Detecting Disease Specific Pathway Substructures through an Integrated Systems Biology Approach




Abstract
:1. Introduction
2. Results
3. Discussion
3.1. Analysis of BRCA Results Using SPECifIC
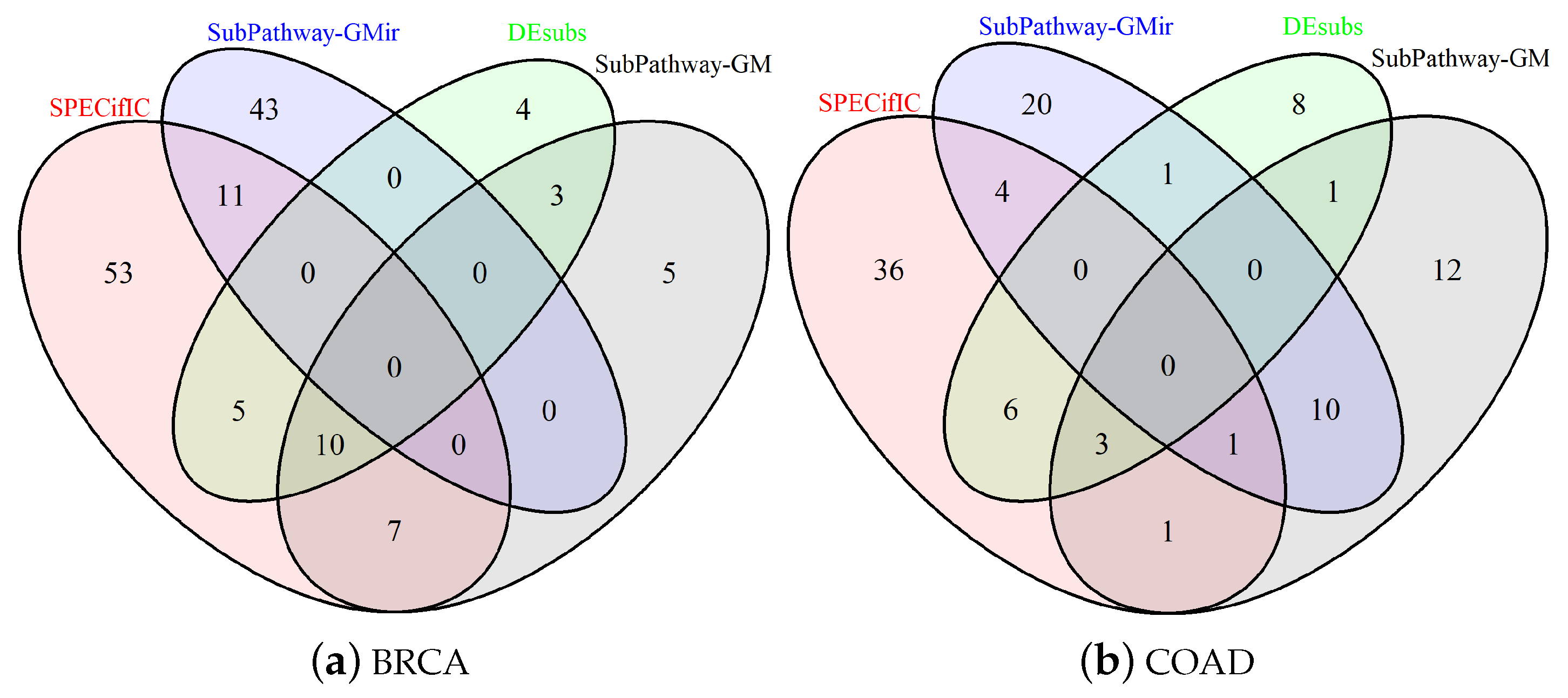
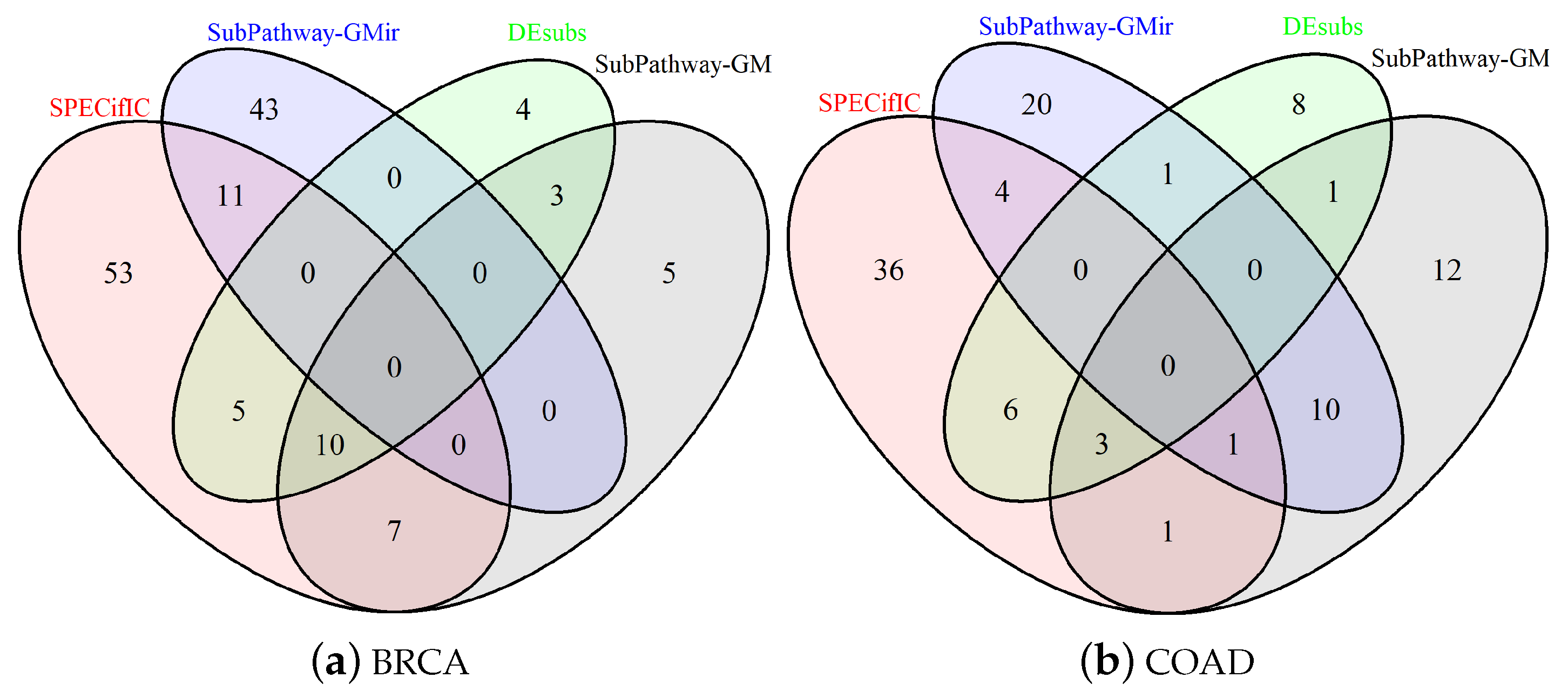
3.2. Comparison for BRCA-Related Pathways
3.3. Analysis of COAD Results Using SPECifIC
3.4. Comparison for COAD-Related Pathways
4. Materials and Methods
4.1. Overview of the Methodology
4.1.1. Extending MITHrIL
4.1.2. Subpathway Extraction
4.2. NoIs Selection
Subpathway Enrichment Analysis
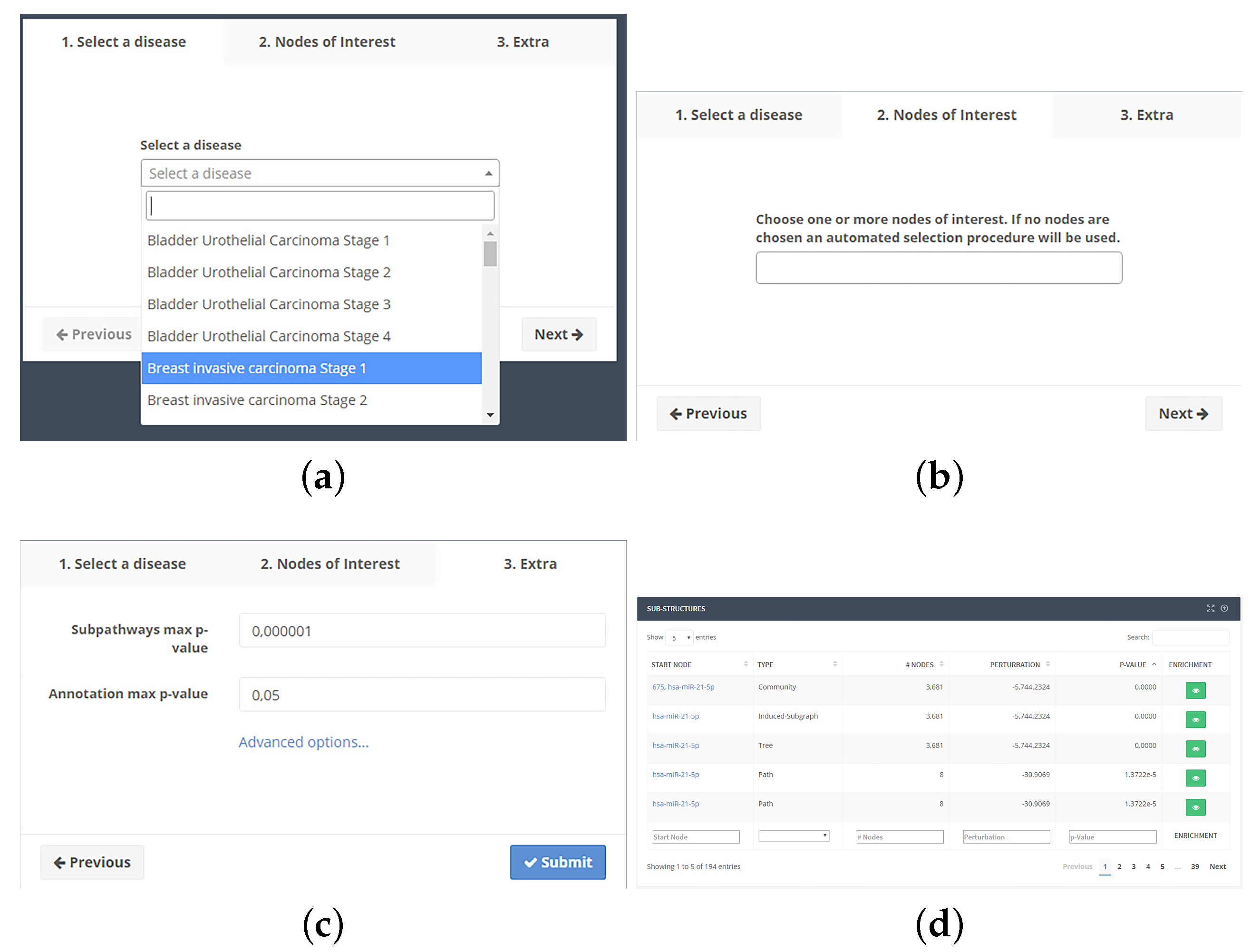
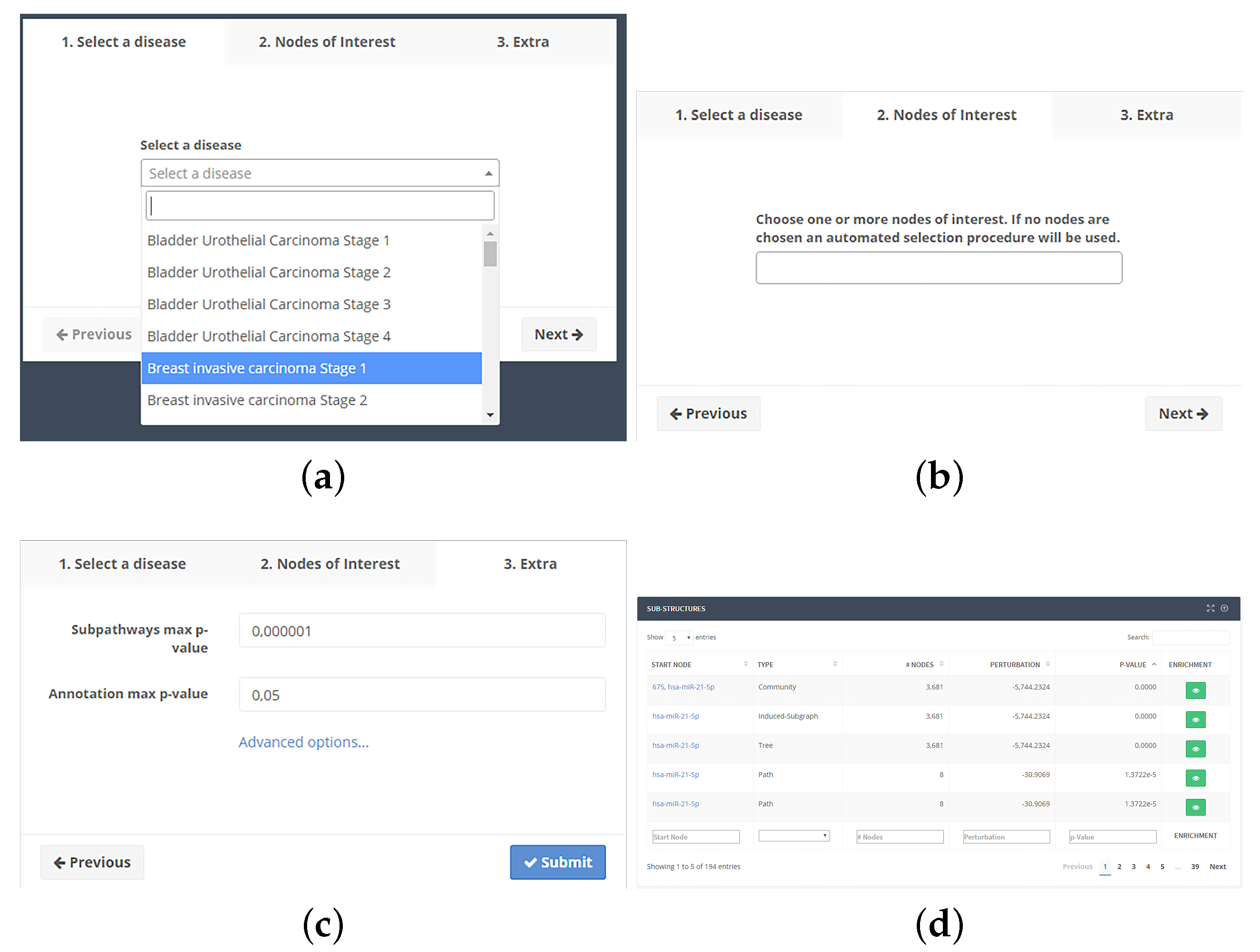
4.3. Web Interface
4.3.1. Expression Data Sources
4.3.2. Analysis Workflow
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TCGA | The Cancer Genome Atlas |
| CRC | Colorectal Carcinoma |
| BRCA | Breast Invasive Carcinoma |
| COAD | Colon Adenocarcinoma |
| READ | Rectal Adenocarcinoma |
| PF | Perturbation Factor |
| MITHrIL | Mirna enrIched paTHway Impact anaLysis |
| SPECifIC | SubPathway ExtraCtor and enrICher |
| SPIA | Signaling Pathway Impact Analysis |
| TEAK | Topology Enrichment Analysis frameworK |
| PARADIGM | PAthway Representation and Analysis by Direct Reference on Graphical Models |
| DEG | Differentially Expressed Gene |
| IF | Impact Factor |
References
- Khatri, P.; Sirota, M.; Butte, A.J. Ten years of pathway analysis: Current approaches and outstanding challenges. PLoS Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2015, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.G.; Gross, B.E.; Demir, E.; Rodchenkov, I.; Babur, Ö.; Anwar, N.; Schultz, N.; Bader, G.D.; Sander, C. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2011, 39, D685–D690. [Google Scholar] [CrossRef] [PubMed]
- Khatri, P.; Draghici, S.; Ostermeier, G.C.; Krawetz, S.A. Profiling gene expression using onto-express. Genomics 2002, 79, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Draghici, S.; Khatri, P.; Martins, R.P.; Ostermeier, G.C.; Krawetz, S.A. Global functional profiling of gene expression. Genomics 2003, 81, 98–104. [Google Scholar] [PubMed]
- Berriz, G.F.; King, O.D.; Bryant, B.; Sander, C.; Roth, F.P. Characterizing gene sets with FuncAssociate. Bioinformatics 2003, 19, 2502–2504. [Google Scholar] [CrossRef] [PubMed]
- Beißbarth, T.; Speed, T.P. GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics 2004, 20, 1464–1465. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Davis, C.I.; Hartl, D.L. GeneMerge—Post-genomic analysis, data mining, and hypothesis testing. Bioinformatics 2003, 19, 891–892. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Brun, C.; Remy, E.; Mouren, P.; Thieffry, D.; Jacq, B. GOToolBox: Functional analysis of gene datasets based on Gene Ontology. Genome Biol. 2004, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Doniger, S.W.; Salomonis, N.; Dahlquist, K.D.; Vranizan, K.; Lawlor, S.C.; Conklin, B.R. MAPPFinder: using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biol. 2003, 4, 1. [Google Scholar] [CrossRef]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Greenberg, S.A.; Kong, S.W.; Altschuler, J.; Kohane, I.S.; Park, P.J. Discovering statistically significant pathways in expression profiling studies. Proc. Natl. Acad. Sci. USA 2005, 102, 13544–13549. