
RNA Metabolism and the Role of Small RNAs in Regulating Multiple Aspects of RNA Metabolism

Abstract
:1. Introduction
2. Eukaryotic Transcriptional Machinery and sRNA-Mediated Regulation of Transcriptional Dynamics
3. Intersections Between mRNA Decay and sRNA-Mediated Gene Silencing Pathways
4. Crosstalk Between Nascent RNA Processing, RNA Modification, and sRNA Pathways
5. RNA Editing, Retrograde Signaling, and Potential Links to sRNA Pathways
6. The Role of RNA Metabolism and sRNA-Mediated Gene Regulation in Plant Growth, Development, and Stress Responses
7. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gregory, B.D.; O’Malley, R.C.; Lister, R.; Urich, M.A.; Tonti-Filippini, J.; Chen, H.; Millar, A.H.; Ecker, J.R. A link between RNA metabolism and silencing affecting Arabidopsis development. Dev. Cell 2008, 14, 854–866. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Liu, Q.; A Smith, N.; Liang, G.; Wang, M.-B. RNA silencing in plants: Mechanisms, technologies and applications in horticultural crops. Curr. Genom. 2016, 17, 476–489. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.-D.; Lin, Y.; Ren, Q.-P.; Wang, Y.-Y.; Xiong, F.; Wang, X.-L. RNA splicing of flc modulates the transition to flowering. Front. Plant Sci. 2019, 10, 1625. [Google Scholar] [CrossRef]
- Parker, M.T.; Knop, K.; Simpson, G.G. Making a mark: The role of RNA modifications in plant biology. Biochemist 2020, 42, 26–30. [Google Scholar] [CrossRef]
- Yoshinaga, M.; Takeuchi, O. RNA metabolism governs immune function and response. Basic Immunol. Its Clin. Appl. 2024, 1444, 145–161. [Google Scholar]
- Ali, N.A.; Song, W.; Huang, J.; Wu, D.; Zhao, X. Recent advances and biotechnological applications of RNA metabolism in plant chloroplasts and mitochondria. Crit. Rev. Biotechnol. 2024, 44, 1552–1573. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Yao, J.; Zhou, S.; Yang, J.; Zhang, Y.; Yang, X.; Li, L.; Zhang, Y.; Zhuang, Y.; Yang, Y. Spatiotemporal control of RNA metabolism and CRISPR–Cas functions using engineered photoswitchable RNA-binding proteins. Nat. Protoc. 2024, 19, 374–405. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chen, X. Intercellular and systemic trafficking of RNAs in plants. Nat. Plants 2018, 4, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Shankar, N.; Nath, U. Advantage looping: Gene regulatory circuits between microRNAs and their target transcription factors in plants. Plant Physiol. 2024, 196, kiae462. [Google Scholar] [CrossRef] [PubMed]
- Moazed, D. Small RNAs in transcriptional gene silencing and genome defence. Nature 2009, 457, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, T.; Tomari, Y. MicroRNAs mediate gene silencing via multiple different pathways in Drosophila. Mol. Cell 2012, 48, 825–836. [Google Scholar] [CrossRef] [PubMed]
- El-Sappah, A.H.; Yan, K.; Huang, Q.; Islam, M.M.; Li, Q.; Wang, Y.; Khan, M.S.; Zhao, X.; Mir, R.R.; Li, J. Comprehensive mechanism of gene silencing and its role in plant growth and development. Front. Plant Sci. 2021, 12, 705249. [Google Scholar] [CrossRef]
- Reyer, M.A.; Chennakesavalu, S.; Heideman, E.M.; Ma, X.; Bujnowska, M.; Hong, L.; Dinner, A.R.; Vanderpool, C.K.; Fei, J. Kinetic modeling reveals additional regulation at co-transcriptional level by post-transcriptional sRNA regulators. Cell Rep. 2021, 36, 109764. [Google Scholar] [CrossRef]
- Axtell, M.J. Classification and comparison of small RNAs from plants. Annu. Rev. Plant Biol. 2013, 64, 137–159. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, R.M.; Picard, C.L. RNA-directed DNA methylation. PLoS Genet. 2020, 16, e1009034. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Rechavi, O. Plant and animal small RNA communications between cells and organisms. Nat. Rev. Mol. Cell Biol. 2022, 23, 185–203. [Google Scholar] [CrossRef]
- Cuerda-Gil, D.; Slotkin, R.K. Non-canonical RNA-directed DNA methylation. Nat. Plants 2016, 2, 16163. [Google Scholar] [CrossRef]
- Castel, S.E.; Martienssen, R.A. RNA interference in the nucleus: Roles for small RNAs in transcription, epigenetics and beyond. Nat. Rev. Genet. 2013, 14, 100–112. [Google Scholar] [CrossRef]
- Corona-Gomez, J.A.; Coss-Navarrete, E.L.; Garcia-Lopez, I.J.; Klapproth, C.; Pérez-Patiño, J.A.; Fernandez-Valverde, S.L. Transcriptome-guided annotation and functional classification of long non-coding RNAs in Arabidopsis thaliana. Sci. Rep. 2022, 12, 14063. [Google Scholar] [CrossRef]
- Franco-Zorrilla, J.M.; Valli, A.; Todesco, M.; Mateos, I.; Puga, M.I.; Rubio-Somoza, I.; Leyva, A.; Weigel, D.; García, J.A.; Paz-Ares, J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 2007, 39, 1033–1037. [Google Scholar] [CrossRef]
- Grelet, S.; Link, L.A.; Howley, B.; Obellianne, C.; Palanisamy, V.; Gangaraju, V.K.; Diehl, J.A.; Howe, P.H. A regulated PNUTS mRNA to lncRNA splice switch mediates EMT and tumour progression. Nat. Cell Biol. 2017, 19, 1105–1115. [Google Scholar] [CrossRef]
- Hu, W.L.; Jin, L.; Xu, A.; Wang, Y.F.; Thorne, R.F.; Zhang, X.D.; Wu, M. GUARDIN is a p53-responsive long non-coding RNA that is essential for genomic stability. Nat. Cell Biol. 2018, 20, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Gebert, L.F.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Hudzik, C.; Maguire, S.; Guan, S.; Held, J.; Axtell, M.J. Trans-species microRNA loci in the parasitic plant Cuscuta campestris have a U6-like snRNA promoter. Plant Cell 2023, 35, 1834–1847. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Dey, C.; Chakraborty, S.; Sengupta, A. RNA Interference (RNAi): A Boon to Medical Biotechnology. In Exploring Medical Biotechnology-In Vivo, In Vitro, In Silico; CRC Press: Boca Raton, FL, USA, 2024; pp. 231–259. [Google Scholar]
- Niu, J.; Chen, R.; Wang, J.J. RNA interference in insects: The link between antiviral defense and pest control. Insect Sci. 2024, 31, 2–12. [Google Scholar] [CrossRef]
- Wu, X.; Chen, S.; Zhang, Z.; Zhou, W.; Sun, T.; Ning, K.; Xu, M.; Ke, X.; Xu, P. A viral small interfering RNA-host plant mRNA pathway modulates virus-induced drought tolerance by enhancing autophagy. Plant Cell 2024, 36, 3219–3236. [Google Scholar] [CrossRef]
- Buchon, N.; Vaury, C. RNAi: A defensive RNA-silencing against viruses and transposable elements. Heredity 2006, 96, 195–202. [Google Scholar] [CrossRef]
- Azad, M.F.; de Silva Weligodage, H.; Dhingra, A.; Dawar, P.; Rock, C.D. Grain development and crop productivity: Role of small RNA. In Plant Small RNA in Food Crops; Elsevier: Amsterdam, The Netherlands, 2023; pp. 385–468. [Google Scholar]
- Singh, D.; Chaudhary, S.; Kumar, R.; Sirohi, P.; Mehla, K.; Sirohi, A.; Kumar, S.; Chand, P.; Singh, P.K. RNA interference technology—Applications and limitations. RNA Interference. Nature 2016, 418, 21–36. [Google Scholar]
- Kuhn, J.M.; Breton, G.; Schroeder, J.I. mRNA metabolism of flowering-time regulators in wild-type Arabidopsis revealed by a nuclear cap binding protein mutant, abh1. Plant J. 2007, 50, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Huang, J.; Chory, J. Unraveling the linkage between retrograde signaling and RNA metabolism in plants. Trends Plant Sci. 2020, 25, 141–147. [Google Scholar] [CrossRef]
