CirComPara: A Multi‐Method Comparative Bioinformatics Pipeline to Detect and Study circRNAs from RNA‐seq Data




Abstract
:1. Introduction
2. Results
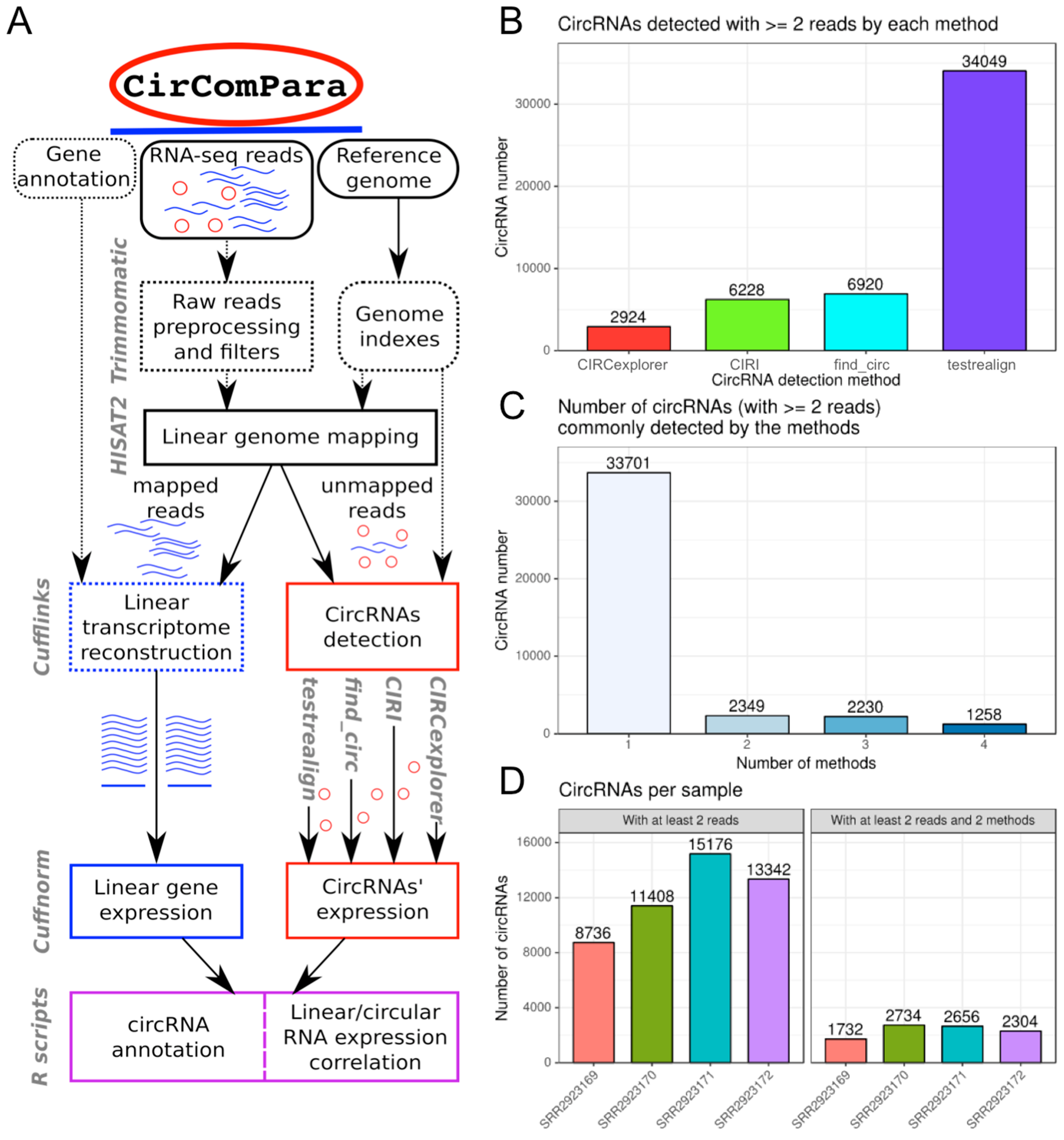
2.1. CirComPara Provides circRNA Detection, Quantification, Annotation and Correlation with Gene Expression
2.2. CirComPara’s Default Workflow and Usage
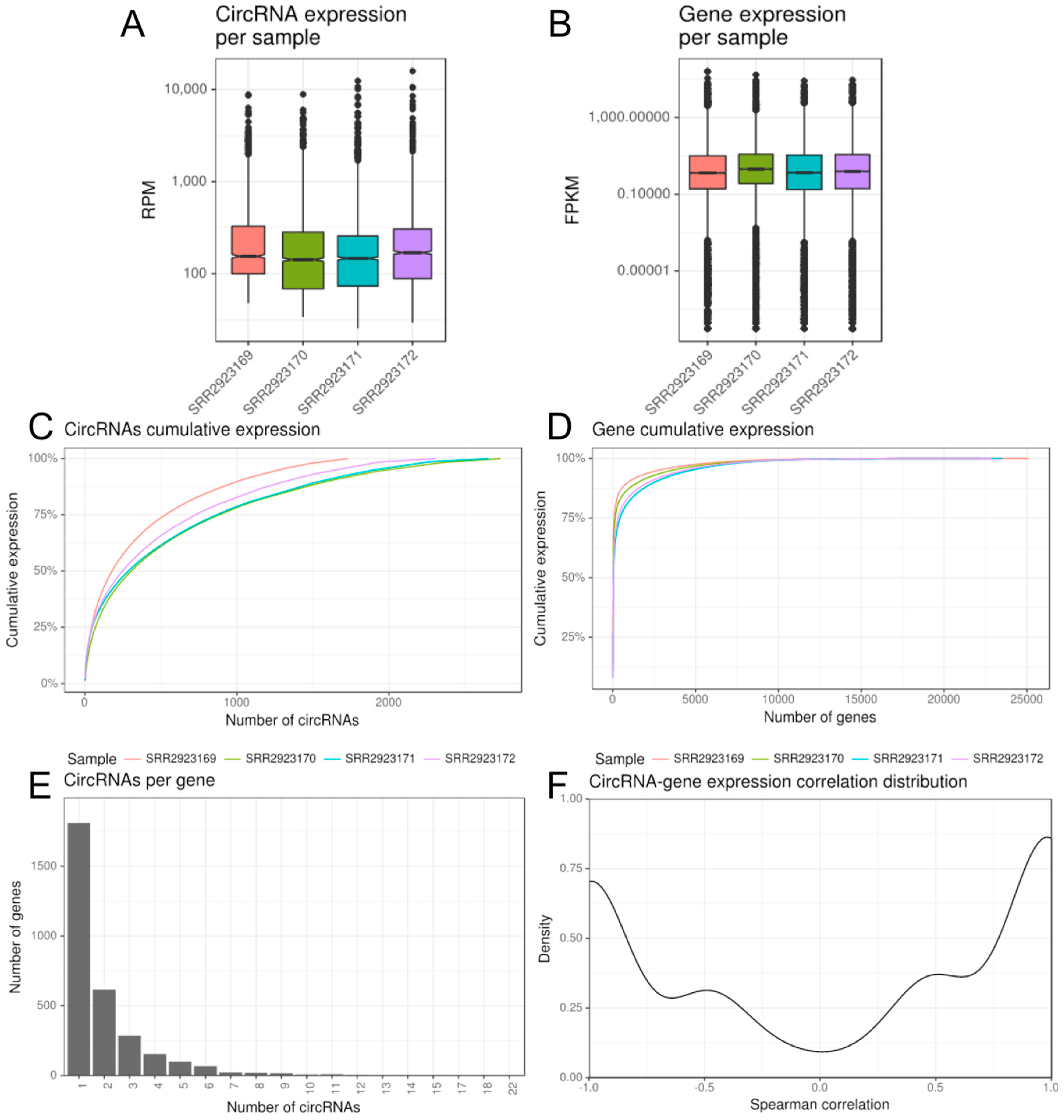
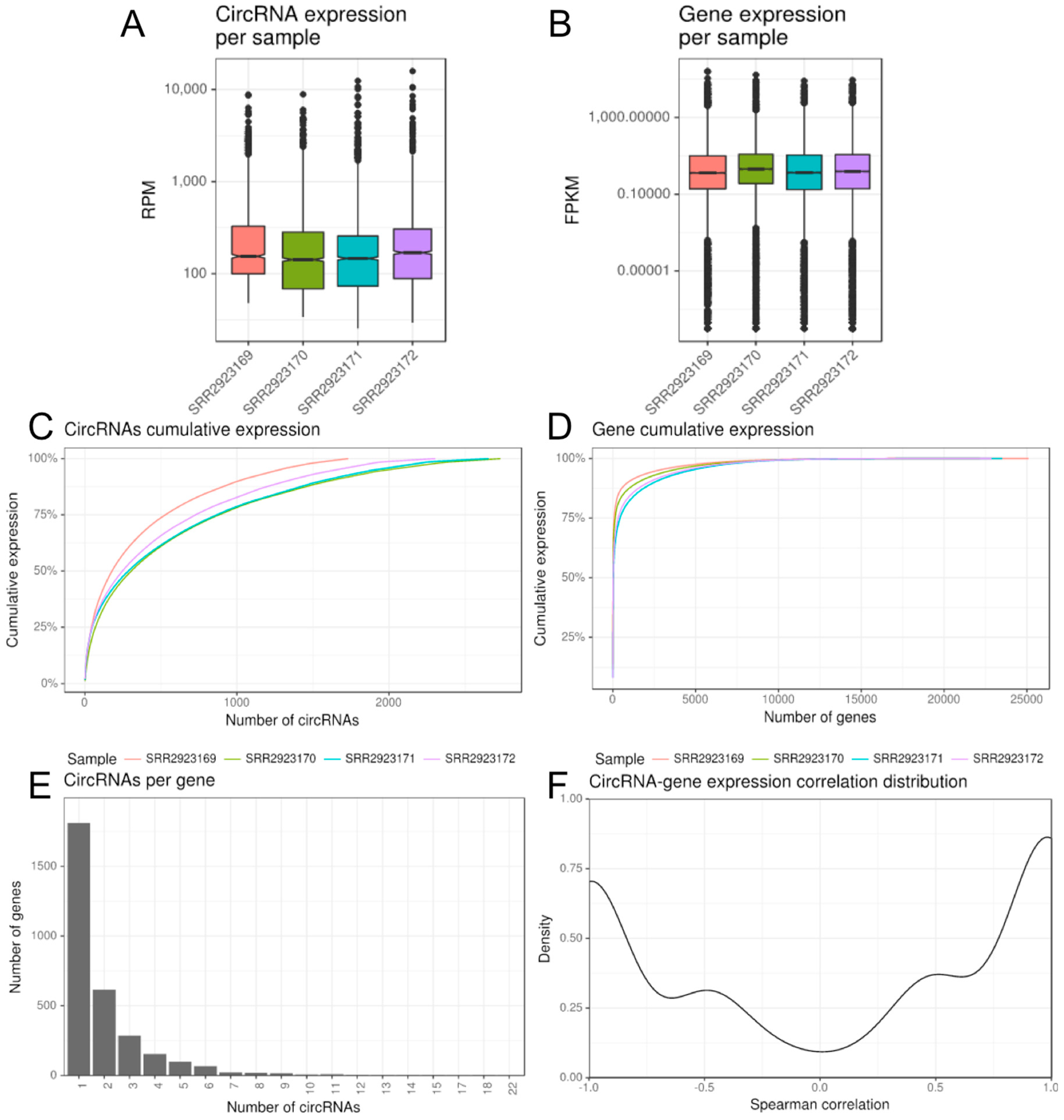
2.2.1. CirComPara’s Output
2.2.2. Additional Options and Features
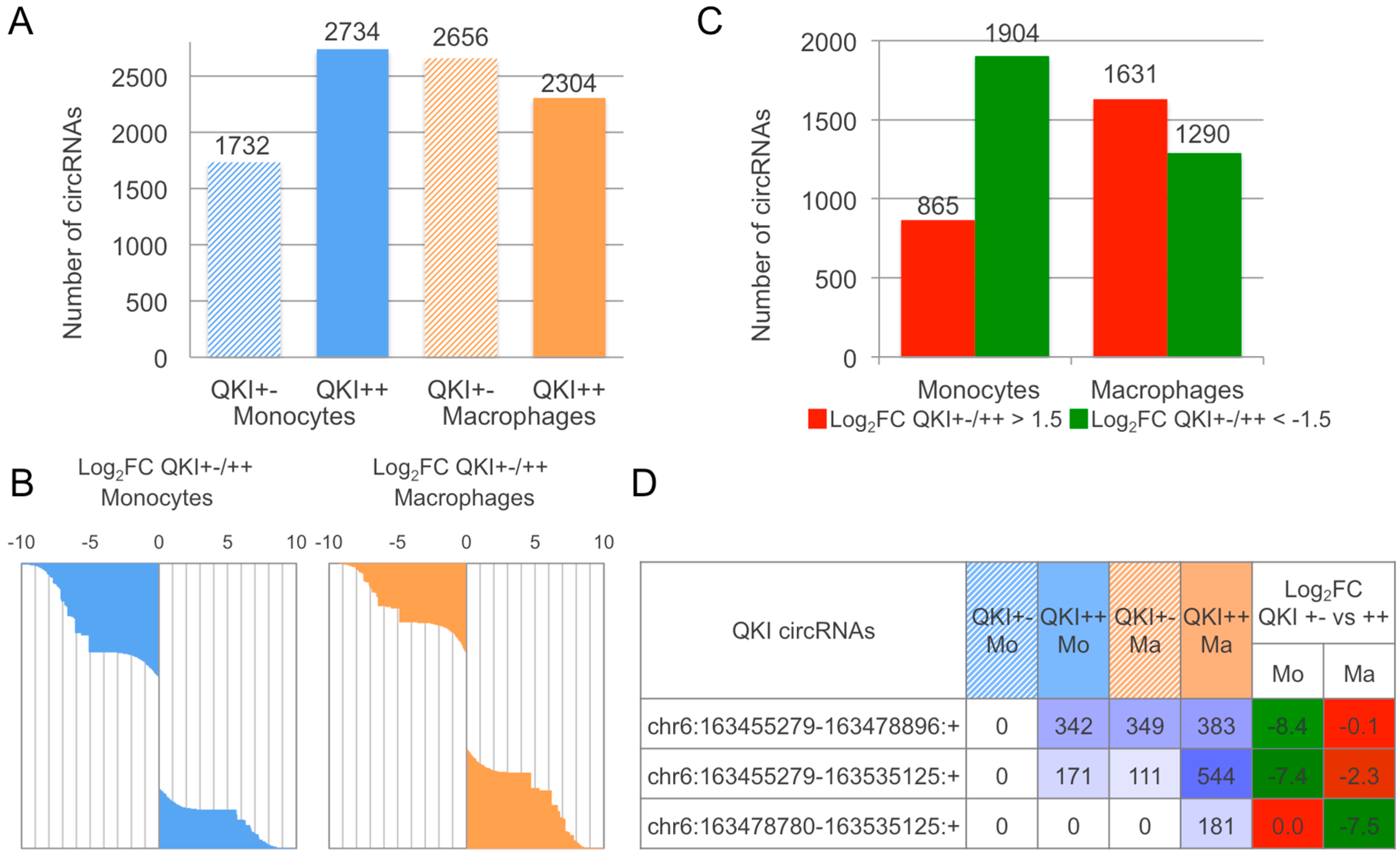
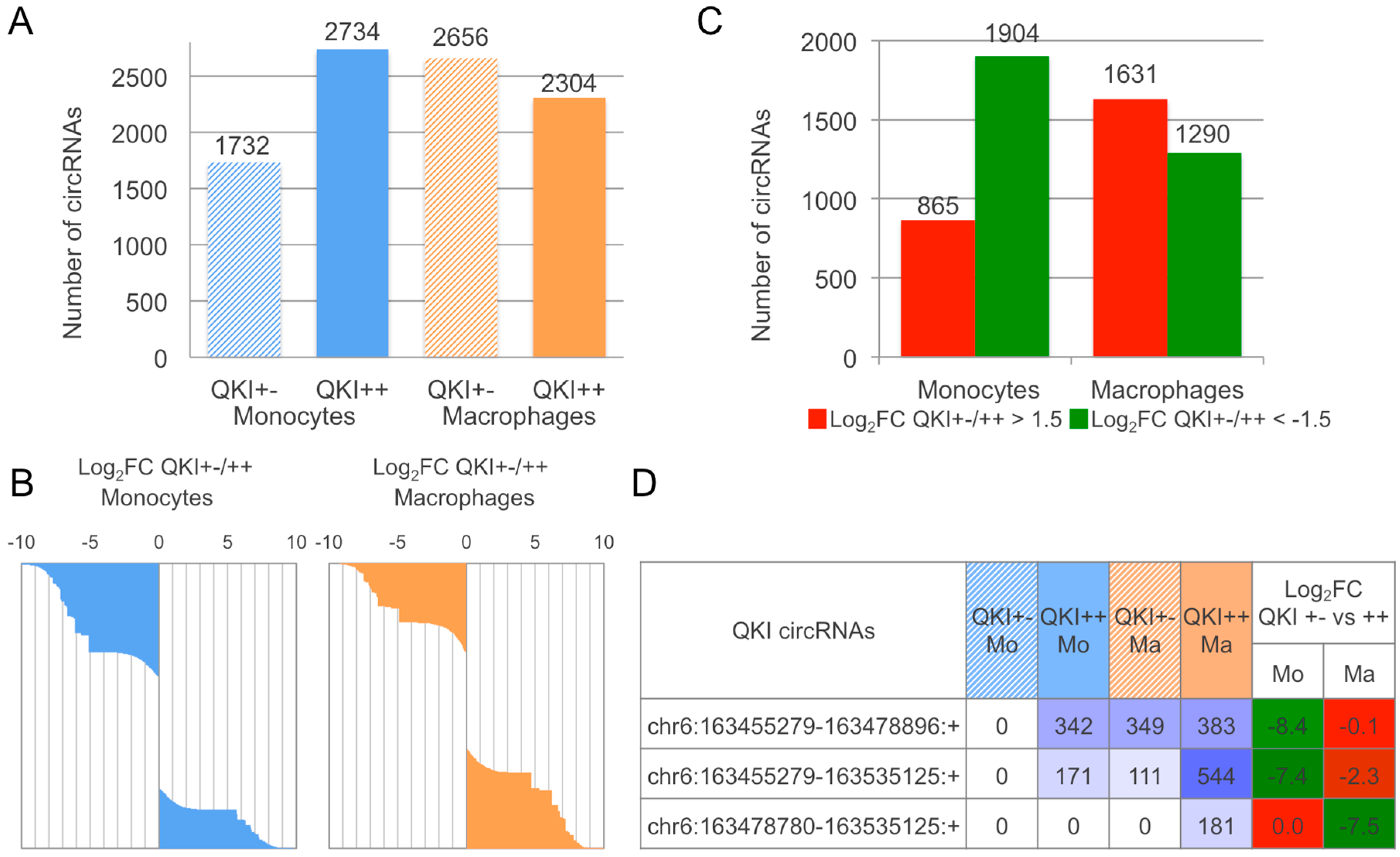
2.3. CirComPara Predicts Thousands of circRNAs Expressed in Monocytes and Macrophages of a QKI Haploinsufficient Patient and Her Sibling
3. Discussion
4. Materials and Methods
4.1. CirComPara’s Implementation Details
4.1.1. Automation and Parallelism: Scons
4.1.2. CirComPara’s Software Tools
4.2. Quaking Haploinsufficiency Dataset and Analysis Parameters
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lasda, E.; Parker, R. Circular RNAs: Diversity of form and function. RNA 2014, 20, 1829–1842. [Google Scholar] [CrossRef]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Bachmayr-Heyda, A.; Reiner, A.T.; Auer, K.; Sukhbaatar, N.; Aust, S.; Bachleitner-Hofmann, T.; Mesteri, I.; Grunt, T.W.; Zeillinger, R.; Pils, D. Correlation of circular RNA abundance with proliferation—Exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci. Rep. 2015, 5, 8057. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J.; Chen, R.E.; Olsen, M.N.; Wang, P.L.; Brown, P.O. Cell-Type Specific Features of Circular RNA Expression. PLoS Genet. 2013, 9, e1003777. [Google Scholar] [CrossRef]