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H. Non-linear tests for identifying differentially expressed genes or genetic networks. Bioinformatics 2006, 22, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Draghici, S.; Khatri, P.; Tarca, A.L.; Amin, K.; Done, A.; Voichita, C.; Georgescu, C.; Romero, R. A systems biology approach for pathway level analysis. Genome Res. 2007, 17, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Tarca, A.L.; Draghici, S.; Khatri, P.; Hassan, S.S.; Mittal, P.; Kim, J.S.; Kim, C.J.; Kusanovic, J.P.; Romero, R. A novel signaling pathway impact analysis. Bioinformatics 2009, 25, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Vaske, C.J.; Benz, S.C.; Sanborn, J.Z.; Earl, D.; Szeto, C.; Zhu, J.; Haussler, D.; Stuart, J.M. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 2010, 26, i237–i245. [Google Scholar] [CrossRef] [PubMed]
- Sedgewick, A.J.; Benz, S.C.; Rabizadeh, S.; Soon-Shiong, P.; Vaske, C.J. Learning subgroup-specific regulatory interactions and regulator independence with PARADIGM. Bioinformatics 2013, 29, i62–i70. [Google Scholar] [CrossRef] [PubMed]
- Calura, E.; Martini, P.; Sales, G.; Beltrame, L.; Chiorino, G.; D’Incalci, M.; Marchini, S.; Romualdi, C. Wiring miRNAs to pathways: A topological approach to integrate miRNA and mRNA expression profiles. Nucleic Acids Res. 2014, 42, e96. [Google Scholar] [CrossRef] [PubMed]
- Alaimo, S.; Giugno, R.; Acunzo, M.; Veneziano, D.; Ferro, A.; Pulvirenti, A. Post-transcriptional knowledge in pathway analysis increases the accuracy of phenotypes classification. Oncotarget 2016, 7, 54572–54582. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Shang, D.; Wang, Y.; Li, J.; Han, J.; Wang, S.; Yao, Q.; Wang, Y.; Zhang, Y.; Zhang, C.; et al. Characterizing the network of drugs and their affected metabolic subpathways. PLoS ONE 2012, 7, e47326. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, C.; Shang, D.; Li, J.; Han, J.; Miao, Y.; Wang, Y.; Wang, Q.; Li, W.; Wu, C.; et al. The implications of relationships between human diseases and metabolic subpathways. PLoS ONE 2011, 6, e21131. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Han, J.; Yao, Q.; Zou, C.; Xu, Y.; Zhang, C.; Shang, D.; Zhou, L.; Zou, C.; Sun, Z.; et al. Subpathway-GM: Identification of metabolic subpathways via joint power of interesting genes and metabolites and their topologies within pathways. Nucleic Acids Res. 2013, 41, E101. [Google Scholar] [CrossRef] [PubMed]
- Judeh, T.; Johnson, C.; Kumar, A.; Zhu, D. TEAK: Topology enrichment analysis framework for detecting activated biological subpathways. Nucleic Acids Res. 2013, 41, 1425–1437. [Google Scholar] [CrossRef] [PubMed]
- Vrahatis, A.G.; Balomenos, P.; Tsakalidis, A.K.; Bezerianos, A. DEsubs: An R package for flexible identification of differentially expressed subpathways using RNA-seq experiments. Bioinformatics 2016, 32, 3844–3846. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Xu, Y.; Zhang, Y.; Sun, Z.; Han, J.; Zhang, C.; Yang, H.; Shang, D.; Su, F.; Shi, X.; et al. Subpathway-GMir: Identifying miRNA-mediated metabolic subpathways by integrating condition-specific genes, microRNAs, and pathway topologies. Oncotarget 2015, 6, 39151. [Google Scholar] [PubMed]
- Vrahatis, A.G.; Dimitrakopoulou, K.; Balomenos, P.; Tsakalidis, A.K.; Bezerianos, A. CHRONOS: A time-varying method for microRNA-mediated subpathway enrichment analysis. Bioinformatics 2016, 32, 884–892. [Google Scholar] [CrossRef] [PubMed]
- SPECifIC. Sub-Pathway Extractor and Enricher. Available online: https://alpha.dmi.unict.it/specific/ (accessed on 28 December 2016).