- Gilbert, S.F. Mechanisms for the environmental regulation of gene expression: Ecological aspects of animal development. J. Biosci. 2005, 30, 65–74. [Google Scholar] [CrossRef]
- Kaufmann, K.; Pajoro, A.; Angenent, G.C. Regulation of transcription in plants: Mechanisms controlling developmental switches. Nat. Rev. Genet. 2010, 11, 830–842. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.; Huang, X.; Jauhari, S.; Jiang, Q.; Li, X. Chromatin Hubs: A biological and computational outlook. Comput. Struct. Biotechnol. J. 2022, 20, 3796–3813. [Google Scholar] [CrossRef] [PubMed]
- Zecchini, V.; Mills, I.G. Putting chromatin immunoprecipitation into context. J. Cell. Biochem. 2009, 107, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Brkljacic, J.; Grotewold, E. Combinatorial control of plant gene expression. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2017, 1860, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Müller, F.; Zaucker, A.; Tora, L. Developmental regulation of transcription initiation: More than just changing the actors. Curr. Opin. Genet. Dev. 2010, 20, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Kirchmaier, S.; Lust, K.; Wittbrodt, J. Golden GATEway cloning–a combinatorial approach to generate fusion and recombination constructs. PLoS ONE 2013, 8, e76117. [Google Scholar] [CrossRef]
- Osman, S.; Cramer, P. Structural biology of RNA polymerase II transcription: 20 years on. Annu. Rev. Cell Dev. Biol. 2020, 36, 1–34. [Google Scholar] [CrossRef]
- Proudfoot, N.J. Transcriptional termination in mammals: Stopping the RNA polymerase II juggernaut. Science 2016, 352, aad9926. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.D.; West, S. Termination of transcription by RNA polymerase II: BOOM! Trends Genet. 2020, 36, 664–675. [Google Scholar] [CrossRef]
- Nagarajan, V.K.; Jones, C.I.; Newbury, S.F.; Green, P.J. XRN 5′ → 3′ exoribonucleases: Structure, mechanisms and functions. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2013, 1829, 590–603. [Google Scholar] [CrossRef]
- Mo, W.; Liu, B.; Zhang, H.; Jin, X.; Lu, D.; Yu, Y.; Liu, Y.; Jia, J.; Long, Y.; Deng, X. Landscape of transcription termination in Arabidopsis revealed by single-molecule nascent RNA sequencing. Genome Biol. 2021, 22, 322. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D. Mediator and the mechanism of transcriptional activation. Trends Biochem. Sci. 2005, 30, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, L.; Qu, L.J. Plant Mediator complex and its critical functions in transcription regulation. J. Integr. Plant Biol. 2016, 58, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Buendía-Monreal, M.; Gillmor, C.S. Mediator: A key regulator of plant development. Dev. Biol. 2016, 419, 7–18. [Google Scholar] [CrossRef]
- Youn, D.Y.; Xiaoli, A.M.; Pessin, J.E.; Yang, F. Regulation of metabolism by the Mediator complex. Biophys. Rep. 2016, 2, 69–77. [Google Scholar] [CrossRef]
- Pai, D.A.; Kaplan, C.D.; Kweon, H.K.; Murakami, K.; Andrews, P.C.; Engelke, D.R. RNAs nonspecifically inhibit RNA polymerase II by preventing binding to the DNA template. RNA 2014, 20, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Grueter, C.E.; Van Rooij, E.; Johnson, B.A.; DeLeon, S.M.; Sutherland, L.B.; Qi, X.; Gautron, L.; Elmquist, J.K.; Bassel-Duby, R.; Olson, E.N. A cardiac microRNA governs systemic energy homeostasis by regulation of MED13. Cell 2012, 149, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Miyoshi, T.; Kugou, K.; Hoffman, C.S.; Shibata, T.; Ohta, K. Stepwise chromatin remodelling by a cascade of transcription initiation of non-coding RNAs. Nature 2008, 456, 130–134. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, X.; Lin, F.; He, G.; Terzaghi, W.; Zhu, D.; Deng, X.W. Arabidopsis noncoding RNA mediates control of photomorphogenesis by red light. Proc. Natl. Acad. Sci. USA 2014, 111, 10359–10364. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.B.; Sung, S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science 2011, 331, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Henson, R.; Wehbe–Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-g.; Wang, J.-j.; Zhao, F.; Liu, Q.; Jiang, K.; Yang, G.-h. MicroRNA-21 (miR-21) represses tumor suppressor PTEN and promotes growth and invasion in non-small cell lung cancer (NSCLC). Clin. Chim. Acta 2010, 411, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.F.L.; Agarwal, V.; Mansfield, J.H.; Denans, N.; Schwartz, M.G.; Prosser, H.M.; Pourquié, O.; Bartel, D.P.; Tabin, C.J.; McGlinn, E. Independent regulation of vertebral number and vertebral identity by microRNA-196 paralogs. Proc. Natl. Acad. Sci. USA 2015, 112, E4884–E4893. [Google Scholar] [CrossRef] [PubMed]
- Hornstein, E.; Mansfield, J.H.; Yekta, S.; Hu, J.K.-H.; Harfe, B.D.; McManus, M.T.; Baskerville, S.; Bartel, D.P.; Tabin, C.J. The microRNA miR-196 acts upstream of Hoxb8 and Shh in limb development. Nature 2005, 438, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhang, Y.; Zhang, L.; Weakley, S.M.; Yao, Q. MicroRNA-196: Critical roles and clinical applications in development and cancer. J. Cell. Mol. Med. 2011, 15, 14–23. [Google Scholar] [CrossRef]
- Lund, A. miR-10 in development and cancer. Cell Death Differ. 2010, 17, 209–214. [Google Scholar] [CrossRef]
- Xia, R.; Xu, J.; Meyers, B.C. The emergence, evolution, and diversification of the miR390-TAS3-ARF pathway in land plants. Plant Cell 2017, 29, 1232–1247. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.-J.; Mittal, A.; Jia, F.; Rock, C.D. An autoregulatory feedback loop involving PAP1 and TAS4 in response to sugars in Arabidopsis. Plant Mol. Biol. 2012, 80, 117–129. [Google Scholar] [CrossRef]
- Bonar, N.; Liney, M.; Zhang, R.; Austin, C.; Dessoly, J.; Davidson, D.; Stephens, J.; McDougall, G.; Taylor, M.; Bryan, G.J. Potato miR828 is associated with purple tuber skin and flesh color. Front. Plant Sci. 2018, 9, 1742. [Google Scholar] [CrossRef]
- Ma, J.; Zhao, P.; Liu, S.; Yang, Q.; Guo, H. The control of developmental phase transitions by microRNAs and their targets in seed plants. Int. J. Mol. Sci. 2020, 21, 1971. [Google Scholar] [CrossRef]
- Xu, M.; Hu, T.; Zhao, J.; Park, M.-Y.; Earley, K.W.; Wu, G.; Yang, L.; Poethig, R.S. Developmental functions of mir156-regulated squamosa promoter binding protein-like (spl) genes in Arabidopsis thaliana. PLoS Genet. 2016, 12, e1006263. [Google Scholar] [CrossRef] [PubMed]
- Jerome Jeyakumar, J.M.; Ali, A.; Wang, W.-M.; Thiruvengadam, M. Characterizing the role of the miR156-SPL network in plant development and stress response. Plants 2020, 9, 1206. [Google Scholar] [CrossRef]
- Millar, A.A.; Lohe, A.; Wong, G. Biology and function of miR159 in plants. Plants 2019, 8, 255. [Google Scholar] [CrossRef]
- Fu, T.; Wang, C.; Yang, Y.; Yang, X.; Wang, J.; Zhang, L.; Wang, Z.; Wang, Y. Function identification of miR159a, a positive regulator during poplar resistance to drought stress. Hortic. Res. 2023, 10, uhad221. [Google Scholar] [CrossRef]
