turner, M.J.; Han, B.; Provenzale, D.; Choi, S.S.; Moylan, C.A.; Hunt, C.M. Risk factors for biopsy-proven advanced non-alcoholic fatty liver disease in the Veterans Health Administration. Aliment. Pharmacol. Ther. 2018, 47, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Deutsch, R.; Kahen, T.; Lavine, J.E.; Stanley, C.; Behling, C. Prevalence of fatty liver in children and adolescents. Pediatrics 2006, 118, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Pirola, C.J. Genetic predisposition in nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2017, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; George, J. Genetic and epigenetic mechanisms of NASH. Hepatol. Int. 2016, 10, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Magee, N.; Zou, A.; Zhang, Y. Pathogenesis of nonalcoholic steatohepatitis: Interactions between liver parenchymal and nonparenchymal cells. Biomed. Res. Int. 2016, 2016, 5170402. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Calabro, S.R.; Lee, A.; Maczurek, A.E.; Budzinska, M.A.; Warner, F.J.; McLennan, S.V.; Shackel, N.A. Hepatocytes in liver injury: Victim, bystander, or accomplice in progressive fibrosis? J. Gastroenterol. Hepatol. 2015, 30, 1696–1704. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, H.F.; Verhoofstad, W.A.; Brouwer, A.; de Leeuw, A.M.; Knook, D.L. Perisinusoidal fat-storing cells are the main vitamin A storage sites in rat liver. Exp. Cell. Res. 1985, 160, 138–149. [Google Scholar] [CrossRef]
- Friedman, S.L. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem. 2000, 275, 2247–2250. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015, 149, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Hagstrom, H.; Nasr, P.; Fredrikson, M.; Stal, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Luo, Y.; Liang, B.; Ye, L.; Lu, G.; He, W. Potential applications of MEG3 in cancer diagnosis and prognosis. Oncotarget 2017, 8, 73282–73295. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wu, Y.T.; Huang, C.; Meng, X.M.; Ma, T.T.; Wu, B.M.; Xu, F.Y.; Zhang, L.; Lv, X.W.; Li, J. Inhibitory effects of long noncoding RNA MEG3 on hepatic stellate cells activation and liver fibrogenesis. Biochim. Biophys. Acta 2014, 1842, 2204–2215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, Z.; Trottier, J.; Barbier, O.; Wang, L. Long noncoding RNA MEG3 induces cholestatic liver injury by interaction with PTBP1 to facilitate shp mRNA decay. Hepatology 2017, 65, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Negishi, M.; Wongpalee, S.P.; Sarkar, S.; Park, J.; Lee, K.Y.; Shibata, Y.; Reon, B.J.; Abounader, R.; Suzuki, Y.; Sugano, S.; et al. A new lncRNA, APTR, associates with and represses the CDKN1A/p21 promoter by recruiting polycomb proteins. PLoS ONE 2014, 9, e95216. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Zheng, J.; Mao, Y.; Dong, P.; Li, G.; Lu, Z.; Guo, C.; Liu, Z.; Fan, X. Long non-coding RNA APTR promotes the activation of hepatic stellate cells and the progression of liver fibrosis. Biochem. Biophys. Res. Commun. 2015, 463, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, A.; Stadler, P.F. Evolutionary clues in lncRNAs. Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, J.; Zhang, K.; Feng, B.; Wang, R.; Chen, L. Progress and prospects of long noncoding RNAs (lncRNAs) in hepatocellular carcinoma. Cell. Physiol. Biochem. 2015, 36, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Lu, Z.; Cai, J.; Huang, K.; Chen, B.; Li, G.; Dong, P.; Zheng, J. MALAT1 functions as a competing endogenous RNA to mediate Rac1 expression by sequestering miR-101b in liver fibrosis. Cell Cycle 2015, 14, 3885–3896. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Chen, J.; Chen, N. Long noncoding RNA MALAT1 promotes hepatic steatosis and insulin resistance by increasing nuclear SREBP-1c protein stability. Sci. Rep. 2016, 6, 22640. [Google Scholar] [CrossRef] [PubMed]
- Leti, F.; Legendre, C.; Still, C.D.; Chu, X.; Petrick, A.; Gerhard, G.S.; DiStefano, J.K. Altered expression of MALAT1 lncRNA in nonalcoholic steatohepatitis fibrosis regulates CXCL5 in hepatic stellate cells. Transl. Res. J. Lab. Clin. Med. 2017, 190, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Luo, P.; Wang, Q.; Ye, Y.; Wang, B. lncRNA PVT1 in cancer: A review and meta-analysis. Clin. Chim. Acta 2017, 474, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.L.; Khosroheidari, M.; Eddy, E.; Kiefer, J.; DiStefano, J.K. Correction: Role of microRNA 1207-5P and its host gene, the long non-coding RNA Pvt1, as mediators of extracellular matrix accumulation in the kidney: Implications for diabetic nephropathy. PLoS ONE 2016, 11, e0168353. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Yu, F.; Dong, P.; Wu, L.; Zhang, Y.; Hu, Y.; Zheng, L. Long non-coding RNA PVT1 activates hepatic stellate cells through competitively binding microRNA-152. Oncotarget 2016, 7, 62886–62897. [Google Scholar] [CrossRef] [PubMed]