- Béroud, C.; Letovsky, S.I.; Braastad, C.D.; Caputo, S.M.; Beaudoux, O.; Bignon, Y.J.; Paillerets, B.; Bronner, M.; Buell, C.M.; Collod-Béroud, G.; et al. BRCA Share: A Collection of Clinical BRCA Gene Variants. Hum. Mutat. 2016, 37, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Hollestelle, A.; Nagel, J.H.; Smid, M.; Lam, S.; Elstrodt, F.; Wasielewski, M.; Ng, S.S.; French, P.J.; Peeters, J.K.; Rozendaal, M.J.; et al. Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Res. Treat. 2010, 121, 53–64. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global Cancer Statistics, 2012. CA: A Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Minsky, B.D. Unique considerations in the patient with rectal cancer. Semin. Oncol. 2011, 4, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network; Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [PubMed]
- Piñero, J.; Queralt-Rosinach, N.; Bravo, À.; Deu-Pons, J.; Bauer-Mehren, A.; Baron, M.; Sanz, F.; Furlong, L.I. DisGeNET: A discovery platform for the dynamical exploration of human diseases and their genes. Database 2015, 2015, bav028. [Google Scholar] [CrossRef] [PubMed]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.G.; Barnes, K.C.; Bright, T.J.; Wang, S.A. The genetic association database. Nat. Genet. 2004, 36, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Stern, H.M.; Ge, L.; O’Brien, C.; Haydu, L.; Honchell, C.D.; Haverty, P.M.; Peters, B.A.; Wu, T.D.; Amler, L.C.; et al. Genetic Alterations and Oncogenic Pathways Associated with Breast Cancer Subtypes. Mol. Cancer Res. 2009, 7, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Schiff, R. Mechanisms of Endocrine Resistance in Breast Cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef] [PubMed]
- LaPensee, E.W.; Ben-Jonathan, N. Novel roles of prolactin and estrogens in breast cancer: Resistance to chemotherapy. Endocr.-Relat. Cancer 2010, 17, R91–R107. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Campiglio, M.; Maiello, M.R.; De Luca, A.; Mancino, M.; Gallo, M.; D’Alessio, A.; Menard, S. Breast cancer cells with acquired resistance to the EGFR tyrosine kinase inhibitor gefitinib show persistent activation of MAPK signaling. Breast Cancer Res. Treat. 2008, 112, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Bulun, S.E.; Chen, D.; Moy, I.; Brooks, D.C.; Zhao, H. Aromatase, breast cancer and obesity: A complex interaction. Trends Endocrinol. Metab. 2012, 23, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.C.; Tu, S.H.; Huang, C.S.; Chen, C.S.; Ho, C.T.; Lin, H.W.; Lee, C.H.; Chang, H.W.; Chang, C.H.; Wu, C.H.; et al. Human breast cancer cell metastasis is attenuated by lysyl oxidase inhibitors through down-regulation of focal adhesion kinase and the paxillin-signaling pathway. Breast Cancer Res. Treat. 2012, 134, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- Emery, L.A.; Tripathi, A.; King, C.; Kavanah, M.; Mendez, J.; Stone, M.D.; de las Morenas, A.; Sebastiani, P.; Rosenberg, C.L. Early Dysregulation of Cell Adhesion and Extracellular Matrix Pathways in Breast Cancer Progression. Am. J. Pathol. 2009, 175, 1292–1302. [Google Scholar] [CrossRef] [PubMed]
- Lal, I.; Dittus, K.; Holmes, C.E. Platelets, coagulation and fibrinolysis in breast cancer progression. Breast Cancer Res. 2013, 15, 207. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.B.; Jokar, I.; Zou, A.; Lambert, D. CCL2/CCR2 chemokine signaling coordinates survival and motility of breast cancer cells through Smad3 protein-and p42/44 mitogen-activated protein kinase (MAPK)-dependent mechanisms. J. Biol. Chem. 2012, 287, 36593–36608. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Rodrik, V.; Foster, D.A. Alternative phospholipase D|[sol]|mTOR survival signal in human breast cancer cells. Oncogene 2005, 24, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Nair, S.A.; Pillai, M.R. Biology of PPAR 947; in Cancer: A Critical Review on Existing Lacunae. Curr. Mol. Med. 2007, 7, 532–540. [Google Scholar] [CrossRef]