- Yang, T.; Wang, Y.; Teotia, S.; Wang, Z.; Shi, C.; Sun, H.; Gu, Y.; Zhang, Z.; Tang, G. The interaction between miR160 and miR165/166 in the control of leaf development and drought tolerance in Arabidopsis. Sci. Rep. 2019, 9, 2832. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Lu, Q.; Wang, J.; Wang, L.; Xiang, F.; Liu, Z. MiR160 and its target genes ARF10, ARF16 and ARF17 modulate hypocotyl elongation in a light, BRZ, or PAC-dependent manner in Arabidopsis: miR160 promotes hypocotyl elongation. Plant Sci. 2021, 303, 110686. [Google Scholar] [CrossRef] [PubMed]
- Hao, K.; Wang, Y.; Zhu, Z.; Wu, Y.; Chen, R.; Zhang, L. miR160: An indispensable regulator in plant. Front. Plant Sci. 2022, 13, 833322. [Google Scholar] [CrossRef] [PubMed]
- Hibara, K.-I.; Karim, M.R.; Takada, S.; Taoka, K.-i.; Furutani, M.; Aida, M.; Tasaka, M. Arabidopsis CUP-SHAPED COTYLEDON3 regulates postembryonic shoot meristem and organ boundary formation. Plant Cell 2006, 18, 2946–2957. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.; Greb, T.; Peaucelle, A.; Blein, T.; Laufs, P.; Theres, K. Interplay of miR164, CUP-SHAPED COTYLEDON genes and LATERAL SUPPRESSOR controls axillary meristem formation in Arabidopsis thaliana. Plant J. 2008, 55, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Wei, W.; Li, Y.; Kan, L.; Wang, F.; Zhang, X.; Li, F.; Liu, Z.; Kang, C. Conserved and novel roles of miR164-CUC 2 regulatory module in specifying leaf and floral organ morphology in strawberry. New Phytol. 2019, 224, 480–492. [Google Scholar] [CrossRef]
- Yan, J.; Zhao, C.; Zhou, J.; Yang, Y.; Wang, P.; Zhu, X.; Tang, G.; Bressan, R.A.; Zhu, J.-K. The miR165/166 mediated regulatory module plays critical roles in ABA homeostasis and response in Arabidopsis thaliana. PLoS Genet. 2016, 12, e1006416. [Google Scholar] [CrossRef]
- Merelo, P.; Ram, H.; Pia Caggiano, M.; Ohno, C.; Ott, F.; Straub, D.; Graeff, M.; Cho, S.K.; Yang, S.W.; Wenkel, S. Regulation of MIR165/166 by class II and class III homeodomain leucine zipper proteins establishes leaf polarity. Proc. Natl. Acad. Sci. USA 2016, 113, 11973–11978. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Kumar, S.; Verma, R.; Lata, C.; Sanyal, I.; Rai, S.P. microRNA 166: An evolutionarily conserved stress biomarker in land plants targeting HD-ZIP family. Physiol. Mol. Biol. Plants 2021, 27, 2471–2485. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Chen, J.; Zhou, J.; Yu, H.; Ge, C.; Zhang, M.; Gao, X.; Dai, X.; Yang, Z.-N.; Zhao, Y. An essential role for miRNA167 in maternal control of embryonic and seed development. Plant Physiol. 2019, 180, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Caruana, J.C.; Dhar, N.; Raina, R. Overexpression of Arabidopsis microRNA167 induces salicylic acid-dependent defense against Pseudomonas syringae through the regulation of its targets ARF6 and ARF8. Plant Direct 2020, 4, e00270. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Hu, B.; Zhang, C. microRNAs and their roles in plant development. Front. Plant Sci. 2022, 13, 824240. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Y.; Zhang, L.; Li, W.W.; Hu, X.L.; Wang, M.-B.; Fan, Y.L.; Zhang, C.Y.; Wang, L. Stress-induced early flowering is mediated by miR169 in Arabidopsis thaliana. J. Exp. Bot. 2014, 65, 89–101. [Google Scholar] [CrossRef]
- Sorin, C.; Declerck, M.; Christ, A.; Blein, T.; Ma, L.; Lelandais-Brière, C.; Njo, M.F.; Beeckman, T.; Crespi, M.; Hartmann, C. A mi R 169 isoform regulates specific NF-YA targets and root architecture in A rabidopsis. New Phytol. 2014, 202, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Balyan, S.; Jha, S.; Mathur, S. Novel insights into expansion and functional diversification of MIR169 family in tomato. Planta 2020, 251, 55. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Hu, X.; Cai, W.; Huang, W.; Zhou, X.; Luo, Q.; Yang, H.; Wang, J.; Huang, J. Arabidopsis miR171-targeted scarecrow-like proteins bind to GT cis-elements and mediate gibberellin-regulated chlorophyll biosynthesis under light conditions. PLoS Genet. 2014, 10, e1004519. [Google Scholar] [CrossRef]
- Pei, L.L.; Zhang, L.L.; Liu, X.; Jiang, J. Role of microRNA miR171 in plant development. PeerJ 2023, 11, e15632. [Google Scholar] [CrossRef]
- Werner, S.; Bartrina, I.; Schmülling, T. Cytokinin regulates vegetative phase change in Arabidopsis thaliana through the miR172/TOE1-TOE2 module. Nat. Commun. 2021, 12, 5816. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Wang, L.; Ma, N.; Zhou, C.-M.; Han, L.; Zhang, T.-Q.; Wang, J.-W. Redundant and specific roles of individual MIR172 genes in plant development. PLoS Biol. 2021, 19, e3001044. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, X. Secrets of the MIR172 family in plant development and flowering unveiled. PLoS Biol. 2021, 19, e3001099. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Sato, F.; Ohme-Takagi, M. Roles of miR319 and TCP transcription factors in leaf development. Plant Physiol. 2017, 175, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Bresso, E.G.; Chorostecki, U.; Rodriguez, R.E.; Palatnik, J.F.; Schommer, C. Spatial control of gene expression by miR319-regulated TCP transcription factors in leaf development. Plant Physiol. 2018, 176, 1694–1708. [Google Scholar] [CrossRef]
- Fang, Y.; Zheng, Y.; Lu, W.; Li, J.; Duan, Y.; Zhang, S.; Wang, Y. Roles of miR319-regulated TCPs in plant development and response to abiotic stress. Crop J. 2021, 9, 17–28. [Google Scholar] [CrossRef]
- Jian, C.; Hao, P.; Hao, C.; Liu, S.; Mao, H.; Song, Q.; Zhou, Y.; Yin, S.; Hou, J.; Zhang, W. The miR319/TaGAMYB3 module regulates plant architecture and improves grain yield in common wheat (Triticum aestivum). New Phytol. 2022, 235, 1515–1530. [Google Scholar] [CrossRef]
- Jiang, J.; Zhu, H.; Li, N.; Batley, J.; Wang, Y. The miR393-target module regulates plant development and responses to biotic and abiotic stresses. Int. J. Mol. Sci. 2022, 23, 9477. [Google Scholar] [CrossRef]
- Mecchia, M.A.; Debernardi, J.M.; Rodriguez, R.E.; Schommer, C.; Palatnik, J.F. MicroRNA miR396 and RDR6 synergistically regulate leaf development. Mech. Dev. 2013, 130, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Szczygieł-Sommer, A.; Gaj, M.D. The miR396–GRF regulatory module controls the embryogenic response in Arabidopsis via an auxin-related pathway. Int. J. Mol. Sci. 2019, 20, 5221. [Google Scholar] [CrossRef]
- Debernardi, J.M.; Rodriguez, R.E.; Mecchia, M.A.; Palatnik, J.F. Functional specialization of the plant miR396 regulatory network through distinct microRNA–target interactions. PLoS Genet. 2012, 8, e1002419. [Google Scholar] [CrossRef]
