
Promoter-Associated RNAs Regulate HSPC152 Gene Expression in Malignant Melanoma


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Melanoma Biopsies
2.2. RNA Purification and Enhancement
2.3. Deep Sequencing (NGS)
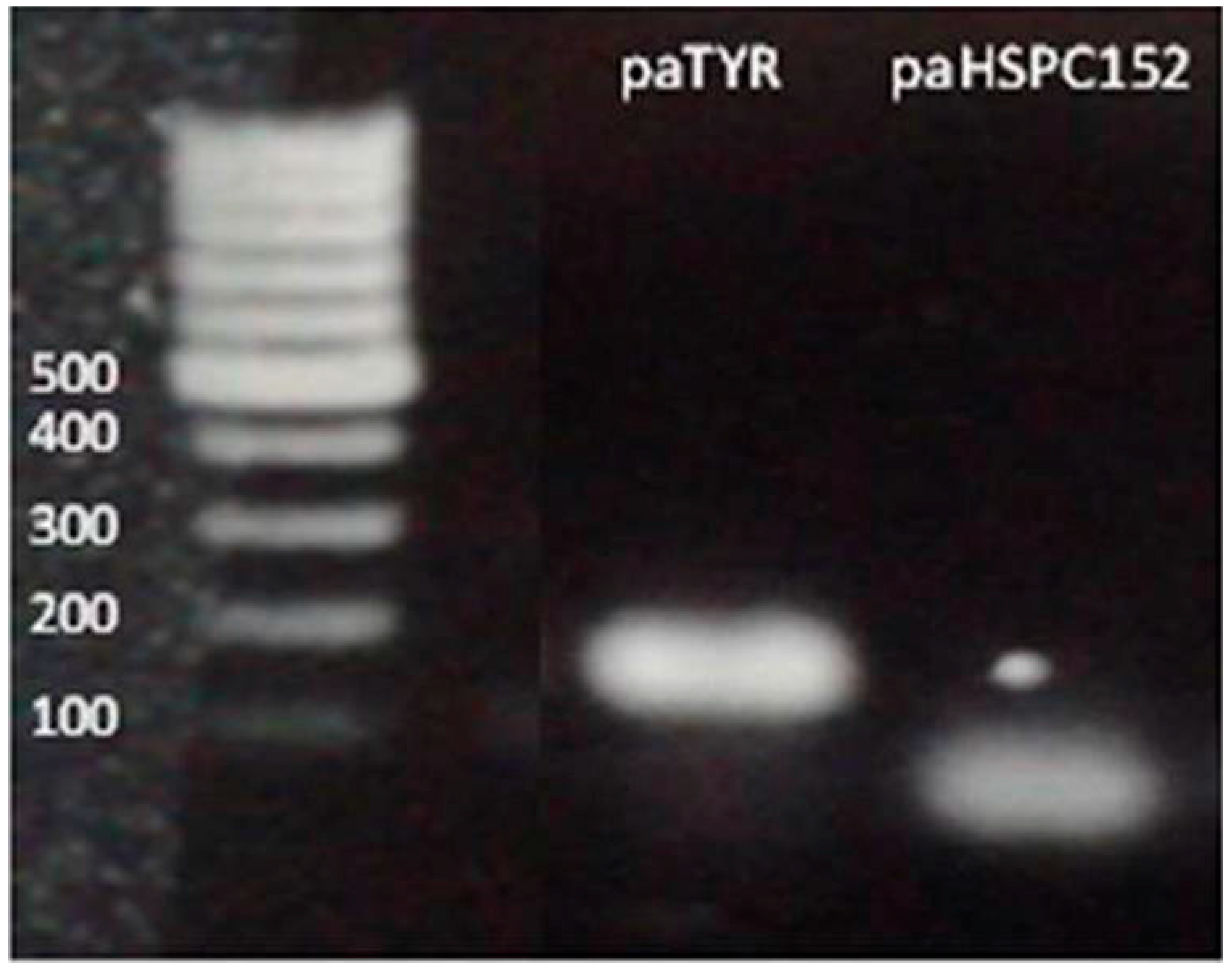
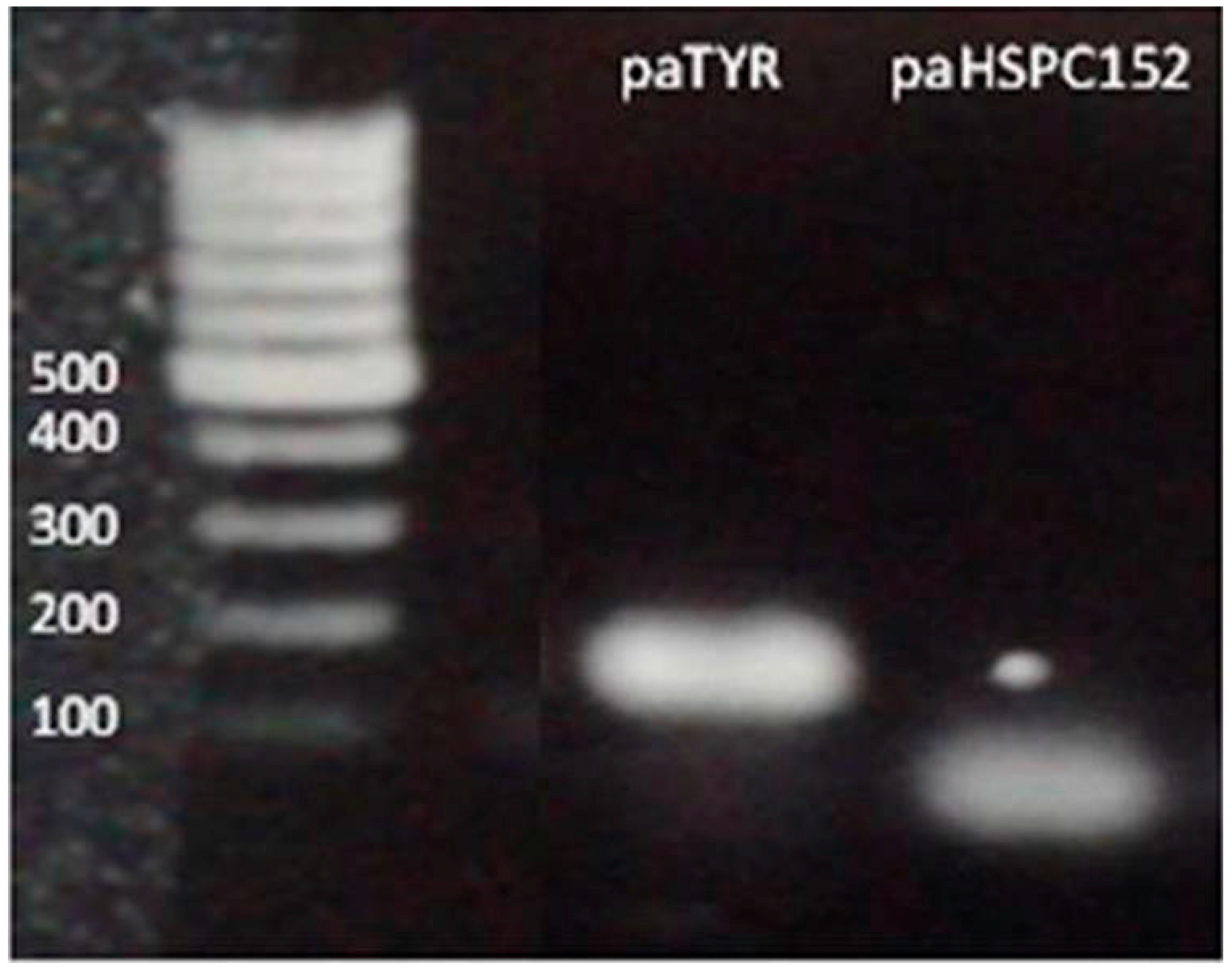
2.4. Cloning and Plasmids
- HSPC152 F-5′-CGACGGTGTTAGGCGC 3′
- HSPC152 R-5′-CGGGTACCTGGAGGCG 3′
- TYR F-5′-CATTTGCAAGGTCAAATCATC 3′
- TYR R-5′-AGTACAAAACAGCCAGGAGC 3′
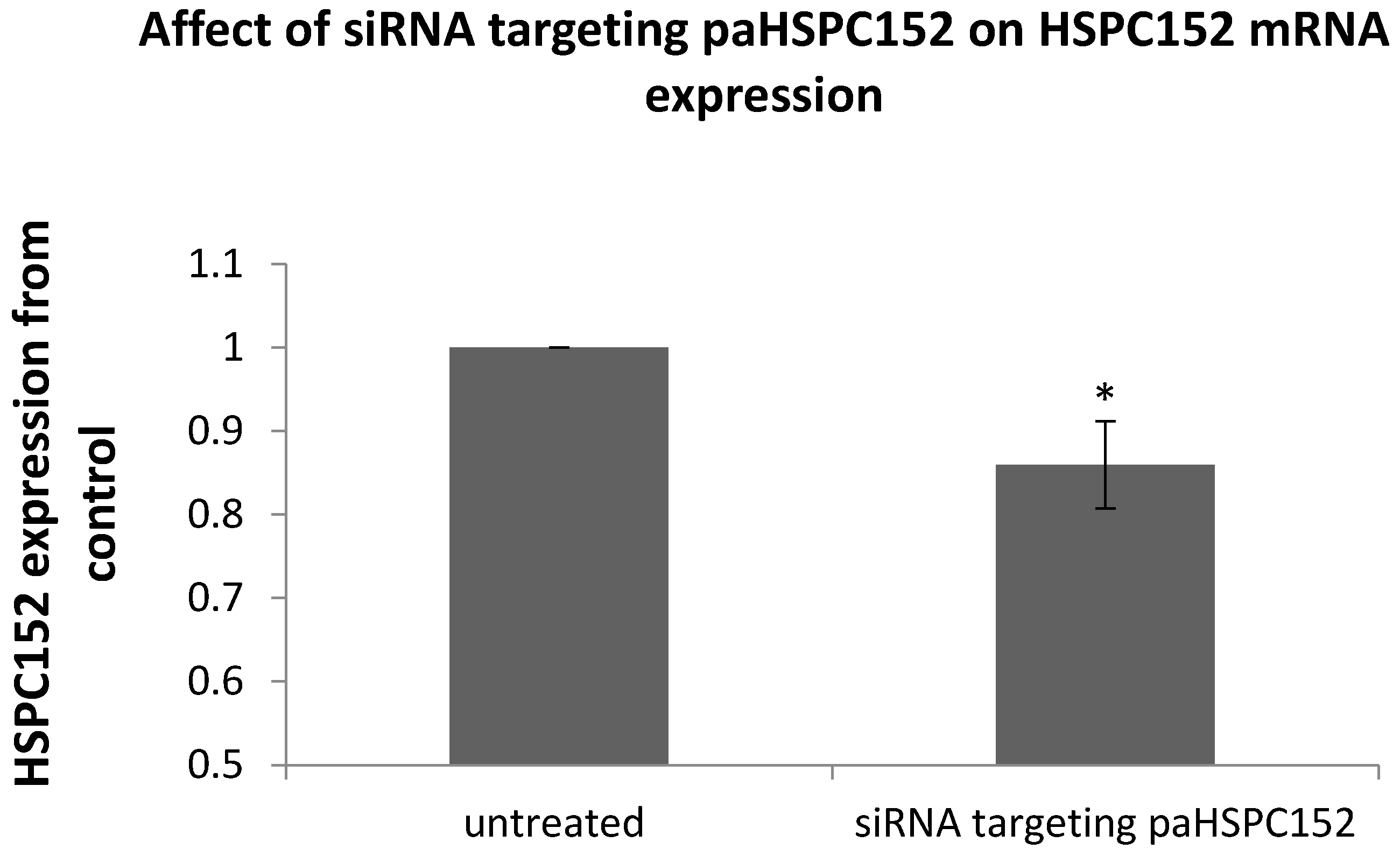
2.5. siRNA Transfection
2.6. Plasmid Transfection
2.7. Chromatin-Immunoprecipitation (ChIP) Assay
2.8. Cytosine Methylation
- BS F3: 5′-TTGTATATGATTTGTATTTTACGAAGAA
- BS R4: 5′-AATAAAAAACCTATCGAATCACG
- BS F6: 5′-AGCGGAGGACGATTTTTT
- BS R6: 5′-CATGCGAGCTCAGCAGA
3. Results
3.1. Screening for paRNAs Differentially Expressed in Normal Melanocytes and Melanoma Cells by Deep-Sequencing Analysis
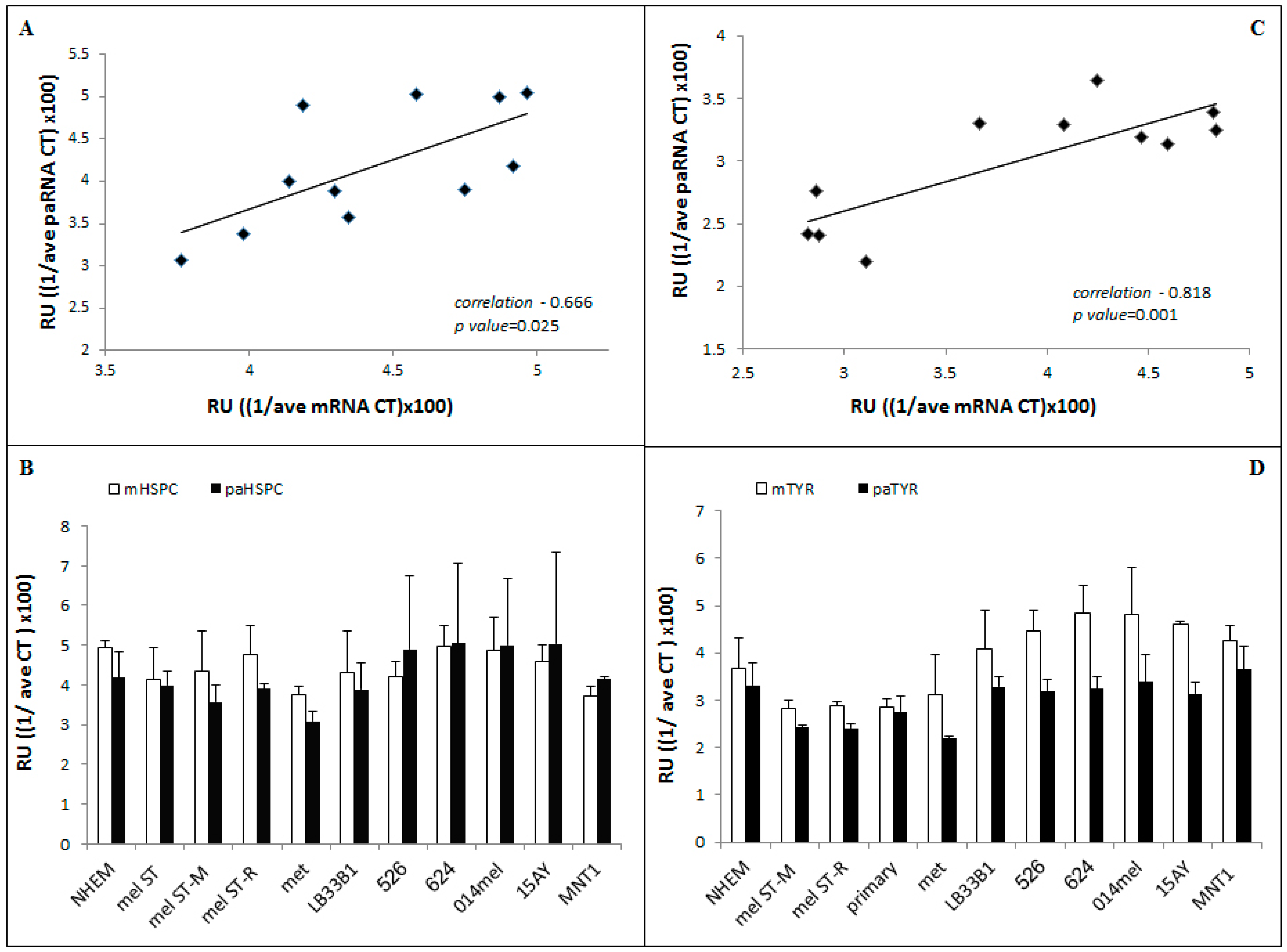
3.2. PaRNA Detection and Correlation to Their Corresponding Gene Expression
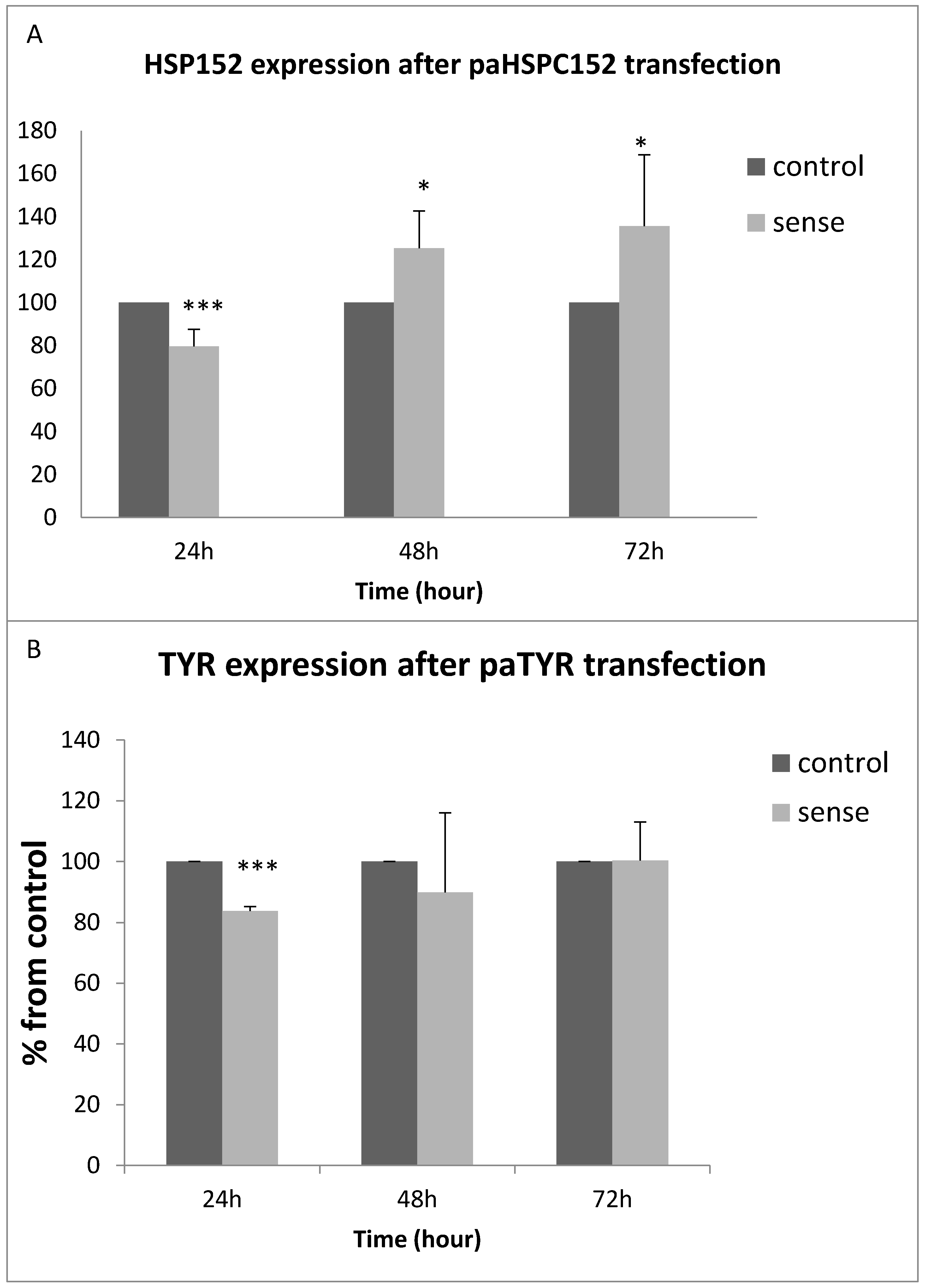
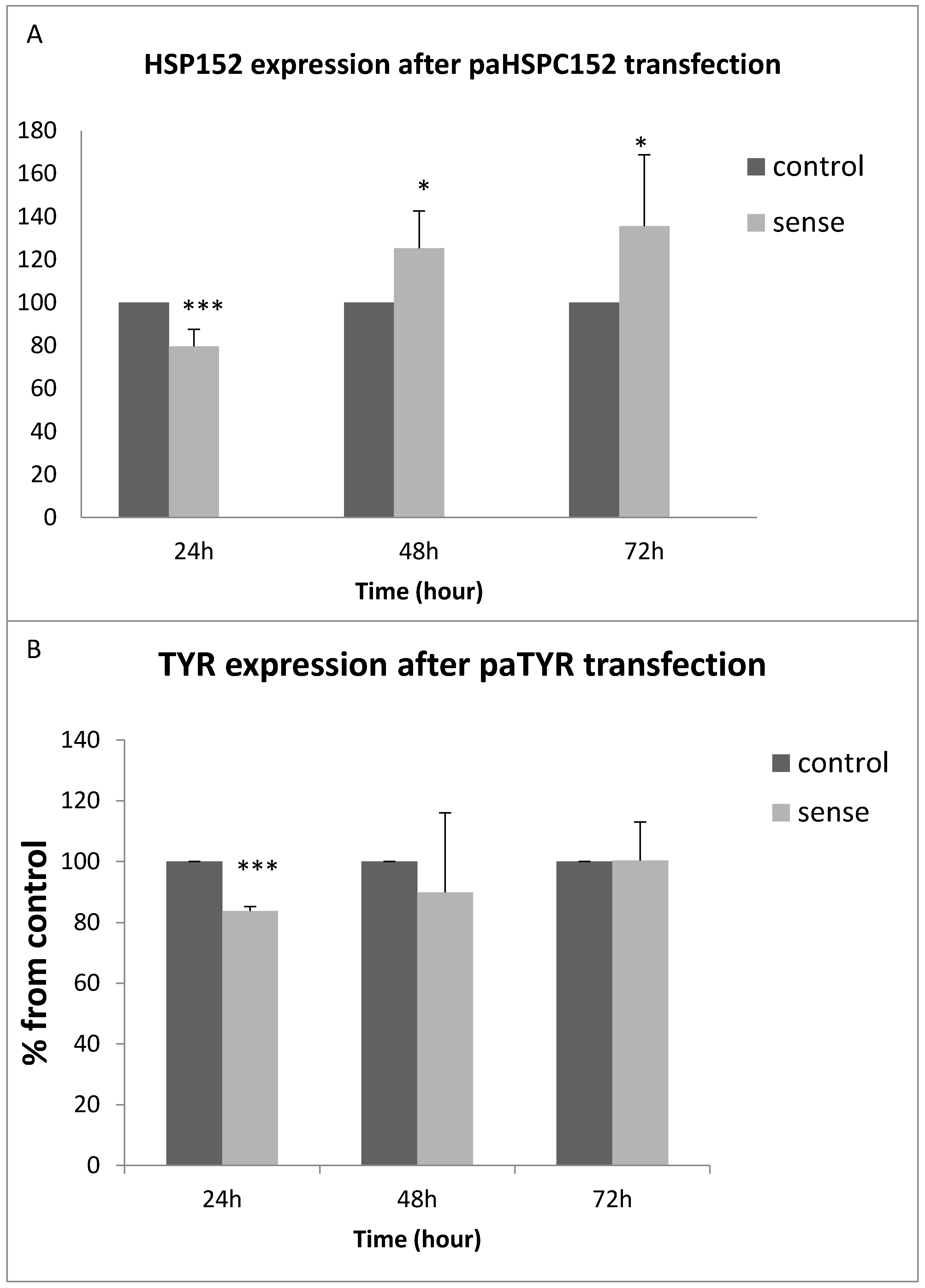
3.3. PaRNA Effect on the Homologous Gene Expression
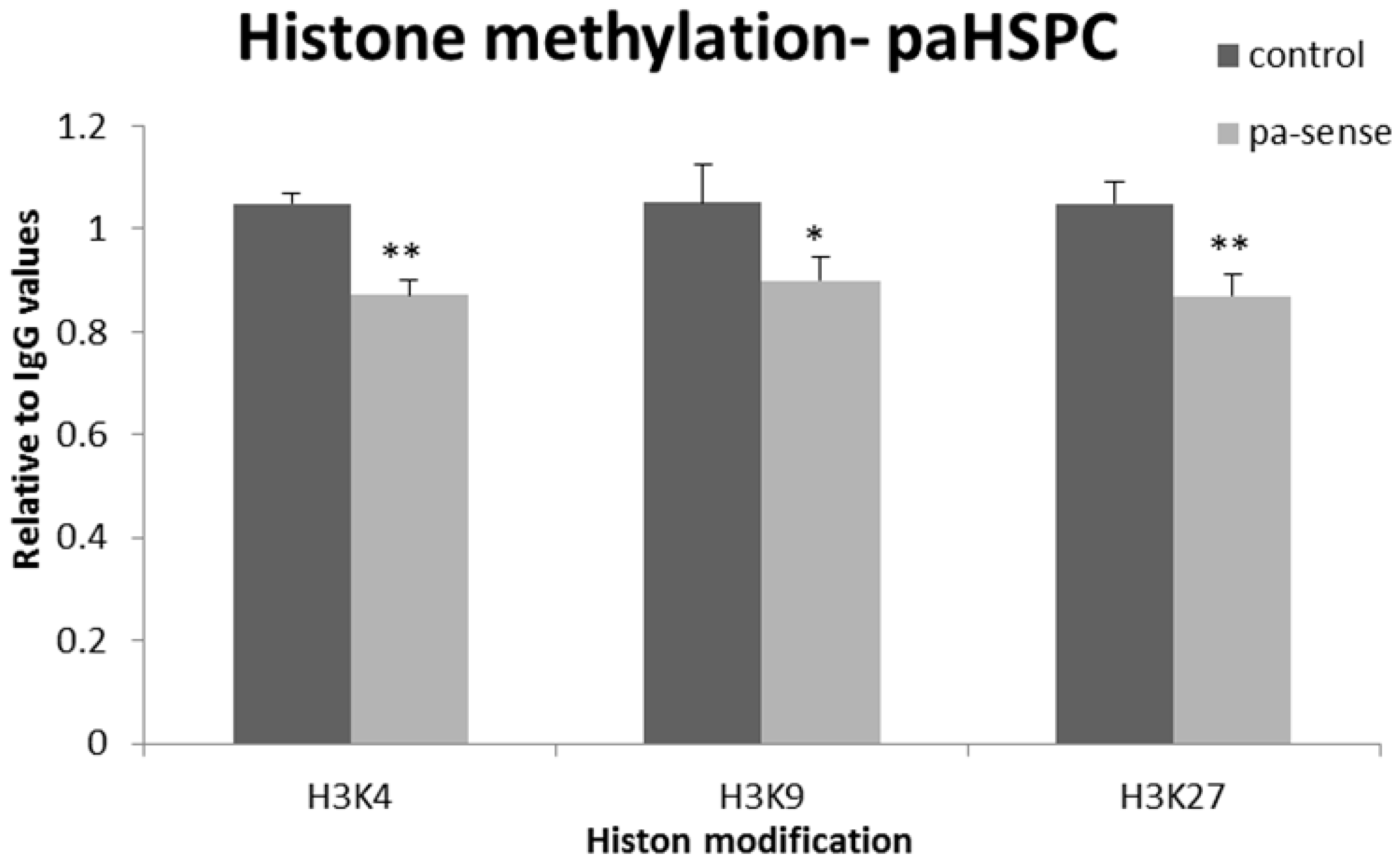
3.4. Epigenetic Modifications in Gene Promoters
3.5. The Phenotypic Effects of paHSPC152 on Melanoma Cell Lines and Immortalized Melanocytes
4. Discussion
Supplementary Files
Supplementary File 1Author Contributions
Conflicts of Interest
References