- Do, M.T.; Kim, H.G.; Tran, T.T.P.; Khanal, T.; Choi, J.H.; Chung, Y.C.; Jeong, T.C.; Jeong, H.G. Metformin suppresses CYP1A1 and CYP1B1 expression in breast cancer cells by down-regulating aryl hydrocarbon receptor expression. Toxicol. Appl. Pharmacol. 2014, 280, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Hakkola, J.; Pasanen, M.; Pelkonen, O.; Hukkanen, J.; Evisalmi, S.; Anttila, S.; Rane, A.; Mäntylä, M.; Purkunen, R.; Saarikoski, S.; et al. Expression of CYP1B1 in human adult and fetal tissues and differential inducibility of CYP1B1 and CYP1A1 by Ah receptor ligands in human placenta and cultured cells. Carcinogenesis 1997, 18, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, D.; Yee, D. The IGF system and breast cancer. Endocr.-Relat. Cancer 2001, 8, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Pust, S.; Klokk, T.I.; Musa, N.; Jenstad, M.; Risberg, B.; Erikstein, B.; Tcatchoff, L.; Liestøl, K.; Danielsen, H.E.; van Deurs, B.; et al. Flotillins as regulators of ErbB2 levels in breast cancer. Oncogene 2013, 32, 3443–3451. [Google Scholar] [CrossRef] [PubMed]
- Patani, N.; Jiang, W.G.; Mokbel, K. Brain-derived neurotrophic factor expression predicts adverse pathological & clinical outcomes in human breast cancer. Cancer Cell Int. 2011, 11, 23. [Google Scholar] [PubMed]
- Dolle, L.; Adriaenssens, E.; Yazidi-Belkoura, I.E.; Bourhis, X.L.; Nurcombe, V.; Hondermarck, H. Nerve Growth Factor Receptors and Signaling in Breast Cancer. Curr. Cancer Drug Targets 2004, 4, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.J.; Dahiya, S.; Richardson, E.; Erlander, M.; Sgroi, D.C. Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res. 2009, 11, R7. [Google Scholar] [CrossRef] [PubMed]
- Turton, N.J.; Judah, D.J.; Riley, J.; Davies, R.; Lipson, D. Gene expression and amplification in breast carcinoma cells with intrinsic and acquired doxorubicin resistance. Oncogene 2001, 20, 1300–1306. [Google Scholar] [CrossRef] [PubMed]
- Viglietto, G.; Motti, M.L.; Bruni, P.; Melillo, R.M.; D’Alessio, A.; Califano, D.; Vinci, F.; Chiappetta, G.; Tsichlis, P.; Bellacosa, A.; et al. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27Kip1 by PKB/Akt-mediated phosphorylation in breast cancer. Nat. Med. 2002, 8, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Hoover, K.B.; Liao, S.Y.; Bryant, P.J. Loss of the Tight Junction MAGUK ZO-1 in Breast Cancer. Am. J. Pathol. 1998, 153, 1767–1773. [Google Scholar] [CrossRef]
- Kominsky, S.L.; Argani, P.; Korz, D.; Evron, E.; Raman, V.; Garrett, E.; Rein, A.; Sauter, G.; Kallioniemi, O.P.; Sukumar, S. Loss of the tight junction protein claudin-7 correlates with histological grade in both ductal carcinoma in situ and invasive ductal carcinoma of the breast. Oncogene 2003, 22, 2021–2033. [Google Scholar] [CrossRef] [PubMed]
- Pierceall, W.E.; Woodard, A.S.; Morrow, J.S.; Rimm, D.; Fearon, E.R. Frequent alterations in E-cadherin and alpha- and beta-catenin expression in human breast cancer cell lines. Oncogene 1995, 11, 1319–1326. [Google Scholar] [PubMed]