- Yuan, S.; Zhao, J.; Li, Z.; Hu, Q.; Yuan, N.; Zhou, M.; Xia, X.; Noorai, R.; Saski, C.; Li, S. MicroRNA396-mediated alteration in plant development and salinity stress response in creeping bentgrass. Hortic. Res. 2019, 6, 48. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Li, Z.; Yuan, N.; Hu, Q.; Zhou, M.; Zhao, J.; Li, D.; Luo, H. MiR396 is involved in plant response to vernalization and flower development in Agrostis stolonifera. Hortic. Res. 2020, 7, 173. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Pang, M.; Nah, G.; Shi, X.; Ye, W.; Stelly, D.M.; Chen, Z.J. miR828 and miR858 regulate homoeologous MYB2 gene functions in Arabidopsis trichome and cotton fibre development. Nat. Commun. 2014, 5, 3050. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, J.; Danzeng, P.; Danzeng, C.; Song, S.; Wang, L.; Zhao, L.; Xu, W.; Zhang, C.; Ma, C. VvMYB114 mediated by miR828 negatively regulates trichome development of Arabidopsis. Plant Sci. 2021, 309, 110936. [Google Scholar] [CrossRef]
- Yamagishi, M.; Sakai, M. The microRNA828/MYB12 module mediates bicolor pattern development in Asiatic hybrid lily (Lilium spp.) flowers. Front. Plant Sci. 2020, 11, 590791. [Google Scholar] [CrossRef]
- Wang, X.; Yao, S.; Htet, W.P.P.M.; Yue, Y.; Zhang, Z.; Sun, K.; Chen, S.; Luo, K.; Fan, D. MicroRNA828 negatively regulates lignin biosynthesis in stem of Populus tomentosa through MYB targets. Tree Physiol. 2022, 42, 1646–1661. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Tiwari, M.; Pandey, A.; Bhatia, C.; Sharma, A.; Trivedi, P.K. MicroRNA858 is a potential regulator of phenylpropanoid pathway and plant development. Plant Physiol. 2016, 171, 944–959. [Google Scholar] [CrossRef] [PubMed]
- Lange, H.; Gagliardi, D. Catalytic activities, molecular connections, and biological functions of plant RNA exosome complexes. Plant Cell 2022, 34, 967–988. [Google Scholar] [CrossRef] [PubMed]
- Reverdatto, S.V.; Dutko, J.A.; Chekanova, J.A.; Hamilton, D.A.; Belostotsky, D.A. mRNA deadenylation by PARN is essential for embryogenesis in higher plants. RNA 2004, 10, 1200–1214. [Google Scholar] [CrossRef]
- Liang, W.; Li, C.; Liu, F.; Jiang, H.; Li, S.; Sun, J.; Wu, X.; Li, C. The Arabidopsis homologs of CCR4-associated factor 1 show mRNA deadenylation activity and play a role in plant defence responses. Cell Res. 2009, 19, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.B. Deadenylation: Enzymes, regulation, and functional implications. Wiley Interdiscip. Rev. RNA 2014, 5, 421–443. [Google Scholar] [CrossRef]
- Goldstrohm, A.C.; Wickens, M. Multifunctional deadenylase complexes diversify mRNA control. Nat. Rev. Mol. Cell Biol. 2008, 9, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Jensen, T.H. The exosome: A multipurpose RNA-decay machine. Trends Biochem. Sci. 2008, 33, 501–510. [Google Scholar] [CrossRef]
- Liu, H.; Kiledjian, M. Scavenger decapping activity facilitates 5′ to 3′ mRNA decay. Mol. Cell. Biol. 2005, 25, 9764–9772. [Google Scholar] [CrossRef]
- Xu, J.; Yang, J.-Y.; Niu, Q.-W.; Chua, N.-H. Arabidopsis DCP2, DCP1, and VARICOSE form a decapping complex required for postembryonic development. Plant Cell 2006, 18, 3386–3398. [Google Scholar] [CrossRef] [PubMed]
- German, M.A.; Luo, S.; Schroth, G.; Meyers, B.C.; Green, P.J. Construction of Parallel Analysis of RNA Ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nat. Protoc. 2009, 4, 356–362. [Google Scholar] [CrossRef]
- Addo-Quaye, C.; Eshoo, T.W.; Bartel, D.P.; Axtell, M.J. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 2008, 18, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Willmann, M.R.; Anderson, S.J.; Gregory, B.D. Genome-wide mapping of uncapped and cleaved transcripts reveals a role for the nuclear mRNA cap-binding complex in cotranslational RNA decay in Arabidopsis. Plant Cell 2016, 28, 2385–2397. [Google Scholar] [CrossRef]
- Chouaib, R.; Safieddine, A.; Pichon, X.; Imbert, A.; Kwon, O.S.; Samacoits, A.; Traboulsi, A.-M.; Robert, M.-C.; Tsanov, N.; Coleno, E. A dual protein-mRNA localization screen reveals compartmentalized translation and widespread co-translational RNA targeting. Dev. Cell 2020, 54, 773–791.e775. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Guo, H. mRNA decay in plants: Both quantity and quality matter. Curr. Opin. Plant Biol. 2017, 35, 138–144. [Google Scholar] [CrossRef]
- Huntzinger, E.; Kashima, I.; Fauser, M.; Saulière, J.; Izaurralde, E. SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA 2008, 14, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Kufel, J.; Bousquet-Antonelli, C.; Beggs, J.D.; Tollervey, D. Nuclear pre-mRNA decapping and 5′ degradation in yeast require the Lsm2-8p complex. Mol. Cell. Biol. 2004, 24, 9646–9657. [Google Scholar] [CrossRef]
- Orban, T.I.; Izaurralde, E. Decay of mRNAs targeted by RISC requires XRN1, the Ski complex, and the exosome. RNA 2005, 11, 459–469. [Google Scholar] [CrossRef]
- Souret, F.F.; Kastenmayer, J.P.; Green, P.J. AtXRN4 degrades mRNA in Arabidopsis and its substrates include selected miRNA targets. Mol. Cell 2004, 15, 173–183. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Han, Y.-W.; Fujii, H.; Aizawa, S.; Nishino, T.; Ishikawa, M. Cooperative recruitment of RDR6 by SGS3 and SDE5 during small interfering RNA amplification in Arabidopsis. Proc. Natl. Acad. Sci. USA 2021, 118, e2102885118. [Google Scholar] [CrossRef] [PubMed]
- Parent, J.-S.; Jauvion, V.; Bouché, N.; Béclin, C.; Hachet, M.; Zytnicki, M.; Vaucheret, H. Post-transcriptional gene silencing triggered by sense transgenes involves uncapped antisense RNA and differs from silencing intentionally triggered by antisense transgenes. Nucleic Acids Res. 2015, 43, 8464–8475. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chen, X. RNA quality control as a key to suppressing RNA silencing of endogenous genes in plants. Mol. Plant 2016, 9, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, Y.; Liu, X.; Hong, X.; Xu, Y.; Zhu, P.; Shen, Y.; Wu, H.; Ji, Y.; Wen, X. Suppression of endogenous gene silencing by bidirectional cytoplasmic RNA decay in Arabidopsis. Science 2015, 348, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Martínez de Alba, A.E.; Moreno, A.B.; Gabriel, M.; Mallory, A.C.; Christ, A.; Bounon, R.; Balzergue, S.; Aubourg, S.; Gautheret, D.; Crespi, M.D. In plants, decapping prevents RDR6-dependent production of small interfering RNAs from endogenous mRNAs. Nucleic Acids Res. 2015, 43, 2902–2913. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Venhuizen, P.; Wu, M.-T.; Chiou, P.; Chang, C.-L.; Kalyna, M.; Matzke, A.J.; Matzke, M. A collection of pre-mRNA splicing mutants in Arabidopsis thaliana. G3 Genes Genomes Genet. 2020, 10, 1983–1996. [Google Scholar] [CrossRef]
- Iwakawa, H.-o.; Tomari, Y. The functions of microRNAs: mRNA decay and translational repression. Trends Cell Biol. 2015, 25, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Tellier, M.; Maudlin, I.; Murphy, S. Transcription and splicing: A two-way street. Wiley Interdiscip. Rev. RNA 2020, 11, e1593. [Google Scholar] [CrossRef]
- Schor, I.E.; Gómez Acuña, L.I.; Kornblihtt, A.R. Coupling between transcription and alternative splicing. RNA Cancer 2013, 158, 1–24. [Google Scholar]
- Sims, R.J.; Millhouse, S.; Chen, C.-F.; Lewis, B.A.; Erdjument-Bromage, H.; Tempst, P.; Manley, J.L.; Reinberg, D. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol. Cell 2007, 28, 665–676. [Google Scholar] [CrossRef]