- Bonizzato, A.; Gaffo, E.; te Kronnie, G.; Bortoluzzi, S. CircRNAs in hematopoiesis and hematological malignancies. Blood Cancer J. 2016, 6, e483. [Google Scholar] [CrossRef] [PubMed]
- Rybak-Wolf, A.; Stottmeister, C.; Glažar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.; Greenman, C.; Cook, P.R.; Papantonis, A. Exon skipping is correlated with exon circularization. J. Mol. Biol. 2015, 427, 2414–2417. [Google Scholar] [CrossRef] [PubMed]
- Conn, S.J.; Pillman, K.A.; Toubia, J.; Conn, V.M.; Salmanidis, M.; Phillips, C.A.; Roslan, S.; Schreiber, A.W.; Gregory, P.A.; Goodall, G.J. The RNA Binding Protein Quaking Regulates Formation of circRNAs. Cell 2015, 160, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Zhu, H.; Shi, Y.; Wu, W.; Cai, H.; Chen, X. cir-ITCH plays an inhibitory role in colorectal cancer by regulating the Wnt/β-catenin pathway. PLoS ONE 2015, 10, e0131225. [Google Scholar] [CrossRef]
- Li, F.; Zhang, L.; Li, W.; Deng, J.; Zheng, J.; An, M.; Lu, J.; Zhou, Y. Circular RNA ITCH has inhibitory effect on ESCC by suppressing the Wnt/β-catenin pathway. Oncotarget 2015, 6, 6001–6013. [Google Scholar] [CrossRef] [PubMed]
- Du, W.W.; Yang, W.; Liu, E.; Yang, Z.; Dhaliwal, P.; Yang, B.B. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 2016, 44, 2846–2858. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-O.; Wang, H.-B.; Zhang, Y.; Lu, X.; Chen, L.-L.; Yang, L. Complementary Sequence-Mediated Exon Circularization. Cell 2014, 159, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Westholm, J.O.; Miura, P.; Olson, S.; Shenker, S.; Joseph, B.; Sanfilippo, P.; Celniker, S.E.; Graveley, B.R.; Lai, E.C. Genome-wide Analysis of Drosophila Circular RNAs Reveals Their Structural and Sequence Properties and Age-Dependent Neural Accumulation. Cell Rep. 2014, 9, 1966–1980. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Otto, C.; Doose, G.; Tanzer, A.; Langenberger, D.; Christ, S.; Kunz, M.; Holdt, L.M.; Teupser, D.; Hackermüller, J.; et al. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing and fusion detection. Genome Biol. 2014, 15, R34. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Szabo, L.; Morey, R.; Palpant, N.J.; Wang, P.L.; Afari, N.; Jiang, C.; Parast, M.M.; Murry, C.E.; Laurent, L.C.; Salzman, J. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 2015, 16, 126. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhang, N.; Han, P.; Moon, B.-S.; Lai, R.K.; Wang, K.; Lu, W. Circular RNA profile in gliomas revealed by identification tool UROBORUS. Nucleic Acids Res. 2016, 44, e87. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.-J.; Wu, C.-S.; Chen, C.-Y.; Hung, L.-Y.; Chiang, T.-W.; Yang, M.-Y. NCLscan: Accurate identification of non-co-linear transcripts (fusion, trans-splicing and circular RNA) with a good balance between sensitivity and precision. Nucleic Acids Res. 2016, 44, e29. [Google Scholar] [CrossRef] [PubMed]
- Izuogu, O.G.; Alhasan, A.A.; Alafghani, H.M.; Santibanez-Koref, M.; Elliott, D.J.; Jackson, M.S. PTESFinder: A computational method to identify post-transcriptional exon shuffling (PTES) events. BMC Bioinform. 2016, 17, 31. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Conrad, T.O. Acfs: Accurate circRNA identification and quantification from RNA-Seq data. Sci. Rep. 2016, 6, 38820. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Singh, D.; Zeng, Z.; Coleman, S.J.; Huang, Y.; Savich, G.L.; He, X.; Mieczkowski, P.; Grimm, S.A.; Perou, C.M.; et al. MapSplice: Accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010, 38, e178. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Venø, M.T.; Damgaard, C.K.; Kjems, J. Comparison of circular RNA prediction tools. Nucleic Acids Res. 2015, 44, e58. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, R.G.; Shiue, L.; Prins, J.; de Boer, H.C.; Singh, A.; Fagg, W.S.; van Gils, J.M.; Duijs, J.M.; Katzman, S.; Kraaijeveld, A.O.; et al. Quaking promotes monocyte differentiation into pro-atherogenic macrophages by controlling pre-mRNA splicing and gene expression. Nat. Commun. 2016, 7, 10846. [Google Scholar] [CrossRef] [PubMed]
- Glažar, P.; Papavasileiou, P.; Rajewsky, N. circBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Bao, C.; Guo, W.; Li, S.; Chen, J.; Chen, B.; Luo, Y.; Lyu, D.; Li, Y.; Shi, G.; et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat. Commun. 2016, 7, 11215. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Chen, G.; Zhu, Z.; Shen, Z.; Du, C.; Zang, R.; Su, Y.; Xie, H.; Li, H.; Xu, X.; et al. Circular RNA ZNF609 functions as a competitive endogenous RNA to regulate AKT3 expression by sponging miR-150-5p in Hirschsprung’s disease. Oncotarget 2016, 8, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Hung, L.-H.; Schreiner, S.; Starke, S.; Eckhof, H.; Rossbach, O.; Reich, S.; Medenbach, J.; Bindereif, A. CircRNA-protein complexes: IMP3 protein component defines subfamily of circRNPs. Sci. Rep. 2016, 6, 31313. [Google Scholar] [CrossRef] [PubMed]
- Starke, S.; Jost, I.; Rossbach, O.; Schneider, T.; Schreiner, S.; Hung, L.-H.; Bindereif, A. Exon circularization requires canonical splice signals. Cell Rep. 2015, 10, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protocols 2016, 11, 1650–1667. [Google Scholar] [PubMed]