- McLachlan, E.; Shao, Q.; Laird, D.W. Connexins and Gap Junctions in Mammary Gland Development and Breast Cancer Progression. J. Membr. Biol. 2007, 218, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Enomoto, A.; Takahashi, M. Cell biology of the movement of breast cancer cells: Intracellular signalling and the actin cytoskeleton. Cancer Lett. 2009, 284, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Warren, R.Q.; Ivy, S.P. Breast cancer growth and metastasis: interplay between cancer stem cells, embryonic signaling pathways and epithelial-to-mesenchymal transition. Breast Cancer Res. 2011, 13, 211. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Subramanyam, D.; Dey, D.; Kumar, R.V.; Rangarajan, A. Cooperation of Notch and Ras/MAPK signaling pathways in human breast carcinogenesis. Mol. Cancer 2009, 8, 128. [Google Scholar] [CrossRef] [PubMed]
- Ebi, H.; Costa, C.; Faber, A.C.; Nishtala, M.; Kotani, H.; Juric, D.; Della Pelle, P.; Song, Y.; Yano, S.; Mino-Kenudson, M.; et al. PI3K regulates MEK/ERK signaling in breast cancer via the Rac-GEF, P-Rex1. Proc. Natl. Acad. Sci. USA 2013, 110, 21124–21129. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, M.P.; Pauley, R.; Heppner, G. Host microenvironment in breast cancer development: Extracellular matrix–stromal cell contribution to neoplastic phenotype of epithelial cells in the breast. Breast Cancer Res. 2003, 5, 130. [Google Scholar] [CrossRef] [PubMed]
- Emons, G.; Grndker, C.G.; Grnthert, A.R.G.; Westphalen, S.; Kavanagh, J.; Verschraegen, C. GnRH antagonists in the treatment of gynecological and breast cancers. Endocr.-Relat. Cancer 2003, 10, 291–299. [Google Scholar] [CrossRef] [PubMed]
- McMahon, G. VEGF Receptor Signaling in Tumor Angiogenesis. Oncologist 2000, 5, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.R.; Brown, K.A. Minireview: Obesity and Breast Cancer: A Tale of Inflammation and Dysregulated Metabolism. Mol. Endocrinol. 2013, 27, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Dean, S.J.; Rhodes, A. Triple negative breast cancer: the role of metabolic pathways. Malays J. Pathol. 2014, 6, 155–162. [Google Scholar]
- Zeestraten, E.; Benard, A.; Reimers, M.S. The prognostic value of the apoptosis pathway in colorectal cancer: A review of the literature on biomarkers identified by immunohistochemistry. Biomark. Cancer 2013, 5, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Gulhati, P.; Rampy, B.A.; Han, Y.; Rychahou, P.G.; Doan, H.Q.; Weiss, H.L.; Evers, B.M. Novel Expression Patterns of PI3K/Akt/mTOR Signaling Pathway Components in Colorectal Cancer. J. Am. Coll. Surg. 2010, 210, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Benvenuti, S.; Sartore-Bianchi, A.; Di Nicolantonio, F.; Zanon, C.; Moroni, M.; Veronese, S.; Siena, S.; Bardelli, A. Oncogenic Activation of the RAS/RAF Signaling Pathway Impairs the Response of Metastatic Colorectal Cancers to Anti–Epidermal Growth Factor Receptor Antibody Therapies. Cancer Res. 2007, 67, 2643–2648. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Iwadate, M.; Higashimoto, M. Expression of phospholipase D2 in human colorectal carcinoma. Oncol. Rep. 2007, 18, 1329–1334. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.W.; Min, D.S. Positive Feedback Regulation between Phospholipase D and Wnt Signaling Promotes Wnt-Driven Anchorage-Independent Growth of Colorectal Cancer Cells. PLoS ONE 2010, 5, e12109. [Google Scholar] [CrossRef] [PubMed]
- Zong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C. Overexpression of Hypoxia inducible Factor 1alpha in common Human Cancer and their Metastasis. Cancer Res. 1999, 59, 5830–5835. [Google Scholar]
- Kaidi, A.; Qualtrough, D.; Williams, A.C.; Paraskeva, C. Direct Transcriptional Up-regulation of Cyclooxygenase- 2 by Hypoxia-Inducible Factor (HIF)-1 Promotes Colorectal Tumor Cell Survival and Enhances HIF-1 Transcriptional Activity during Hypoxia. Cancer Res. 2006, 66, 6683–6691. [Google Scholar] [CrossRef] [PubMed]