- Lee, J.T. Epigenetic regulation by long noncoding RNAs. Science 2012, 338, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Bajic, V.B.; Zhang, Z. On the classification of long non-coding RNAs. RNA Biol. 2013, 10, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Taft, R.J.; Kaplan, C.D.; Simons, C.; Mattick, J.S. Evolution, biogenesis and function of promoter-associated RNAs. Cell Cycle 2009, 8, 2332–2338. [Google Scholar] [CrossRef] [PubMed]
- Taft, R.J.; Glazov, E.A.; Cloonan, N.; Simons, C.; Stephen, S.; Faulkner, G.J.; Lassmann, T.; Forrest, A.R.; Grimmond, S.M.; Schroder, K.; et al. Tiny RNAs associated with transcription start sites in animals. Nat. Genet. 2009, 41, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Napoli, S.; Pastori, C.; Magistri, M.; Carbone, G.M.; Catapano, C.V. Promoter-specific transcriptional interference and c-myc gene silencing by siRNAs in human cells. EMBO J. 2009, 28, 1708–1719. [Google Scholar] [CrossRef] [PubMed]
- Seila, A.C.; Calabrese, J.M.; Levine, S.S.; Yeo, G.W.; Rahl, P.B.; Flynn, R.A.; Young, R.A.; Sharp, P.A. Divergent transcription from active promoters. Science 2008, 322, 1849–1851. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kim, D.; Morris, K.V. Promoter-associated RNA is required for RNA-directed transcriptional gene silencing in human cells. Proc. Natl. Acad. Sci. USA 2007, 104, 12422–12427. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.X.; Ma, J.X. Promoter-associated RNAs and promoter-targeted RNAs. Cell Mol. Life Sci. 2012, 69, 2833–2842. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Sunkin, S.M.; Mehler, M.F.; Mattick, J.S. Specific expression of long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. USA 2008, 105, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Davidow, L.S.; Warshawsky, D. Tsix, a gene antisense to Xist at the X-inactivation centre. Nat. Genet. 