- Kelemen, O.; Convertini, P.; Zhang, Z.; Wen, Y.; Shen, M.; Falaleeva, M.; Stamm, S. Function of alternative splicing. Gene 2013, 514, 1–30. [Google Scholar] [CrossRef]
- Bayne, E.H.; Portoso, M.; Kagansky, A.; Kos-Braun, I.C.; Urano, T.; Ekwall, K.; Alves, F.; Rappsilber, J.; Allshire, R.C. Splicing factors facilitate RNAi-directed silencing in fission yeast. Science 2008, 322, 602–606. [Google Scholar] [CrossRef]
- Lockhart, S.R.; Rymond, B.C. Commitment of yeast pre-mRNA to the splicing pathway requires a novel Ul small nuclear ribonucleoprotein polypeptide, Prp39p. Mol. Cell. Biol. 1994, 14, 3623–3633. [Google Scholar] [CrossRef] [PubMed]
- Kallgren, S.P.; Andrews, S.; Tadeo, X.; Hou, H.; Moresco, J.J.; Tu, P.G.; Yates III, J.R.; Nagy, P.L.; Jia, S. The proper splicing of RNAi factors is critical for pericentric heterochromatin assembly in fission yeast. PLoS Genet. 2014, 10, e1004334. [Google Scholar] [CrossRef] [PubMed]
- Bielewicz, D.; Kalak, M.; Kalyna, M.; Windels, D.; Barta, A.; Vazquez, F.; Szweykowska-Kulinska, Z.; Jarmolowski, A. Introns of plant pri-miRNAs enhance miRNA biogenesis. EMBO Rep. 2013, 14, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Liu, P.; Wu, C.-A.; Yang, G.-D.; Xu, R.; Guo, Q.-H.; Huang, J.-G.; Zheng, C.-C. Stress-induced alternative splicing provides a mechanism for the regulation of microRNA processing in Arabidopsis thaliana. Mol. Cell 2012, 48, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.; Rock, C.D. MIR846 and MIR842 comprise a cistronic MIRNA pair that is regulated by abscisic acid by alternative splicing in roots of Arabidopsis. Plant Mol. Biol. 2013, 81, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Christie, M.; Croft, L.J.; Carroll, B.J. Intron splicing suppresses RNA silencing in Arabidopsis. Plant J. 2011, 68, 159–167. [Google Scholar] [CrossRef]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.-J.; Chen, Q. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 2017, 541, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Moshitch-Moshkovitz, S.; Han, D.; Kol, N.; Amariglio, N.; Rechavi, G.; Dominissini, D.; He, C. Nm-seq maps 2′-O-methylation sites in human mRNA with base precision. Nat. Methods 2017, 14, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Li, X.; Xiong, X.; Wang, K.; Wang, L.; Shu, X.; Ma, S.; Yi, C. Transcriptome-wide mapping reveals reversible and dynamic N 1-methyladenosine methylome. Nat. Chem. Biol. 2016, 12, 311–316. [Google Scholar] [CrossRef]
- Duan, H.-C.; Wei, L.-H.; Zhang, C.; Wang, Y.; Chen, L.; Lu, Z.; Chen, P.R.; He, C.; Jia, G. ALKBH10B is an RNA N 6-methyladenosine demethylase affecting Arabidopsis floral transition. Plant Cell 2017, 29, 2995–3011. [Google Scholar] [CrossRef] [PubMed]
- Squires, J.E.; Patel, H.R.; Nousch, M.; Sibbritt, T.; Humphreys, D.T.; Parker, B.J.; Suter, C.M.; Preiss, T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012, 40, 5023–5033. [Google Scholar] [CrossRef]
- Delatte, B.; Wang, F.; Ngoc, L.V.; Collignon, E.; Bonvin, E.; Deplus, R.; Calonne, E.; Hassabi, B.; Putmans, P.; Awe, S. Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science 2016, 351, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Levanon, E.Y.; Eisenberg, E.; Yelin, R.; Nemzer, S.; Hallegger, M.; Shemesh, R.; Fligelman, Z.Y.; Shoshan, A.; Pollock, S.R.; Sztybel, D. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 2004, 22, 1001–1005. [Google Scholar] [CrossRef]
- Carlile, T.M.; Rojas-Duran, M.F.; Zinshteyn, B.; Shin, H.; Bartoli, K.M.; Gilbert, W.V. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 2014, 515, 143–146. [Google Scholar] [CrossRef]
- Chang, H.; Lim, J.; Ha, M.; Kim, V.N. TAIL-seq: Genome-wide determination of poly (A) tail length and 3′ end modifications. Mol. Cell 2014, 53, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Chen, X.; Yu, B. Small RNAs meet their targets: When methylation defends miRNAs from uridylation. RNA Biol. 2014, 11, 1099–1104. [Google Scholar] [CrossRef]
- Xiong, Q.; Zhang, Y. Small RNA modifications: Regulatory molecules and potential applications. J. Hematol. Oncol. 2023, 16, 64. [Google Scholar] [CrossRef]
- Arribas-Hernández, L.; Brodersen, P. Occurrence and functions of m6A and other covalent modifications in plant mRNA. Plant Physiol. 2020, 182, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Wei, L.; Chen, Z.; Cai, Z.; Lu, Q.; Wang, C.; Tian, E.; Jia, G. m6A readers ECT2/ECT3/ECT4 enhance mRNA stability through direct recruitment of the poly (A) binding proteins in Arabidopsis. Genome Biol. 2023, 24, 103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Theler, D.; Kaminska, K.H.; Hiller, M.; de la Grange, P.; Pudimat, R.; Rafalska, I.; Heinrich, B.; Bujnicki, J.M.; Allain, F.H.-T. The YTH domain is a novel RNA binding domain. J. Biol. Chem. 2010, 285, 14701–14710. [Google Scholar] [CrossRef]
- Yang, Y.; Hsu, P.J.; Chen, Y.-S.; Yang, Y.-G. Dynamic transcriptomic m6A decoration: Writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018, 28, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Guo, J.; Fan, Z. Interactions between m6A modification and miRNAs in malignant tumors. Cell Death Dis. 2021, 12, 598. [Google Scholar] [CrossRef]
- Bhat, S.S.; Bielewicz, D.; Gulanicz, T.; Bodi, Z.; Yu, X.; Anderson, S.J.; Szewc, L.; Bajczyk, M.; Dolata, J.; Grzelak, N. mRNA adenosine methylase (MTA) deposits m6A on pri-miRNAs to modulate miRNA biogenesis in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2020, 117, 21785–21795. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Liu, G.; Wang, W.; Shen, W.; Zhao, Y.; Sun, J.; Yang, Q.; Zhang, Y.; Fan, W.; Pei, S. RNA editing and its roles in plant organelles. Front. Genet. 2021, 12, 757109. [Google Scholar] [CrossRef]
- Popitsch, N.; Huber, C.D.; Buchumenski, I.; Eisenberg, E.; Jantsch, M.; von Haeseler, A.; Gallach, M. A-to-I RNA editing uncovers hidden signals of adaptive genome evolution in animals. Genome Biol. Evol. 2020, 12, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Small, I.D.; Schallenberg-Rüdinger, M.; Takenaka, M.; Mireau, H.; Ostersetzer-Biran, O. Plant organellar RNA editing: What 30 years of research has revealed. Plant J. 2020, 101, 1040–1056. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, Q.; Yin, P. RNA editing machinery in plant organelles. Sci. China Life Sci. 2018, 61, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Van Wijk, K.J.; Bentolila, S.; Leppert, T.; Sun, Q.; Sun, Z.; Mendoza, L.; Li, M.; Deutsch, E.W. Detection and editing of the updated Arabidopsis plastid-and mitochondrial-encoded proteomes through PeptideAtlas. Plant Physiol. 2024, 194, 1411–1430. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Xie, B.; Peng, L.; Wu, Q.; Hu, J. Profiling of RNA editing events in plant organellar transcriptomes with high-throughput sequencing. Plant J. 2024, 118, 345–357. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Jiang, R.-C.; Wang, Y.; Tang, J.-J.; Sun, F.; Yang, Y.-Z.; Tan, B.-C. ZmPPR26, a DYW-type pentatricopeptide repeat protein, is required for C-to-U RNA editing at atpA-1148 in maize chloroplasts. J. Exp. Bot. 2021, 72, 4809–4821. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Yang, S.; Zhang, Y.; Zhiyuan, C.; Tang, K.; Li, G.; Yu, H.; Leng, J.; Wang, Q. GmPGL2, encoding a pentatricopeptide repeat protein, is essential for chloroplast RNA editing and biogenesis in soybean. Front. Plant Sci. 2021, 12, 690973. [Google Scholar] [CrossRef]