- Kumarakulasingham, M.; Rooney, P.H.; Dundas, S.R.; Telfer, C.; Melvin, W.T.; Curran, S.; Murray, G.I. Cytochrome P450 Profile of Colorectal Cancer: Identification of Markers of Prognosis. Clin. Cancer Res. 2005, 11, 3758–3765. [Google Scholar] [CrossRef] [PubMed]
- Tamási, V.; Monostory, K.; Prough, R.A.; Falus, A. Role of xenobiotic metabolism in cancer: Involvement of transcriptional and miRNA regulation of P450s. Cell. Mol. Life Sci. 2011, 68, 1131–1146. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET Receptor Drives Resistance to Anti-EGFR Therapies in Colorectal Cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef] [PubMed]
- Szachowicz-Petelska, B.; Sulkowski, S.; Figaszewski, Z.A. Altered membrane free unsaturated fatty acid composition in human colorectal cancer tissue. Mol. Cell. Biochem. 2007, 294, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Luo, J.; Hao, H.; Hu, J.; Xie, S.K.; Ren, D.; Rao, B. MicroRNA-100 regulates SW620 colorectal cancer cell proliferation and invasion by targeting RAP1B. Oncol. Rep. 2014, 31, 2055–2062. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Hu, X.; Ge, S.; Qian, G.; Zhang, J. Regulation of RAP1B by miR-139 suppresses human colorectal carcinoma cell proliferation. Int. J. Biochem. Cell Biol. 2012, 44, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Sainz, J.; Rudolph, A.; Hein, R.; Hoffmeister, M.; Buch, S.; von Schönfels, W.; Hampe, J.; Schafmayer, C.; Völzke, H.; Frank, B.; et al. Association of genetic polymorphisms in ESR2, HSD17B1, ABCB1, and SHBG genes with colorectal cancer risk. Endocr.-Relat. Cancer 2011, 18, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Boursi, B.; Haynes, K.; Mamtani, R.; Yang, Y.X. Thyroid Dysfunction, Thyroid Hormone Replacement and Colorectal Cancer Risk. JNCI J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, I.P.M.; Carvajal-Carmona, L.G.; Dobbins, S.E.; Tenesa, A.; Jones, A.M.; Howarth, K.; Palles, C.; Broderick, P.; Jaeger, E.E.M.; Farrington, S.; et al. Multiple Common Susceptibility Variants near BMP Pathway Loci GREM1, BMP4, and BMP2 Explain Part of the Missing Heritability of Colorectal Cancer. PLoS Genet. 2011, 7, e1002105. [Google Scholar] [CrossRef] [PubMed]
- Catalano, V.; Dentice, M.; Ambrosio, R.; Luongo, C.; Carollo, R.; Benfante, A.; Todaro, M.; Stassi, G.; Salvatore, D. Activated thyroid hormone promotes differentiation and chemotherapeutic sensitization of colorectal cancer stem cells by regulating Wnt and BMP4 signaling. Cancer Res. 2016, 76, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Sarraf, P.; Mueller, E.; Jones, D.; King, F.J.; DeAngelo, D.J.; Partridge, J.B.; Holden, S.A.; Chen, L.B.; Singer, S.; Fletcher, C.; et al. Differentiation and reversal of malignant changes in colon cancer through PPAR|[ggr]|. Nat. Med. 1998, 4, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Wahli, W. PPARs Mediate Lipid Signaling in Inflammation and Cancer. PPAR Res. 2008, 2008, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hollande, F.; Papin, M. Tight Junctions in Colorectal Cancer. In Tight Junctions in Cancer Metastasis; Martin, T.A., Jiang, W.G., Eds.; Springer: Dordrecht, The Netherlands, 2013; pp. 149–167. [Google Scholar]
- Albasri, A.; Fadhil, W.; Scholefield, J.H.; Durrant, L.G.; Ilyas, M. Nuclear expression of phosphorylated focal adhesion kinase is associated with poor prognosis in human colorectal cancer. Anticancer Res. 2014, 34, 3969–3974. [Google Scholar] [PubMed]
- Watson, A.J.M. Apoptosis and colorectal cancer. Gut 2004, 53, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.Y.; Richardson, B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327. [Google Scholar] [CrossRef]