1999, 21, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Loewer, S.; Cabili, M.N.; Guttman, M.; Loh, Y.H.; Thomas, K.; Park, I.H.; Garber, M.; Curran, M.; Onder, T.; Agarwal, S.; et al. Large intergenic non-coding RNA-ROR modulates reprogramming of human induced pluripotent stem cells. Nat. Genet. 2010, 42, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.; Chang, H.Y. Long noncoding RNA in genome regulation: Prospects and mechanisms. RNA Biol. 2010, 7, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, F.; Marchand, M.; Hainaut, P.; Pouillart, P.; Sastre, X.; Ikeda, H.; Boon, T.; Coulie, P.G. Differences in the antigens recognized by cytolytic T cells on two successive metastases of a melanoma patient are consistent with immune selection. Eur. J. Immunol. 1995, 25, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.P.; Marincola, F.M.; Kawakami, Y.; Taubenberger, J.; Yannelli, J.R.; Rosenberg, S.A. Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J. Natl. Cancer Inst. 1996, 88, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, M.; Nicotra, M.R.; Apollonj, C.; Fraioli, R.; Giacomini, P.; Natali, P.G. Production and characterization of the murine monoclonal antibody 2G10 to a human T4-tyrosinase epitope. J. Investig. Dermatol. 1991, 96, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Kuperwasser, C.; Brunet, J.P.; Ramaswamy, S.; Kuo, W.L.; Gray, J.W.; Naber, S.P.; Weinberg, R.A. The melanocyte differentiation program predisposes to metastasis after neoplastic transformation. Nat. Genet. 