- Bian, E.B.; Wang, Y.Y.; Yang, Y.; Wu, B.M.; Xu, T.; Meng, X.M.; Huang, C.; Zhang, L.; Lv, X.W.; Xiong, Z.G.; et al. Hotair facilitates hepatic stellate cells activation and fibrogenesis in the liver. Biochim. Biophys. Acta 2017, 1863, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.W.; Jang, J.Y.; Lee, S.H.; Kim, S.G.; Cheon, Y.K.; Kim, Y.S.; Cho, Y.D.; Kim, H.S.; Lee, J.S.; Jin, S.Y.; et al. Increased expression of cyclooxygenase-2 is associated with the progression to cirrhosis. Korean J. Intern. Med. 2010, 25, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Tong, Q.; Gong, A.Y.; Zhang, X.T.; Lin, C.; Ma, S.; Chen, J.; Hu, G.; Chen, X.M. LincRNA-Cox2 modulates TNF-α-induced transcription of Il12b gene in intestinal epithelial cells through regulation of Mi-2/NuRD-mediated epigenetic histone modifications. FASEB J. 2016, 30, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Mang, Y.; Li, L.; Ran, J.; Zhang, S.; Liu, J.; Li, L.; Chen, Y.; Liu, J.; Gao, Y.; Ren, G. Long noncoding RNA NEAT1 promotes cell proliferation and invasion by regulating hnRNP A2 expression in hepatocellular carcinoma cells. OncoTargets Ther. 2017, 10, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Jiang, Z.; Chen, B.; Dong, P.; Zheng, J. NEAT1 accelerates the progression of liver fibrosis via regulation of microRNA-122 and Kruppel-like factor 6. J. Mol. Med. 2017, 95, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Han, X.; Zhang, Z.; Zheng, L.; Hu, Z.; Yao, Q.; Cui, H.; Shu, G.; Si, M.; Li, C.; et al. The liver-enriched lnc-LFAR1 promotes liver fibrosis by activating TGFβ and Notch pathways. Nat. Commun. 2017, 8, 144. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Tang, J.; Xiang, T.; Lin, J.; Deng, C.; Peng, Y.; Zheng, J.; Hu, G. Genomewide identification of long noncoding RNAs in CCl4-induced liver fibrosis via RNA sequencing. Mol. Med. Rep. 2018, 18, 299–307. [Google Scholar] [PubMed]
- Delire, B.; Starkel, P.; Leclercq, I. Animal models for fibrotic liver diseases: What we have, what we need, and what is under development. J. Clin. Transl. Hepatol. 2015, 3, 53–66. [Google Scholar] [PubMed]
- Dong, S.; Chen, Q.L.; Song, Y.N.; Sun, Y.; Wei, B.; Li, X.Y.; Hu, Y.Y.; Liu, P.; Su, S.B. Mechanisms of CCl4-induced liver fibrosis with combined transcriptomic and proteomic analysis. J. Toxicol. Sci. 2016, 41, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Liu, X.; Yi, Z.; Xiao, X.; Yang, M.; Hu, G.; Liu, H.; Liao, L.; Huang, F. Genome-wide analysis of long noncoding RNA expression profiles in patients with non-alcoholic fatty liver disease. IUBMB Life 2015, 67, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Atanasovska, B.; Rensen, S.S.; van der Sijde, M.R.; Marsman, G.; Kumar, V.; Jonkers, I.; Withoff, S.; Shiri-Sverdlov, R.; Greve, J.W.M.; Faber, K.N.; et al. A liver-specific long noncoding RNA with a role in cell viability is elevated in human nonalcoholic steatohepatitis. Hepatology 2017, 66, 794–808. [Google Scholar] [CrossRef] [PubMed]
- Wobser, H.; Dorn, C.; Weiss, T.S.; Amann, T.; Bollheimer, C.; Buttner, R.; Scholmerich, J.; Hellerbrand, C. Lipid accumulation in hepatocytes induces fibrogenic activation of hepatic stellate cells. Cell Res. 2009, 19, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Colletti, L.M.; Green, M.; Burdick, M.D.; Kunkel, S.L.; Strieter, R.M. Proliferative effects of CXC chemokines in rat hepatocytes in vitro and in vivo. Shock 1998, 10, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Colletti, L.M.; Kunkel, S.L.; Green, M.; Burdick, M.; Strieter, R.M. Hepatic inflammation following 70% hepatectomy may be related to up-regulation of epithelial neutrophil activating protein-78. Shock 1996, 6, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Colletti, L.M.; Kunkel, S.L.; Walz, A.; Burdick, M.D.; Kunkel, R.G.; Wilke, C.A.; Strieter, R.M. The role of cytokine networks in the local liver injury following hepatic ischemia/reperfusion in the rat. Hepatology 1996, 23, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Flichman, D.; Garaycoechea, M.E.; San Martino, J.; Castano, G.O.; Pirola, C.J. Metastasis-associated lung adenocarcinoma transcript 1 as a common molecular driver in the pathogenesis of nonalcoholic steatohepatitis and chronic immune-mediated liver damage. Hepatol. Commun. 2018, 2, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Sumida, Y.; Nakajima, A.; Itoh, Y. Limitations of liver biopsy and non-invasive diagnostic tests for the diagnosis of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2014, 20, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Castera, L.; Pinzani, M. Non-invasive assessment of liver fibrosis: Are we ready? Lancet 2010, 375, 1419–1420. [Google Scholar] [CrossRef]