- Vermeulen, L.; De Sousa E Melo, F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Manna, S.K.; Tanaka, N.; Krausz, K.W.; Haznadar, M.; Xue, X.; Matsubara, T.; Bowman, E.D.; Fearon, E.R.; Harris, C.C.; Shah, Y.M.; et al. Biomarkers of Coordinate Metabolic Reprogramming in Colorectal Tumors in Mice and Humans. Gastroenterology 2014, 146, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, A.; Kami, K.; Sugimoto, M.; Sugawara, M.; Toki, N.; Onozuka, H.; Kinoshita, T.; Saito, N.; Ochiai, A.; Tomita, M.; et al. Quantitative Metabolome Profiling of Colon and Stomach Cancer Microenvironment by Capillary Electrophoresis Time-of-Flight Mass Spectrometry. Cancer Res. 2009, 69, 4918–4925. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tso, V.K.; Slupsky, C.M.; Fedorak, R.N. Metabolomics and detection of colorectal cancer in humans: A systematic review. Future Oncol. 2010, 6, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Vetvik, K.K.; Sonerud, T.; Lindeberg, M.; Lüders, T.; Størkson, R.H.; Jonsdottir, K.; Frengen, E.; Pietiläinen, K.H.; Bukholm, I. Globular adiponectin and its downstream target genes are up-regulated locally in human colorectal tumors: Ex vivo and in vitro studies. Metabolism 2014, 63, 672–681. [Google Scholar] [CrossRef] [PubMed]
- Mazzarelli, P.; Pucci, S.; Bonanno, E.; Sesti, F.; Calvani, M.; Spagnoli, L.G. Carnitine palmitoyltransferase I in human carcinomas: A novel role in histone deacetylation? Cancer Biol. Ther. 2007, 6, 1606–1613. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.P.; Zhu, Y.; Zhang, J.J.; Xu, Z.K.; Qian, Z.Y.; Dai, C.C.; Jiang, K.R.; Wu, J.L.; Gao, W.T.; Li, Q.; et al. Comprehensive analysis of the percentage of surface receptors and cytotoxic granules positive natural killer cells in patients with pancreatic cancer, gastric cancer, and colorectal cancer. J. Transl. Med. 2013, 11, 262. [Google Scholar] [CrossRef] [PubMed]
- Rocca, Y.S.; Roberti, M.P.; Arriaga, J.M.; Amat, M.; Bruno, L.; Pampena, M.B.; Huertas, E.; Loria, F.S.; Pairola, A.; Bianchini, M.; et al. Altered phenotype in peripheral blood and tumor-associated NK cells from colorectal cancer patients. Innate Immunity 2012, 19, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Untersmayr, E.; Bises, G.; Starkl, P.; Bevins, C.L.; Scheiner, O.; Boltz-Nitulescu, G.; Wrba, F.; Jensen-Jarolim, E. The High Affinity IgE Receptor FcεRI Is Expressed by Human Intestinal Epithelial Cells. PLoS ONE 2010, 5, e9023. [Google Scholar] [CrossRef] [PubMed]
- Francescone, R.; Hou, V.; Grivennikov, S.I. Cytokines, IBD and colitis-associated cancer. Inflamm. Bowel Dis. 2015, 21, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Uchibori, R.; Tsukahara, T.; Mizuguchi, H.; Saga, Y.; Urabe, M.; Mizukami, H.; Kume, A.; Ozawa, K. NF-κB Activity Regulates Mesenchymal Stem Cell Accumulation at Tumor Sites. Cancer Res. 2013, 73, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Cormen, T.H. Introduction to Algorithms; MIT Press: Cambridge, MA, USA, 2009. [Google Scholar]
- Poole, W.; Gibbs, D.L.; Shmulevich, I.; Bernard, B.; Knijnenburg, T.A. Combining dependent p-values with an empirical adaptation of Brown’s method. Bioinformatics 2016, 32, i430–i436. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.B. 400: A method for combining non-independent, one-sided tests of significance. Biometrics 1975, 31, 987–992. [Google Scholar] [CrossRef]
- Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar]
- Otwell, T. Laravel. Available online: https://laravel.com (accessed on 28 December 2016).