2005, 37, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Avni, D.; Yang, H.; Martelli, F.; Hofmann, F.; ElShamy, W.M.; Ganesan, S.; Scully, R.; Livingston, D.M. Active localization of the retinoblastoma protein in chromatin and its response to S phase DNA damage. Mol. Cell 2003, 12, 735–746. [Google Scholar] [CrossRef]
- Markel, G.; Ortenberg, R.; Seidman, R.; Sapoznik, S.; Koren-Morag, N.; Besser, M.J.; Bar, J.; Shapira, R.; Kubi, A.; Nardini, G.; et al. Systemic dysregulation of CEACAM1 in melanoma patients. Cancer Immunol. Immunother. CII 2010, 59, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermuller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef] [PubMed]
- Fejes-Toth, K.; Sotirova, V.; Sachidanandam, R.; Assaf, G.; Hannon, G.J.; Kapranov, P.; Foissac, S.; Willingham, A.T.; Duttagupta, R.; Dumais, E.; et al. Post-transcriptional processing generates a diversity of 5′-modified long and short RNAs. Nature 2009, 457, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- DataBase of Transcriptional Start Sites. Available online: http://dbtss.hgc.jp (accessed on 29 February 2012).
- Yamashita, R.; Sathira, N.P.; Kanai, A.; Tanimoto, K.; Arauchi, T.; Tanaka, Y.; Hashimoto, S.; Sugano, S.; Nakai, K.; Suzuki, Y. Genome-wide characterization of transcriptional start sites in humans by integrative transcriptome analysis. Genome Res. 2011, 21, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, R.; Wakaguri, H.; Sugano, S.; Suzuki, Y.; Nakai, K. DBTSS provides a tissue specific dynamic view of Transcription Start Sites. Nucleic Acids Res. 2010, 38, D98–D104. [Google Scholar] [CrossRef] [PubMed]