- Del Fabbro, C.; Scalabrin, S.; Morgante, M.; Giorgi, F.M. An Extensive Evaluation of Read Trimming Effects on Illumina NGS Data Analysis. PLoS ONE 2013, 8, e85024. [Google Scholar] [CrossRef] [PubMed]
- Guarnerio, J.; Bezzi, M.; Jeong, J.C.; Paffenholz, S.V.; Berry, K.; Naldini, M.M.; Lo-Coco, F.; Tay, Y.; Beck, A.H.; Pandolfi, P.P. Oncogenic Role of Fusion-circRNAs Derived from Cancer-Associated Chromosomal Translocations. Cell 2016, 165, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie. Nat. Methods 2012, 9, 357–359. [Google Scholar]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013. [Google Scholar]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| CircRNA ID | Overlapping Gene Ensembl ID | Overlapping Gene Symbol | CircRNA Category | QKI+− Mo (SRR2923169) | QKI++ Mo (SRR2923170) | QKI+−Ma (SRR2923171) | QKI++ Ma (SRR2923172) | RNase R Enrichment [24] | Validated (CircBase) | Other Studies |
|---|---|---|---|---|---|---|---|---|---|---|
| 11:33286413-33287511:+ | ENSG00000110422 | HIPK3 | exonic | 2964 | 8861 | 12386 | 15822 | E | VAL | [27] |
| 2:40428473-40430304:− | ENSG00000183023 | SLC8A1 | exonic | 2519 | 4845 | 10077 | 10604 | U | NA | |
| 12:108652272-108654410:− | ENSG00000110880 | CORO1C | exonic | 3090 | 3625 | 10701 | 10471 | E | VAL | |
| 17:20204333-20205912:+ | ENSG00000128487 | SPECC1 | exonic | 2341 | 3273 | 8062 | 8485 | U | NA | [29] |
| 1:7777160-7778169:+ | ENSG00000049245 | VAMP3 | exonic | 8613 | 4941 | 4105 | 2832 | E|U | NA | |
| 4:143543509-143543972:+ | ENSG00000153147 | SMARCA5 | exonic | 4458 | 6008 | 4288 | 4863 | E|U | VAL | |
| 14:99458279-99465813:− | ENSG00000183576 | SETD3 | exonic | 5399 | 2854 | 5094 | 6114 | E | VAL | |