- Fang, J.; Jiang, X.-H.; Wang, T.-F.; Zhang, X.-J.; Zhang, A.-D. Tissue-specificity of RNA editing in plant: Analysis of transcripts from three tobacco (Nicotiana tabacum) varieties. Plant Biotechnol. Rep. 2021, 15, 471–482. [Google Scholar] [CrossRef]
- Covello, P.S.; Gray, M.W. RNA editing in plant mitochondria. Nature 1989, 341, 662–666. [Google Scholar] [CrossRef]
- Hoch, B.; Maier, R.M.; Appel, K.; Igloi, G.L.; Kössel, H. Editing of a chloroplast mRNA by creation of an initiation codon. Nature 1991, 353, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Barkan, A.; Small, I. Pentatricopeptide repeat proteins in plants. Annu. Rev. Plant Biol. 2014, 65, 415–442. [Google Scholar] [CrossRef]
- Lesch, E.; Schilling, M.T.; Brenner, S.; Yang, Y.; Gruss, O.J.; Knoop, V.; Schallenberg-Rüdinger, M. Plant mitochondrial RNA editing factors can perform targeted C-to-U editing of nuclear transcripts in human cells. Nucleic Acids Res. 2022, 50, 9966–9983. [Google Scholar] [CrossRef] [PubMed]
- Bentolila, S.; Heller, W.P.; Sun, T.; Babina, A.M.; Friso, G.; van Wijk, K.J.; Hanson, M.R. RIP1, a member of an Arabidopsis protein family, interacts with the protein RARE1 and broadly affects RNA editing. Proc. Natl. Acad. Sci. USA 2012, 109, E1453–E1461. [Google Scholar] [CrossRef]
- Zhang, F.; Tang, W.; Hedtke, B.; Zhong, L.; Liu, L.; Peng, L.; Lu, C.; Grimm, B.; Lin, R. Tetrapyrrole biosynthetic enzyme protoporphyrinogen IX oxidase 1 is required for plastid RNA editing. Proc. Natl. Acad. Sci. USA 2014, 111, 2023–2028. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Shi, X.; Friso, G.; Van Wijk, K.; Bentolila, S.; Hanson, M.R. A zinc finger motif-containing protein is essential for chloroplast RNA editing. PLoS Genet. 2015, 11, e1005028. [Google Scholar] [CrossRef]
- Shi, X.; Bentolila, S.; Hanson, M.R. Organelle RNA recognition motif-containing (ORRM) proteins are plastid and mitochondrial editing factors in Arabidopsis. Plant Signal. Behav. 2016, 11, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Gipson, A.B.; Hanson, M.R.; Bentolila, S. The RanBP2 zinc finger domains of chloroplast RNA editing factor OZ1 are required for protein–protein interactions and conversion of C to U. Plant J. 2022, 109, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Huang, J.; Chory, J. GUN1 interacts with MORF2 to regulate plastid RNA editing during retrograde signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 10162–10167. [Google Scholar] [CrossRef] [PubMed]
- De Las Rivas, J.; Lozano, J.J.; Ortiz, A.R. Comparative analysis of chloroplast genomes: Functional annotation, genome-based phylogeny, and deduced evolutionary patterns. Genome Res. 2002, 12, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhang, Y.; Xu, J.; Li, W.; Li, M. Characterization of the complete chloroplast genome sequence of Dalbergia species and its phylogenetic implications. Sci. Rep. 2019, 9, 20401. [Google Scholar] [CrossRef]
- Møller, I.M.; Rasmusson, A.G.; Van Aken, O. Plant mitochondria–past, present and future. Plant J. 2021, 108, 912–959. [Google Scholar] [CrossRef] [PubMed]
- Chi, W.; He, B.; Mao, J.; Jiang, J.; Zhang, L. Plastid sigma factors: Their individual functions and regulation in transcription. Biochim. Biophys. Acta (BBA)-Bioenerg. 2015, 1847, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Savchenko, T.; Baidoo, E.E.; Chehab, W.E.; Hayden, D.M.; Tolstikov, V.; Corwin, J.A.; Kliebenstein, D.J.; Keasling, J.D.; Dehesh, K. Retrograde signaling by the plastidial metabolite MEcPP regulates expression of nuclear stress-response genes. Cell 2012, 149, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Leister, D. Retrograde signaling in plants: From simple to complex scenarios. Front. Plant Sci. 2012, 3, 135. [Google Scholar] [CrossRef]
- Petrillo, E.; Godoy Herz, M.A.; Fuchs, A.; Reifer, D.; Fuller, J.; Yanovsky, M.J.; Simpson, C.; Brown, J.W.; Barta, A.; Kalyna, M. A chloroplast retrograde signal regulates nuclear alternative splicing. Science 2014, 344, 427–430. [Google Scholar] [CrossRef] [PubMed]
- De Souza, A.; Wang, J.-Z.; Dehesh, K. Retrograde signals: Integrators of interorganellar communication and orchestrators of plant development. Annu. Rev. Plant Biol. 2017, 68, 85–108. [Google Scholar] [CrossRef] [PubMed]
- Barreto, P.; Dambire, C.; Sharma, G.; Vicente, J.; Osborne, R.; Yassitepe, J.; Gibbs, D.J.; Maia, I.G.; Holdsworth, M.J.; Arruda, P. Mitochondrial retrograde signaling through UCP1-mediated inhibition of the plant oxygen-sensing pathway. Curr. Biol. 2022, 32, 1403–1411.e4. [Google Scholar] [CrossRef] [PubMed]
- Dietz, K.-J.; Turkan, I.; Krieger-Liszkay, A. Redox-and reactive oxygen species-dependent signaling into and out of the photosynthesizing chloroplast. Plant Physiol. 2016, 171, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Estavillo, G.M.; Crisp, P.A.; Pornsiriwong, W.; Wirtz, M.; Collinge, D.; Carrie, C.; Giraud, E.; Whelan, J.; David, P.; Javot, H. Evidence for a SAL1-PAP chloroplast retrograde pathway that functions in drought and high light signaling in Arabidopsis. Plant Cell 2011, 23, 3992–4012. [Google Scholar] [CrossRef]
- Havaux, M. Carotenoid oxidation products as stress signals in plants. Plant J. 2014, 79, 597–606. [Google Scholar] [CrossRef]
- He, C.; Liew, L.C.; Yin, L.; Lewsey, M.G.; Whelan, J.; Berkowitz, O. The retrograde signaling regulator ANAC017 recruits the MKK9–MPK3/6, ethylene, and auxin signaling pathways to balance mitochondrial dysfunction with growth. Plant Cell 2022, 34, 3460–3481. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, A.; Plazzi, F.; Milani, L.; Ghiselli, F.; Passamonti, M. SmithRNAs: Could mitochondria “bend” nuclear regulation? Mol. Biol. Evol. 2017, 34, 1960–1973. [Google Scholar] [CrossRef]
- Pozzi, A.; Dowling, D.K. New insights into mitochondrial–nuclear interactions revealed through analysis of small RNAs. Genome Biol. Evol. 2022, 14, evac023. [Google Scholar] [CrossRef]
- Habermann, K.; Tiwari, B.; Krantz, M.; Adler, S.O.; Klipp, E.; Arif, M.A.; Frank, W. Identification of small non-coding RNAs responsive to GUN1 and GUN5 related retrograde signals in Arabidopsis thaliana. Plant J. 2020, 104, 138–155. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Zhao, G.; Zhang, S.; Li, Y.; Gu, H.; Li, Y.; Zhao, Q.; Qi, Y. Chloroplast-to-nucleus signaling regulates microRNA biogenesis in Arabidopsis. Dev. Cell 2019, 48, 371–382.e374. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Kapoor, A.; Zhu, J.-K. Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell 2006, 18, 2051–2065. [Google Scholar] [CrossRef]
- Xu, W.B.; Zhao, L.; Liu, P.; Guo, Q.H.; Wu, C.A.; Yang, G.D.; Huang, J.G.; Zhang, S.X.; Guo, X.Q.; Zhang, S.Z. Intronic microRNA-directed regulation of mitochondrial reactive oxygen species enhances plant stress tolerance in Arabidopsis. New Phytol. 2023, 240, 710–726. [Google Scholar] [CrossRef]