- Bittner, M.; Meltzer, P.; Chen, Y.; Jiang, Y.; Seftor, E.; Hendrix, M.; Radmacher, M.; Simon, R.; Yakhini, Z.; Ben-Dor, A.; et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature 2000, 406, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Boratyn, G.M.; Camacho, C.; Cooper, P.S.; Coulouris, G.; Fong, A.; Ma, N.; Madden, T.L.; Matten, W.T.; McGinnis, S.D.; Merezhuk, Y.; et al. BLAST: A more efficient report with usability improvements. Nucleic Acids Res. 2013, 41, W29–W33. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.V. The emerging role of RNA in the regulation of gene transcription in human cells. Semin. Cell Dev. Biol. 2011, 22, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Botchkarev, V.A.; Gdula, M.R.; Mardaryev, A.N.; Sharov, A.A.; Fessing, M.Y. Epigenetic regulation of gene expression in keratinocytes. J. Investig. Dermatol. 2012, 132, 2505–2521. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Schones, D.E.; Zhao, K. Characterization of human epigenomes. Curr. Opin. Genet. Dev. 2009, 19, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.G.; Santoso, S.; Adams, C.; Anest, V.; Morris, K.V. Promoter targeted small RNAs induce long-term transcriptional gene silencing in human cells. Nucleic Acids Res. 2009, 37, 2984–2995. [Google Scholar] [CrossRef] [PubMed]
- Preker, P.; Nielsen, J.; Kammler, S.; Lykke-Andersen, S.; Christensen, M.S.; Mapendano, C.K.; Schierup, M.H.; Jensen, T.H. RNA exosome depletion reveals transcription upstream of active human promoters. Science 2008, 322, 1851–1854. [Google Scholar] [CrossRef] [PubMed]