| 3:196391813-196403019:− | ENSG00000163960| ENSG00000206644| ENSG00000241868 | UBXN7|RNU6-1279P| RN7SL434P | exonic|intergenic spanning gene | 2848 | 5594 | 5553 | 3853 | E | VAL | |
| 8:130152736-130180880:− | ENSG00000153317 | ASAP1 | exonic | 1823 | 2232 | 6744 | 6410 | E | NA | |
| 2:201145378-201149835:+ | ENSG00000003402 | CFLAR | exonic | 5294 | 3675 | 3775 | 3644 | E | NA | |
| 2:61522611-61533903:− | ENSG00000082898 | XPO1 | exonic | 8801 | 3638 | 1503 | 2329 | E | VAL | |
| 1:117402186-117420649:+ | ENSG00000198162 | MAN1A2 | exonic | 3498 | 4779 | 3812 | 4127 | U | VAL | |
| 8:130358017-130361771:− | ENSG00000153317 | ASAP1 | exonic | 0 | 250 | 8392 | 7447 | E | NA | |
| 3:149846011-149921227:+ | ENSG00000082996 | RNF13 | exonic | 1044 | 1278 | 6891 | 6829 | E|U | VAL | |
| 13:32517857-32527532:− | ENSG00000244754 | N4BP2L2 | exonic | 5770 | 3191 | 2788 | 3627 | E | VAL | |
| 18:9182382-9221999:+ | ENSG00000265257| ENSG00000101745 | RP11-21J18.1|ANKRD12 | exonic | 5431 | 3236 | 3041 | 2173 | U | VAL | |
| 21:15762891-15766141:+ | ENSG00000155313 | USP25 | exonic | 3122 | 2630 | 2896 | 4360 | E | VAL | |
| 15:64499293-64500166:+ | ENSG00000180357 | ZNF609 | exonic | 2770 | 4645 | 2109 | 3334 | E | NA | [28] |
| 4:152411303-152412529:− | ENSG00000109670 | FBXW7 | exonic | 3297 | 4365 | 2566 | 2339 | E | NA | |
| 4:87195324-87195690:− | ENSG00000145332 | KLHL8 | exonic | 6312 | 2664 | 2162 | 1246 | E | NA | |
| 9:110972073-110973558:− | ENSG00000198121 | LPAR1 | exonic | 2039 | 1168 | 5094 | 3715 | E | NA | [30] |
| 16:85633914-85634132:+ | ENSG00000131149 | GSE1 | exonic | 3340 | 4629 | 2089 | 1859 | E | NA | |
| 4:37631385-37638504:− | ENSG00000181826 | RELL1 | exonic | 3469 | 2260 | 3336 | 2653 | E | VAL | |
| 6:4891713-4892379:+ | ENSG00000153046 | CDYL | exonic | 1048 | 1845 | 3431 | 4272 | E | NA | [29] |