- Azad, M.F.; Dawar, P.; Esim, N.; Rock, C.D. Role of miRNAs in sucrose stress response, reactive oxygen species, and anthocyanin biosynthesis in Arabidopsis thaliana. Front. Plant Sci. 2023, 14, 1278320. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xu, L.; Hao, C.; Wan, M.; Tao, Y.; Zhuang, Y.; Su, Y.; Li, L. The microRNA408–plantacyanin module balances plant growth and drought resistance by regulating reactive oxygen species homeostasis in guard cells. Plant Cell 2024, 36, koae144. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Chen, C.; Zeng, J.; Yun, Z.; Liu, Y.; Qu, H.; Jiang, Y.; Duan, X.; Xia, R. Micro RNA 528, a hub regulator modulating ROS homeostasis via targeting of a diverse set of genes encoding copper-containing proteins in monocots. New Phytol. 2020, 225, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Mandal, S.N.; Neelam, K.; de Los Reyes, B.G. MicroRNA-mediated host defense mechanisms against pathogens and herbivores in rice: Balancing gains from genetic resistance with trade-offs to productivity potential. BMC Plant Biol. 2022, 22, 351. [Google Scholar] [CrossRef] [PubMed]
- Laubinger, S.; Sachsenberg, T.; Zeller, G.; Busch, W.; Lohmann, J.U.; Rätsch, G.; Weigel, D. Dual roles of the nuclear cap-binding complex and SERRATE in pre-mRNA splicing and microRNA processing in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2008, 105, 8795–8800. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Dhingra, A.; Dawar, P.; Payton, P.; Rock, C.D. The role of microRNAs in responses to drought and heat stress in peanut (Arachis hypogaea). Plant Genome 2023, 16, e20350. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Mandal, S.N.; Pradhan, B.; Kaur, P.; Kaur, K.; Neelam, K. From evolution to revolution: Accelerating crop domestication through genome editing. Plant Cell Physiol. 2022, 63, 1607–1623. [Google Scholar] [CrossRef]
- Herr, A.J.; Molnàr, A.; Jones, A.; Baulcombe, D.C. Defective RNA processing enhances RNA silencing and influences flowering of Arabidopsis. Proc. Natl. Acad. Sci. USA 2006, 103, 14994–15001. [Google Scholar] [CrossRef]
- Baurle, I.; Smith, L.; Baulcombe, D.C.; Dean, C. Widespread role for the flowering-time regulators FCA and FPA in RNA-mediated chromatin silencing. Science 2007, 318, 109–112. [Google Scholar] [CrossRef]
- Zhang, Y.; Gu, L.; Hou, Y.; Wang, L.; Deng, X.; Hang, R.; Chen, D.; Zhang, X.; Zhang, Y.; Liu, C. Integrative genome-wide analysis reveals HLP1, a novel RNA-binding protein, regulates plant flowering by targeting alternative polyadenylation. Cell Res. 2015, 25, 864–876. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhu, D.; Lin, X.; Miao, J.; Gu, L.; Deng, X.; Yang, Q.; Sun, K.; Zhu, D.; Cao, X. RNA binding proteins RZ-1B and RZ-1C play critical roles in regulating pre-mRNA splicing and gene expression during development in Arabidopsis. Plant Cell 2016, 28, 55–73. [Google Scholar] [CrossRef]
- Shi, X.; Germain, A.; Hanson, M.R.; Bentolila, S. RNA recognition motif-containing protein ORRM4 broadly affects mitochondrial RNA editing and impacts plant development and flowering. Plant Physiol. 2016, 170, 294–309. [Google Scholar] [CrossRef]
- Kappel, C.; Trost, G.; Czesnick, H.; Ramming, A.; Kolbe, B.; Vi, S.L.; Bispo, C.; Becker, J.D.; de Moor, C.; Lenhard, M. Genome-wide analysis of PAPS1-dependent polyadenylation identifies novel roles for functionally specialized poly (A) polymerases in Arabidopsis thaliana. PLoS Genet. 2015, 11, e1005474. [Google Scholar] [CrossRef] [PubMed]
- Van Lijsebettens, M.; Grasser, K.D. Transcript elongation factors: Shaping transcriptomes after transcript initiation. Trends Plant Sci. 2014, 19, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Kwak, K.J.; Jung, H.J.; Lee, K.H.; Kim, Y.S.; Kim, W.Y.; Ahn, S.J.; Kang, H. The minor spliceosomal protein U11/U12-31K is an RNA chaperone crucial for U12 intron splicing and the development of dicot and monocot plants. PLoS ONE 2012, 7, e43707. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Cho, H.S.; Nam, H.; Jo, H.; Yoon, J.; Park, C.; Dang, T.V.T.; Kim, E.; Jeong, J.; Park, S. Translational control of phloem development by RNA G-quadruplex–JULGI determines plant sink strength. Nat. Plants 2018, 4, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Bardou, F.; Ariel, F.; Simpson, C.G.; Romero-Barrios, N.; Laporte, P.; Balzergue, S.; Brown, J.W.; Crespi, M. Long noncoding RNA modulates alternative splicing regulators in Arabidopsis. Dev. Cell 2014, 30, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Amor, B.B.; Wirth, S.; Merchan, F.; Laporte, P.; d’Aubenton-Carafa, Y.; Hirsch, J.; Maizel, A.; Mallory, A.; Lucas, A.; Deragon, J.M. Novel long non-protein coding RNAs involved in Arabidopsis differentiation and stress responses. Genome Res. 2009, 19, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Rao, S.; Chang, B.; Wang, X.; Zhang, K.; Hou, X.; Zhu, X.; Wu, H.; Tian, Z.; Zhao, Z. AtLa1 protein initiates IRES-dependent translation of WUSCHEL mRNA and regulates the stem cell homeostasis of Arabidopsis in response to environmental hazards. Plant Cell Environ. 2015, 38, 2098–2114. [Google Scholar] [CrossRef] [PubMed]
- Kant, P.; Kant, S.; Gordon, M.; Shaked, R.; Barak, S. STRESS RESPONSE SUPPRESSOR1 and STRESS RESPONSE SUPPRESSOR2, two DEAD-box RNA helicases that attenuate Arabidopsis responses to multiple abiotic stresses. Plant Physiol. 2007, 145, 814–830. [Google Scholar] [CrossRef]
- Teotia, S.; Tang, G. To bloom or not to bloom: Role of microRNAs in plant flowering. Mol. Plant 2015, 8, 359–377. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Yang, M.; Le, B.H.; He, W.; Hou, Y. The master role of siRNAs in plant immunity. Mol. Plant Pathol. 2022, 23, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Mathioni, S.M.; Hammond, R.; Harkess, A.E.; Kakrana, A.; Arikit, S.; Dusia, A.; Meyers, B.C. Reproductive phasiRNA loci and DICER-LIKE5, but not microRNA loci, diversified in monocotyledonous plants. Plant Physiol. 2021, 185, 1764–1782. [Google Scholar] [CrossRef]
- Sarkar Das, S.; Yadav, S.; Singh, A.; Gautam, V.; Sarkar, A.K.; Nandi, A.K.; Karmakar, P.; Majee, M.; Sanan-Mishra, N. Expression dynamics of miRNAs and their targets in seed germination conditions reveals miRNA-ta-siRNA crosstalk as regulator of seed germination. Sci. Rep. 2018, 8, 1233. [Google Scholar] [CrossRef]
- Liu, Y.; Teng, C.; Xia, R.; Meyers, B.C. PhasiRNAs in plants: Their biogenesis, genic sources, and roles in stress responses, development, and reproduction. Plant Cell 2020, 32, 3059–3080. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Liu, J.; Liu, Z.; Li, X.; Wu, F.; He, Y. HEAT-INDUCED TAS1 TARGET1 mediates thermotolerance via heat stress transcription factor A1a–directed pathways in Arabidopsis. Plant Cell 2014, 26, 1764–1780. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, T.A.; Yoo, S.J.; Fahlgren, N.; Gilbert, S.D.; Howell, M.D.; Sullivan, C.M.; Alexander, A.; Nguyen, G.; Allen, E.; Ahn, J.H. AGO1-miR173 complex initiates phased siRNA formation in plants. Proc. Natl. Acad. Sci. USA 2008, 105, 20055–20062. [Google Scholar] [CrossRef] [PubMed]