- Towns, W.L.; Begley, T.J. Transfer RNA methytransferases and their corresponding modifications in budding yeast and humans: Activities, predications, and potential roles in human health. DNA Cell Biol. 2012, 31, 434–454. [Google Scholar] [CrossRef] [PubMed]
- Figaro, S.; Scrima, N.; Buckingham, R.H.; Heurgue-Hamard, V. HemK2 protein, encoded on human chromosome 21, methylates translation termination factor eRF1. FEBS Lett. 2008, 582, 2352–2356. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; He, H.; Zhang, Y.; Han, Z.; Hou, G.; Zeng, T.; Liu, Q.; Wu, Q. TRMT112 gene expression in mouse embryonic development. Acta Histochem. Cytochem. 2012, 45, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Mazauric, M.H.; Dirick, L.; Purushothaman, S.K.; Bjork, G.R.; Lapeyre, B. Trm112p is a 15-kDa zinc finger protein essential for the activity of two tRNA and one protein methyltransferases in yeast. J. Biol. Chem. 2010, 285, 18505–18515. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Qin, Z.; Wang, M.; Xu, C.; Feng, G.; Liu, J.; Meng, Z.; Hu, Y. The Arabidopsis SMO2, a homologue of yeast TRM112, modulates progression of cell division during organ growth. Plant J. 2010, 61, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, V.; Chen, Y.; Elkahloun, A.; Dutra, A.; Pak, E.; Chandrasekharappa, S. Chromosome 8 BAC array comparative genomic hybridization and expression analysis identify amplification and overexpression of TRMT12 in breast cancer. Genes Chromosomes Cancer 2007, 46, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Jimbow, K.; Hua, C.; Gomez, P.F.; Hirosaki, K.; Shinoda, K.; Salopek, T.G.; Matsusaka, H.; Jin, H.Y.; Yamashita, T. Intracellular vesicular trafficking of tyrosinase gene family protein in EU- and pheomelanosome biogenesis. Pigment Cell Res. 2000, 13 (Suppl. 8), 110–117. [Google Scholar] [CrossRef] [PubMed]