| 5:73074742-73077493:+ | ENSG00000157107 | FCHO2 | exonic | 763 | 1482 | 4361 | 3953 | E | NA | |
| 5:137985257-137988315:− | ENSG00000031003 | FAM13B | exonic | 3018 | 1830 | 2530 | 2753 | E | NA | |
| 14:73147795-73148094:+ | ENSG00000080815 | PSEN1 | exonic | 3881 | 2625 | 2055 | 1408 | E | NA | |
| 12:32598497-32611283:+ | ENSG00000139132 | FGD4 | exonic | 1579 | 1639 | 3884 | 2764 | E|U | NA | |
| 7:158759486-158764853:− | ENSG00000117868 | ESYT2 | exonic | 2734 | 4661 | 1026 | 1042 | E | VAL | |
| 8:37765526-37766355:+ | ENSG00000147471 | PROSC | exonic | 1980 | 2841 | 2712 | 1908 | E | VAL |
| Software Tool | Description | Citation/Website | Version |
|---|---|---|---|
| R | custom scripts | http://cran.r-project.org | 3.2.5 (2016-04-14) |
| Python | custom scripts | http://www.python.org | 2.7.3 |
| Scons | script execution manager | http://www.scons.org | 2.5.0 |
| Trimmomatic | read preprocessing | [38] | 0.36 |
| FASTQC | read statistics | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ | 0.11.5 |
| HISAT2 | linear genome mapping | [34] | 2.0.4 |
| CIRCexplorer | circRNAs detection | [14] | 1.1.10 |
| STAR | reads alignment by CIRCexplorer | [37] | 2.5.2a |
| CIRI | circRNAs detection | [17] | 2.0.2 |
| BWA | reads alignment by CIRI | [36] | 0.7.15-r1140 |
| find_circ | circRNAs detection | [10] | 1.2 |
| Bowtie2 | reads alignment by find_circ | [35] | 2.2.9 |
| testrealign | circRNAs detection | [16] | 0.1 |
| Segemehl | reads alignment by testrealign | [16] | 0.2.0-418 |
| Cufflinks | gene/transcript expression quantification and transcriptome reconstruction | [39] | 2.2.1 |
| BEDtools | genome coordinates comparison | [41] | 2.26.0 |