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Franz, M.; Lopes, C.T.; Huck, G.; Dong, Y.; Sumer, O.; Bader, G.D. Cytoscape. js: A graph theory library for visualisation and analysis. Bioinformatics 2015, 32, 309–311. [Google Scholar] [PubMed]
- Hamosh, A.; Scott, A.F.; Amberger, J.S.; Bocchini, C.A.; McKusick, V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005, 33, D514–D517. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium; Blake, J.A.; Christie, K.R.; Dolan, M.E.; Drabkin, H.J.; Hill, D.P.; Ni, L.; Sitnikov, D.; Burgess, S.; Buza, T.; et al. Gene ontology consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| BRCA | COAD | ||
|---|---|---|---|
| Pathway | p | Pathway | p |
| metabolism of xenobiotics by cytochrome p450 | 0 | metabolism of xenobiotics by cytochrome p450 | 0 |
| steroid hormone biosynthesis | 0 | drug metabolism cytochrome p450 | 0 |
| drug metabolism cytochrome p450 | 0 | chemical carcinogenesis | 0 |
| chemical carcinogenesis | 0 | steroid hormone biosynthesis | 0 |
| drug metabolism other enzymes | 0 | drug metabolism other enzymes | 0 |
| linoleic acid metabolism | 0 | linoleic acid metabolism | 0 |
| longevity regulating pathway | ppar signaling pathway | 0 | |
| egfr tyrosine kinase inhibitor resistance | phenylalanine metabolism | 0 | |
| endocrine resistance | estrogen signaling pathway | ||
| rap1 signaling pathway | chemokine signaling pathway | ||
| progesterone mediated oocyte maturation | erbb signaling pathway | ||
| hif 1 signaling pathway | phospholipase d signaling pathway | ||
| melanogenesis | neurotrophin signaling pathway | ||
| apoptosis | insulin signaling pathway | ||
| platinum drug resistance | egfr tyrosine kinase inhibitor resistance | ||
| phospholipase d signaling pathway | prolactin signaling pathway | ||
| mtor signaling pathway | oxytocin signaling pathway | ||
| ras signaling pathway | platelet activation | ||
| thyroid hormone signaling pathway | endocrine resistance | ||
| erbb signaling pathway | focal adhesion | ||
| # Nodes | # Disease Genes | |||||||
|---|---|---|---|---|---|---|---|---|
| Dataset | Algorithm | All | All | Reachable Pairs | † | ‡ | ||
| KEGG Pathways | 1009 | 7121 | 30 | 104 | 283 | - | 7 | |
| SPECifIC | 466 | 466 | 15 | 15 | 6 | 1.78 | 3 | |
| BRCA | SubPathway-GM | 101 | 214 | 9 | 14 | 6 | 1.89 | 3 |
| SubPathway-Gmir | 142 | 722 | 4 | 8 | 1 | 2.09 | 2 | |
| DESubs | 34 | 34 | 0 | 0 | 0 | 2.48 | - | |
| KEGG Pathways | 1009 | 7121 | 11 | 81 | 490 | - | 9 | |
| SPECifIC | 486 | 486 | 9 | 9 | 6 | 1.67 | 4 | |
| COAD | SubPathway-GM | 59 | 173 | 3 | 8 | 4 | 2.04 | 3 |
| SubPathway-Gmir | 158 | 248 | 4 | 7 | 9 | 2.20 | 2 | |
| DESubs | 6 | 6 | 0 | 0 | 0 | 2.97 | - | |
| Category | Source | # Terms | # Nodes |
|---|---|---|---|
| Diseases | |||
| DisGeNET [37,38] | 7607 | 2978 | |
| GAD [39] | 403 | 1519 | |
| KEGG [2,3,4] Diseases | 1278 | 1234 | |
| OMIM [115] | 89 | 518 | |
| Drugs | |||
| Drugbank [116] Carriers | 247 | 7 | |
| Drugbank [116] Enzymes | 797 | 180 | |
| Drugbank [116] Targets | 4815 | 1494 | |
| Drugbank [116] Transporters | 560 | 18 | |
| KEGG [2,3,4] Drugs | 3793 | 706 | |
| Gene Ontology | |||
| GO [117,118] Biological Processes | 11,386 | 4850 | |
| GO [117,118] Cellular Component | 1545 | 4852 | |
| GO [117,118] Molecular Function | 4146 | 4832 | |
| Pathways | |||
| KEGG [2,3,4] Pathways | 310 | 4904 | |
| Code | Cancer Type | Control Samples | Case Samples | Case Samples Categories |
|---|---|---|---|---|
| BLCA | Bladder Urothelial Carcinoma | 19 | 193 | Stage I, II, III, IV |
| BRCA | Breast invasive carcinoma | 86 | 642 | Stage I, II, III, IV, X |
| COAD | Colon adenocarcinoma | 8 | 389 | Stage I, II, III, IV |
| KICH | Kidney Chromophobe | 25 | 66 | Stage I, II, III, IV |
| KIRC | Kidney renal clear cell carcinoma | 71 | 224 | Stage I, II, III, IV |
| LUAD | Lung adenocarcinoma | 19 | 388 | Stage I, II, III, IV |
| LUSC | Lung squamous cell carcinoma | 37 | 247 | Stage I, II, III, IV |
| PRAD | Prostate adenocarcinoma | 50 | 191 | Category 6, 7, 8, 9, 10 |
| READ | Rectum adenocarcinoma | 3 | 150 | Stage I, II, III, IV |
| UCEC | Uterine Corpus Endometrial Carcinoma | 14 | 231 | Stage I, II, III, IV |
| All Samples | 332 | 2721 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alaimo, S.; Marceca, G.P.; Ferro, A.; Pulvirenti, A. Detecting Disease Specific Pathway Substructures through an Integrated Systems Biology Approach. Non-Coding RNA 2017, 3, 20. https://doi.org/10.3390/ncrna3020020
Alaimo S, Marceca GP, Ferro A, Pulvirenti A. Detecting Disease Specific Pathway Substructures through an Integrated Systems Biology Approach. Non-Coding RNA. 2017; 3(2):20. https://doi.org/10.3390/ncrna3020020
Chicago/Turabian StyleAlaimo, Salvatore, Gioacchino Paolo Marceca, Alfredo Ferro, and Alfredo Pulvirenti. 2017. "Detecting Disease Specific Pathway Substructures through an Integrated Systems Biology Approach" Non-Coding RNA 3, no. 2: 20. https://doi.org/10.3390/ncrna3020020
APA StyleAlaimo, S., Marceca, G. P., Ferro, A., & Pulvirenti, A. (2017). Detecting Disease Specific Pathway Substructures through an Integrated Systems Biology Approach. Non-Coding RNA, 3(2), 20. https://doi.org/10.3390/ncrna3020020