- Felippes, F.F.; Weigel, D. Triggering the formation of tasiRNAs in Arabidopsis thaliana: The role of microRNA miR173. EMBO Rep. 2009, 10, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Meyers, B.C.; Liu, Z.; Beers, E.P.; Ye, S.; Liu, Z. MicroRNA superfamilies descended from miR390 and their roles in secondary small interfering RNA biogenesis in eudicots. Plant Cell 2013, 25, 1555–1572. [Google Scholar] [CrossRef] [PubMed]
- Si-Ammour, A.; Windels, D.; Arn-Bouldoires, E.; Kutter, C.; Ailhas, J.; Meins, F., Jr.; Vazquez, F. miR393 and secondary siRNAs regulate expression of the TIR1/AFB2 auxin receptor clade and auxin-related development of Arabidopsis leaves. Plant Physiol. 2011, 157, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huang, K.; Chen, W. Resolving cellular dynamics using single-cell temporal transcriptomics. Curr. Opin. Biotechnol. 2024, 85, 103060. [Google Scholar] [CrossRef]
- Conte, M.I.; Fuentes-Trillo, A.; Conde, C.D. Opportunities and tradeoffs in single-cell transcriptomic technologies. Trends Genet. 2024, 40, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Peidli, S.; Green, T.D.; Shen, C.; Gross, T.; Min, J.; Garda, S.; Yuan, B.; Schumacher, L.J.; Taylor-King, J.P.; Marks, D.S. scPerturb: Harmonized single-cell perturbation data. Nat. Methods 2024, 21, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Y.; Zhang, T.; Tan, W.T.; Lambert, F.; Darmawan, J.; Huber, R.; Wan, Y. RNA structure profiling at single-cell resolution reveals new determinants of cell identity. Nat. Methods 2024, 21, 411–422. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Z.; Zhuang, Y.; Wang, F.; Cai, T. Small RNA transcriptome analysis using parallel single-cell small RNA sequencing. Sci. Rep. 2023, 13, 7501. [Google Scholar] [CrossRef]
- Erhard, F.; Baptista, M.A.; Krammer, T.; Hennig, T.; Lange, M.; Arampatzi, P.; Jürges, C.S.; Theis, F.J.; Saliba, A.-E.; Dölken, L. scSLAM-seq reveals core features of transcription dynamics in single cells. Nature 2019, 571, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, K.; Lyu, Y.; Pan, H.; Zhang, J.; Stambolian, D.; Susztak, K.; Reilly, M.P.; Hu, G.; Li, M. Deep learning enables accurate clustering with batch effect removal in single-cell RNA-seq analysis. Nat. Commun. 2020, 11, 2338. [Google Scholar] [CrossRef]
- Asada, K.; Takasawa, K.; Machino, H.; Takahashi, S.; Shinkai, N.; Bolatkan, A.; Kobayashi, K.; Komatsu, M.; Kaneko, S.; Okamoto, K. Single-cell analysis using machine learning techniques and its application to medical research. Biomedicines 2021, 9, 1513. [Google Scholar] [CrossRef]
- Hou, N.; Lin, X.; Lin, L.; Zeng, X.; Zhong, Z.; Wang, X.; Cheng, R.; Lin, X.; Yang, C.; Song, J. Artificial intelligence in cell annotation for high-resolution RNA sequencing data. TrAC Trends Anal. Chem. 2024, 178, 117818. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
- Peterson, S.M.; Thompson, J.A.; Ufkin, M.L.; Sathyanarayana, P.; Liaw, L.; Congdon, C.B. Common features of microRNA target prediction tools. Front. Genet. 2014, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Tjaden, B. TargetRNA3: Predicting prokaryotic RNA regulatory targets with machine learning. Genome Biol. 2023, 24, 276. [Google Scholar] [CrossRef] [PubMed]
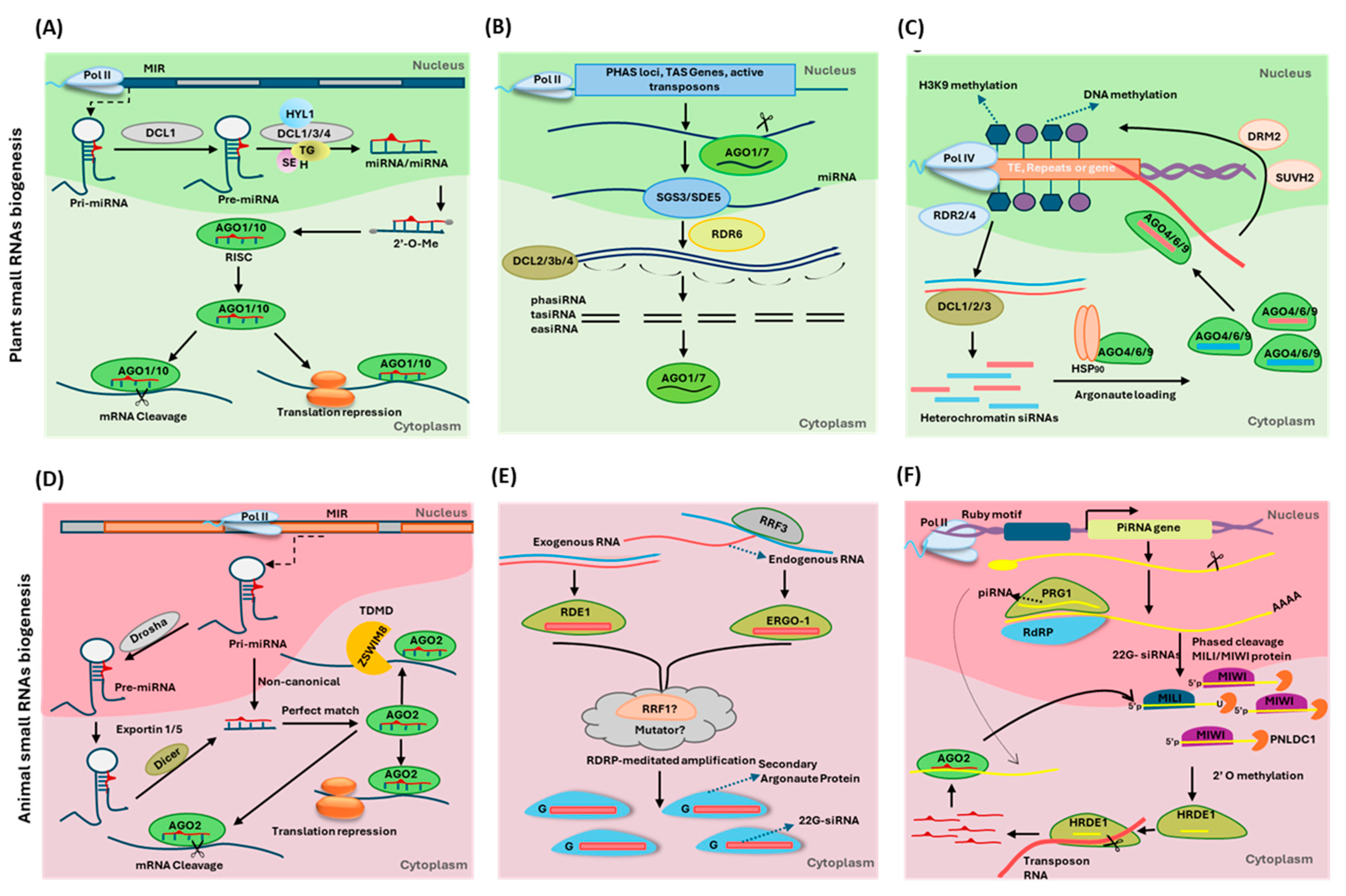
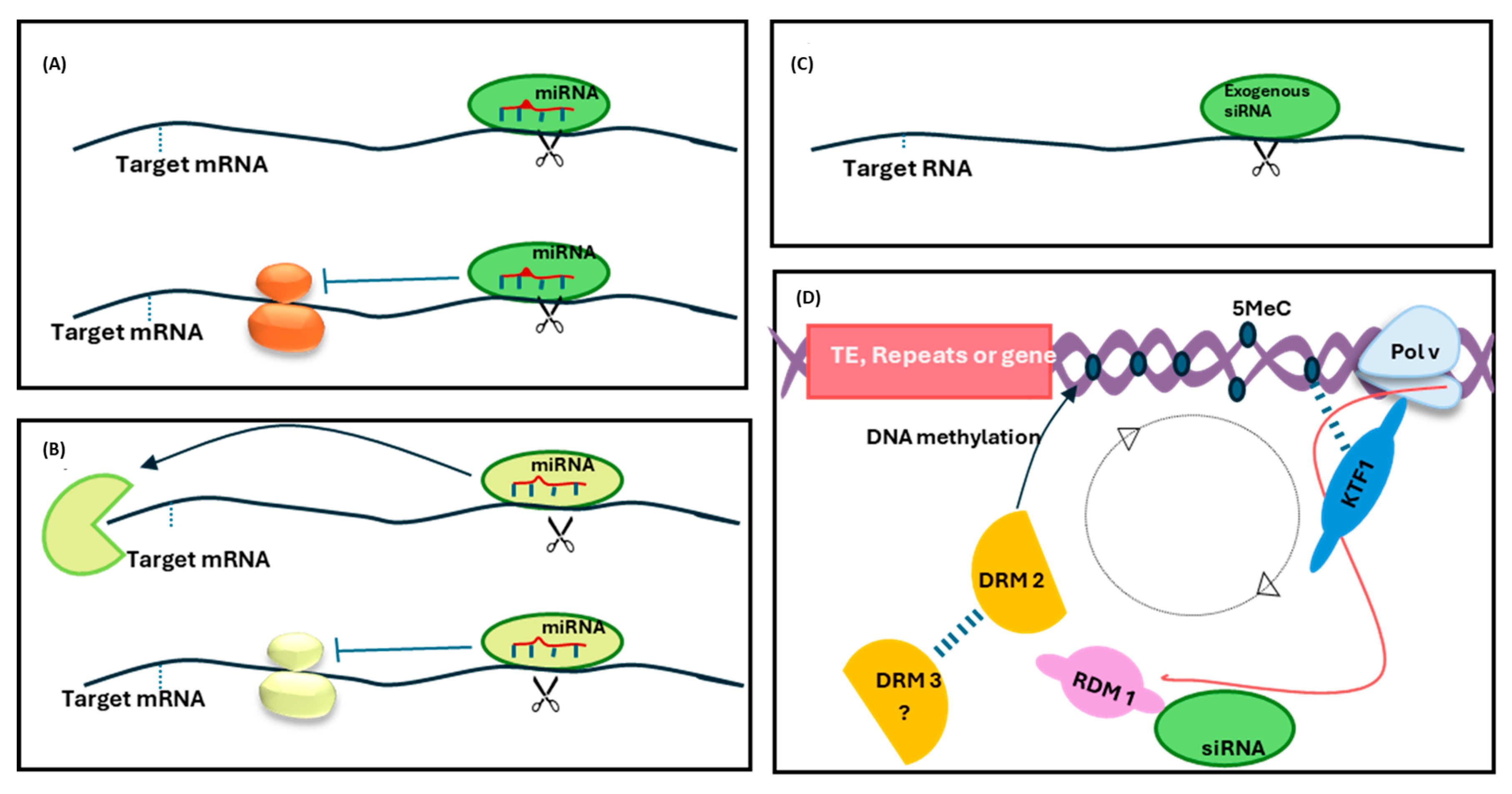
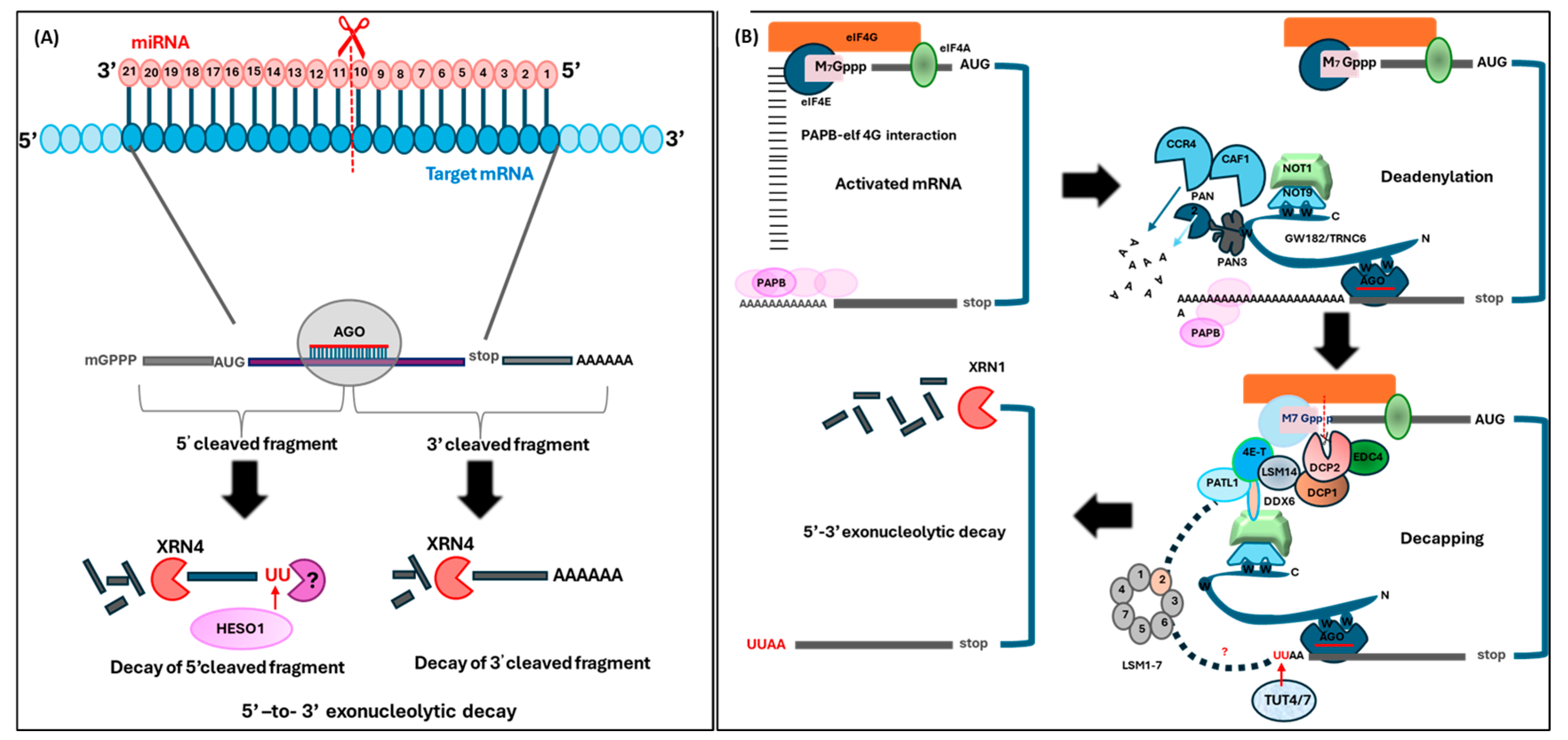

{kind=link}
{kind=link}
{kind=link}
| miRNA | Target Transcription Factor Family | Target Tissue/Cells | Regulatory and Developmental Function | References |
|---|---|---|---|---|
| miR156/157 | SPL gene family | seed, leaf, root, stem, trichome, pistil, and nodule | Vegetative to reproductive phase transition, leaf and root development, abiotic stress response, secondary metabolism | [63,64,65] |
| miR159 | GAMYB or GAMYB-like gene | seed, leaf, root, stamen, and pollen | Seed, leaf, and male reproductive development, drought response | [66,67] |
| miR160 | ARF10/16/17 | seed, leaf, stem, root, stamen, stamen, and pollen | Development of embryos, leaves, and roots, hypocotyl elongation | [68,69,70] |
| miR164 | NAC gene family | seed, leaf, lateral root, shoot apical meristem (SAM), flower, and fruit | Boundary formation, leaf, lateral root, fruit and flower development, auxiliary meristem formation | [71,72,73] |
| miR165/166 | HD-ZIPIII gene family | seed, root, SAM, vascular bundles, and nodule | Maintenance of meristematic cells, adaxial identity of leaves, promotion of the lateral root growth, and procambium development | [74,75,76] |
| miR167 | ARF6/8 | root, stamen, and pollen | Embryo development, root, stem, leaf, and flower formation, flowering time, abiotic stress response, pathogen defense | [77,78,79] |
| miR169 | CBF and NF-YA family | leaf, root, flower, and nodule | Flower and root development, abiotic stress response | [80,81,82] |
| miR170/171 | SCARECROW-like transcription factor genes | embryo, SAM, stem, leaf, and root | leaf, root, and flower development, meristem formation and maintenance, chlorophyll biosynthesis, phase transition | [83,84] |
| miR172 | AP2, TOE1/2/3, SMZ, SNZ | leaf, SAM, and flower | Flower meristem identity and organ development, vegetative to reproductive phase transition | [85,86,87] |
| miR319 | TCP2/3/4/10/24, MYB33/65/81/97/104/120 | SAM, leaf | leaf development and senescence, abiotic stress response, plant architecture, and grain yield | [88,89,90,91] |
| miR393 | AFB | embryo, root, shoot, leaf | Participates in embryo, root, shoot, and leaf development; biotic and abiotic stress response | [92] |
| miR396 | GRFs | embryo, SAM, leaf, and lateral root | somatic embryogenesis, leaf growth, flower development, grain size, panicle branching, biotic and abiotic stress response | [93,94,95,96,97] |
| miR828 and miR858 | MYBs | leaf, stem, flower, and fruit | Fiber development, lignin biosynthesis, trichome development, anthocyanin, and flavanol accumulation | [61,98,99,100,101,102] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dawar, P.; Adhikari, I.; Mandal, S.N.; Jayee, B. RNA Metabolism and the Role of Small RNAs in Regulating Multiple Aspects of RNA Metabolism. Non-Coding RNA 2025, 11, 1. https://doi.org/10.3390/ncrna11010001
Dawar P, Adhikari I, Mandal SN, Jayee B. RNA Metabolism and the Role of Small RNAs in Regulating Multiple Aspects of RNA Metabolism. Non-Coding RNA. 2025; 11(1):1. https://doi.org/10.3390/ncrna11010001
Chicago/Turabian StyleDawar, Pranav, Indra Adhikari, Swarupa Nanda Mandal, and Bhumika Jayee. 2025. "RNA Metabolism and the Role of Small RNAs in Regulating Multiple Aspects of RNA Metabolism" Non-Coding RNA 11, no. 1: 1. https://doi.org/10.3390/ncrna11010001
APA StyleDawar, P., Adhikari, I., Mandal, S. N., & Jayee, B. (2025). RNA Metabolism and the Role of Small RNAs in Regulating Multiple Aspects of RNA Metabolism. Non-Coding RNA, 11(1), 1. https://doi.org/10.3390/ncrna11010001