- DiStefano, J.K. Long noncoding RNAs in the initiation, progression, and metastasis of hepatocellular carcinoma. Non-Coding RNA Res. 2017, 2, 129–136. [Google Scholar] [CrossRef]
- Bolha, L.; Ravnik-Glavac, M.; Glavac, D. Long noncoding RNAs as biomarkers in cancer. Dis. Mark. 2017, 2017, 7243968. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Lei, R.; Ning, Q. Circulating long noncoding RNAs as novel biomarkers of human diseases. Biomark. Med. 2016, 10, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Jiang, R.; Deng, L.; Zhang, X.; Wang, K.; Sun, B. Circulation long non-coding RNAs act as biomarkers for predicting tumorigenesis and metastasis in hepatocellular carcinoma. Oncotarget 2015, 6, 4505–4515. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Sun, Y.; Liu, L.; Zhou, B.; Wang, S.; Gu, D. Circulating lncRNAs serve as diagnostic markers for hepatocellular carcinoma. Cell. Physiol. Biochem. 2017, 44, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Toraih, E.A.; Ellawindy, A.; Fala, S.Y.; Al Ageeli, E.; Gouda, N.S.; Fawzy, M.S.; Hosny, S. Oncogenic long noncoding RNA MALAT1 and HCV-related hepatocellular carcinoma. Biomed. Pharmacother. 2018, 102, 653–669. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.H.; Yang, F.; Wang, F.; Ma, J.Z.; Guo, Y.J.; Tao, Q.F.; Liu, F.; Pan, W.; Wang, T.T.; Zhou, C.C.; et al. A long noncoding RNA activated by TGF-β promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 2014, 25, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Fu, N.; Niu, X.; Wang, Y.; Du, H.; Wang, B.; Du, J.; Li, Y.; Wang, R.; Zhang, Y.; Zhao, S.; et al. Role of lncRNA-activated by transforming growth factor beta in the progression of hepatitis C virus-related liver fibrosis. Discov. Med. 2016, 22, 29–42. [Google Scholar] [PubMed]
- Yu, F.; Zhou, G.; Huang, K.; Fan, X.; Li, G.; Chen, B.; Dong, P.; Zheng, J. Serum lincRNA-p21 as a potential biomarker of liver fibrosis in chronic hepatitis B patients. J. Viral Hepat. 2017, 24, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ross, R.A.; Zhao, S.; Tu, W.; Liangpunsakul, S.; Wang, L. lncRNA AK054921 and AK128652 are potential serum biomarkers and predictors of patient survival with alcoholic cirrhosis. Hepatol. Commun. 2017, 1, 513–523. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| lncRNA | Expression | Replicated in Humans | Ref |
|---|---|---|---|
| MEG3 | downregulated | Yes | [61,62] |
| APTR | upregulated | Yes | [63,64] |
| MALAT1 | upregulated | Yes | [67,68] |
| PVT1 | upregulated | No | [72] |
| HOTAIR | upregulated | Yes | [73] |
| lncRNA-COX2 | upregulated | No | [75] |
| NEAT1 | upregulated | Yes | [76,77] |
| lnc-LFAR1 | upregulated in HSC | No | [78] |
| downregulated in hepatocytes | |||
| NR_002155.1 | downregulated | No | [79] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanson, A.; Wilhelmsen, D.; DiStefano, J.K. The Role of Long Non-Coding RNAs (lncRNAs) in the Development and Progression of Fibrosis Associated with Nonalcoholic Fatty Liver Disease (NAFLD). Non-Coding RNA 2018, 4, 18. https://doi.org/10.3390/ncrna4030018
Hanson A, Wilhelmsen D, DiStefano JK. The Role of Long Non-Coding RNAs (lncRNAs) in the Development and Progression of Fibrosis Associated with Nonalcoholic Fatty Liver Disease (NAFLD). Non-Coding RNA. 2018; 4(3):18. https://doi.org/10.3390/ncrna4030018
Chicago/Turabian StyleHanson, Amanda, Danielle Wilhelmsen, and Johanna K. DiStefano. 2018. "The Role of Long Non-Coding RNAs (lncRNAs) in the Development and Progression of Fibrosis Associated with Nonalcoholic Fatty Liver Disease (NAFLD)" Non-Coding RNA 4, no. 3: 18. https://doi.org/10.3390/ncrna4030018
APA StyleHanson, A., Wilhelmsen, D., & DiStefano, J. K. (2018). The Role of Long Non-Coding RNAs (lncRNAs) in the Development and Progression of Fibrosis Associated with Nonalcoholic Fatty Liver Disease (NAFLD). Non-Coding RNA, 4(3), 18. https://doi.org/10.3390/ncrna4030018