- Pawelek, J.; Korner, A.; Bergstrom, A.; Bologna, J. New regulators of melanin biosynthesis and the autodestruction of melanoma cells. Nature 1980, 286, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Urabe, K.; Aroca, P.; Tsukamoto, K.; Mascagna, D.; Palumbo, A.; Prota, G.; Hearing, V.J. The inherent cytotoxicity of melanin precursors: A revision. Biochim. Biophys. Acta 1994, 1221, 272–278. [Google Scholar] [CrossRef]
- Singh, M.V.; Jimbow, K. Tyrosinase transfection produces melanin synthesis and growth retardation in glioma cells. Melanoma Res. 1998, 8, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Rad, H.H.; Yamashita, T.; Jin, H.Y.; Hirosaki, K.; Wakamatsu, K.; Ito, S.; Jimbow, K. Tyrosinase-related proteins suppress tyrosinase-mediated cell death of melanocytes and melanoma cells. Exp. Cell Res. 2004, 298, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Chourrout, D.; Vervoort, M.; Leblanc, B.; Cavalli, G. Genome regulation by polycomb and trithorax proteins. Cell 2007, 128, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.E.; Moore, H.M.; Li, X.; Toy, K.A.; Huang, W.; Sabel, M.S.; Kidwell, K.M.; Kleer, C.G. EZH2 expands breast stem cells through activation of NOTCH1 signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 3098–3103. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Bi, C.; Clark, B.S.; Mady, R.; Shah, P.; Kohtz, J.D. The Evf-2 noncoding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes Dev. 2006, 20, 1470–1484. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Qu, K.; Zhong, F.L.; Artandi, S.E.; Chang, H.Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 2011, 44, 667–678. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| paRNA/Gene Name | Forward 5′–3′ | Reverse 5′–3′ |
|---|---|---|
| TYR | AGCACCCCACAAATCCTAACTTAC | ATGGCTGTTGTACTCCTCCAATC |
| paTYR | GTGGGATACGAGCCAATT | TGGCTGAGACCTATATAATACCA |
| HSPC152 | CGTATCTGCCCTGTGGAATT | ATCAGACGCAAGTTATCGGC |
| paHSPC | AGCGGAGGACGACCTTTT | CATGCGAGCTCAGCAGATTG |
| RPLPO | CAGATCCGCATGTCCCTTCG | GCAGCAGTTTCTCCAGAGCTGG |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonen, H.; Kol, N.; Shomron, N.; Leibowitz-Amit, R.; Quagliata, L.; Lorber, T.; Sidi, Y.; Avni, D. Promoter-Associated RNAs Regulate HSPC152 Gene Expression in Malignant Melanoma. Non-Coding RNA 2016, 2, 7. https://doi.org/10.3390/ncrna2030007
Bonen H, Kol N, Shomron N, Leibowitz-Amit R, Quagliata L, Lorber T, Sidi Y, Avni D. Promoter-Associated RNAs Regulate HSPC152 Gene Expression in Malignant Melanoma. Non-Coding RNA. 2016; 2(3):7. https://doi.org/10.3390/ncrna2030007
Chicago/Turabian StyleBonen, Hamutal, Nitzan Kol, Noam Shomron, Raya Leibowitz-Amit, Luca Quagliata, Thomas Lorber, Yechezkel Sidi, and Dror Avni. 2016. "Promoter-Associated RNAs Regulate HSPC152 Gene Expression in Malignant Melanoma" Non-Coding RNA 2, no. 3: 7. https://doi.org/10.3390/ncrna2030007
APA StyleBonen, H., Kol, N., Shomron, N., Leibowitz-Amit, R., Quagliata, L., Lorber, T., Sidi, Y., & Avni, D. (2016). Promoter-Associated RNAs Regulate HSPC152 Gene Expression in Malignant Melanoma. Non-Coding RNA, 2(3), 7. https://doi.org/10.3390/ncrna2030007