| Samtools | handle alignment files; extract unmapped reads | http://www.htslib.org | 1.3.1 |
| ggplot2 | R library for analysis report | http://ggplot2.org | 2.2.0 |
| data.table | R library for analysis report | https://cran.r-project.org/web/packages/data.table/index.html | 1.10.0 |
| knitr | R library for analysis report | http://yihui.name/knitr | 1.14.0 |
| Sample ID | GEO ID | QKI Status | Cell Type |
|---|---|---|---|
| SRR2923169 | GSM1939602 | QKI+/− | (CD14+) monocytes from peripheral blood |
| SRR2923170 | GSM1939603 | QKI+/+ | (CD14+) monocytes from peripheral blood |
| SRR2923171 | GSM1939604 | QKI+/− | differentiated CD14+ cells (macrophages) |
| SRR2923172 | GSM1939605 | QKI+/+ | differentiated CD14+ cells (macrophages) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaffo, E.; Bonizzato, A.; Kronnie, G.T.; Bortoluzzi, S. CirComPara: A Multi‐Method Comparative Bioinformatics Pipeline to Detect and Study circRNAs from RNA‐seq Data. Non-Coding RNA 2017, 3, 8. https://doi.org/10.3390/ncrna3010008
Gaffo E, Bonizzato A, Kronnie GT, Bortoluzzi S. CirComPara: A Multi‐Method Comparative Bioinformatics Pipeline to Detect and Study circRNAs from RNA‐seq Data. Non-Coding RNA. 2017; 3(1):8. https://doi.org/10.3390/ncrna3010008
Chicago/Turabian StyleGaffo, Enrico, Annagiulia Bonizzato, Geertruy Te Kronnie, and Stefania Bortoluzzi. 2017. "CirComPara: A Multi‐Method Comparative Bioinformatics Pipeline to Detect and Study circRNAs from RNA‐seq Data" Non-Coding RNA 3, no. 1: 8. https://doi.org/10.3390/ncrna3010008
APA StyleGaffo, E., Bonizzato, A., Kronnie, G. T., & Bortoluzzi, S. (2017). CirComPara: A Multi‐Method Comparative Bioinformatics Pipeline to Detect and Study circRNAs from RNA‐seq Data. Non-Coding RNA, 3(1), 8. https://doi.org/10.3390/ncrna3010008