
The RNAi Machinery in the Fungus Fusarium fujikuroi Is Not Very Active in Synthetic Medium and Is Related to Transposable Elements
 , , , and
, , , and
Abstract

1. Introduction
2. Results
2.1. Small RNA Machinery in F. fujikuroi
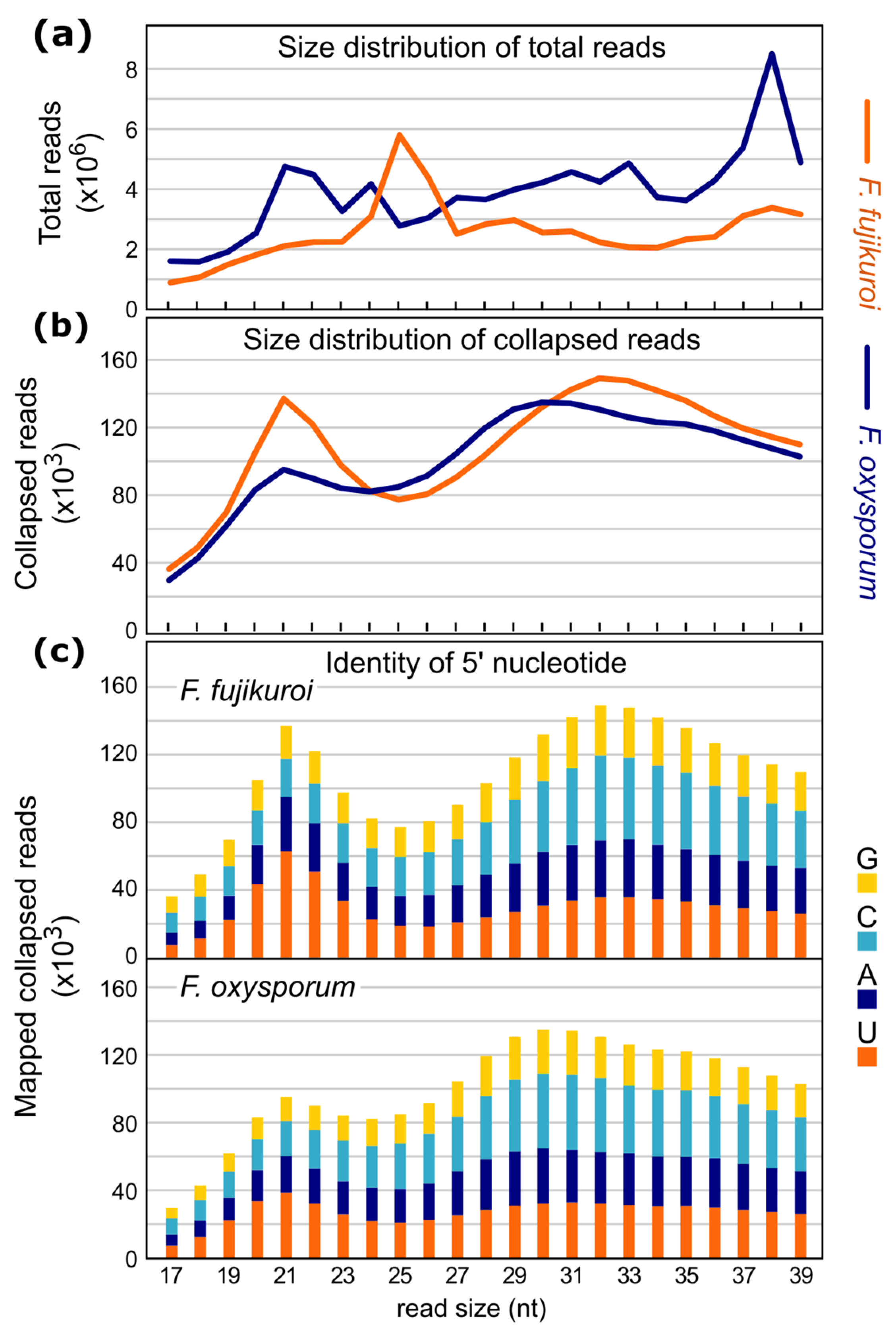
2.2. Sequencing and Characterization of Small RNAs in F. fujikuroi and F. oxysporum
2.3. Origin of sRNAs in F. fujikuroi and F. oxysporum
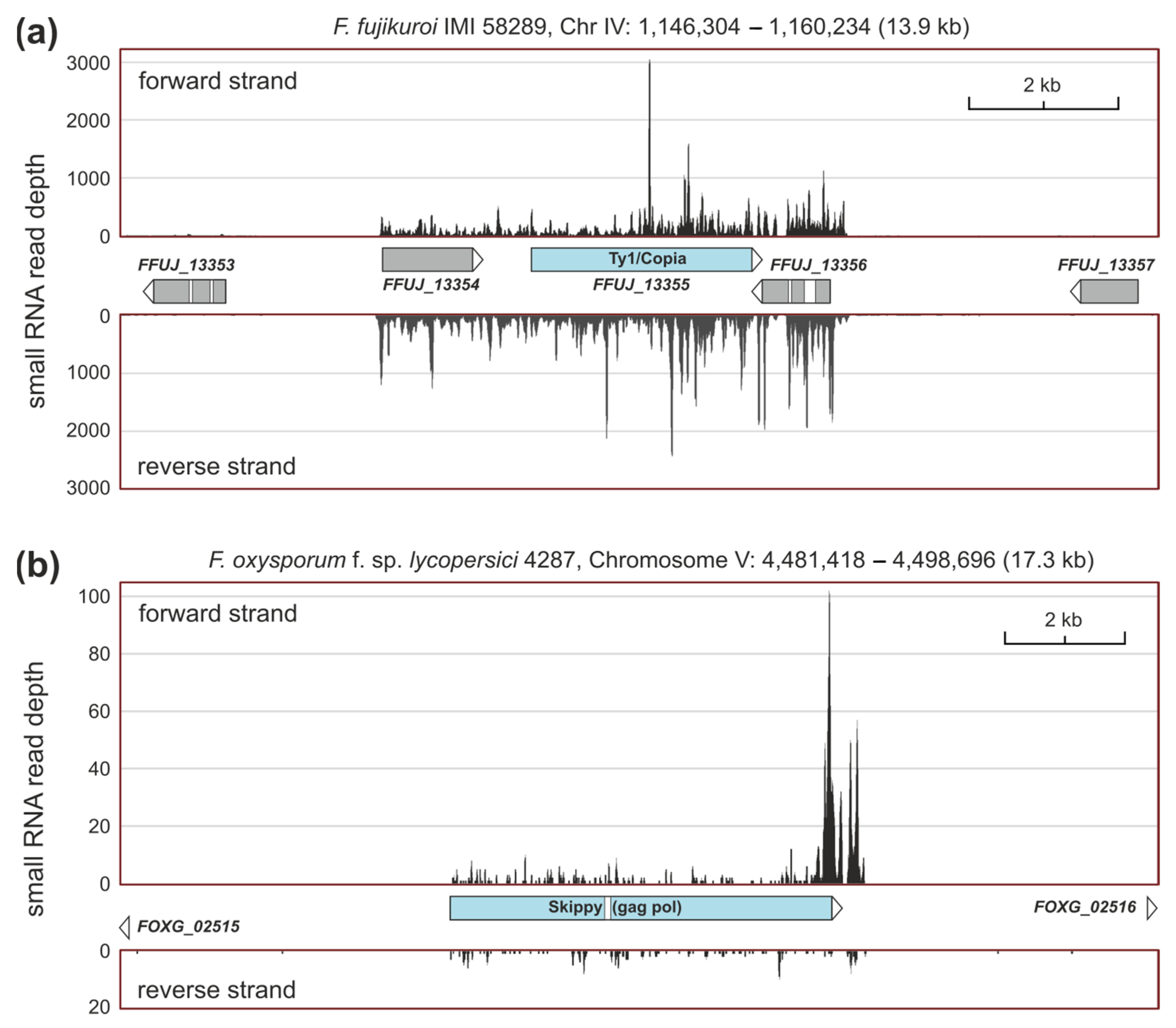
2.4. sRNAs Related to Transposable Elements
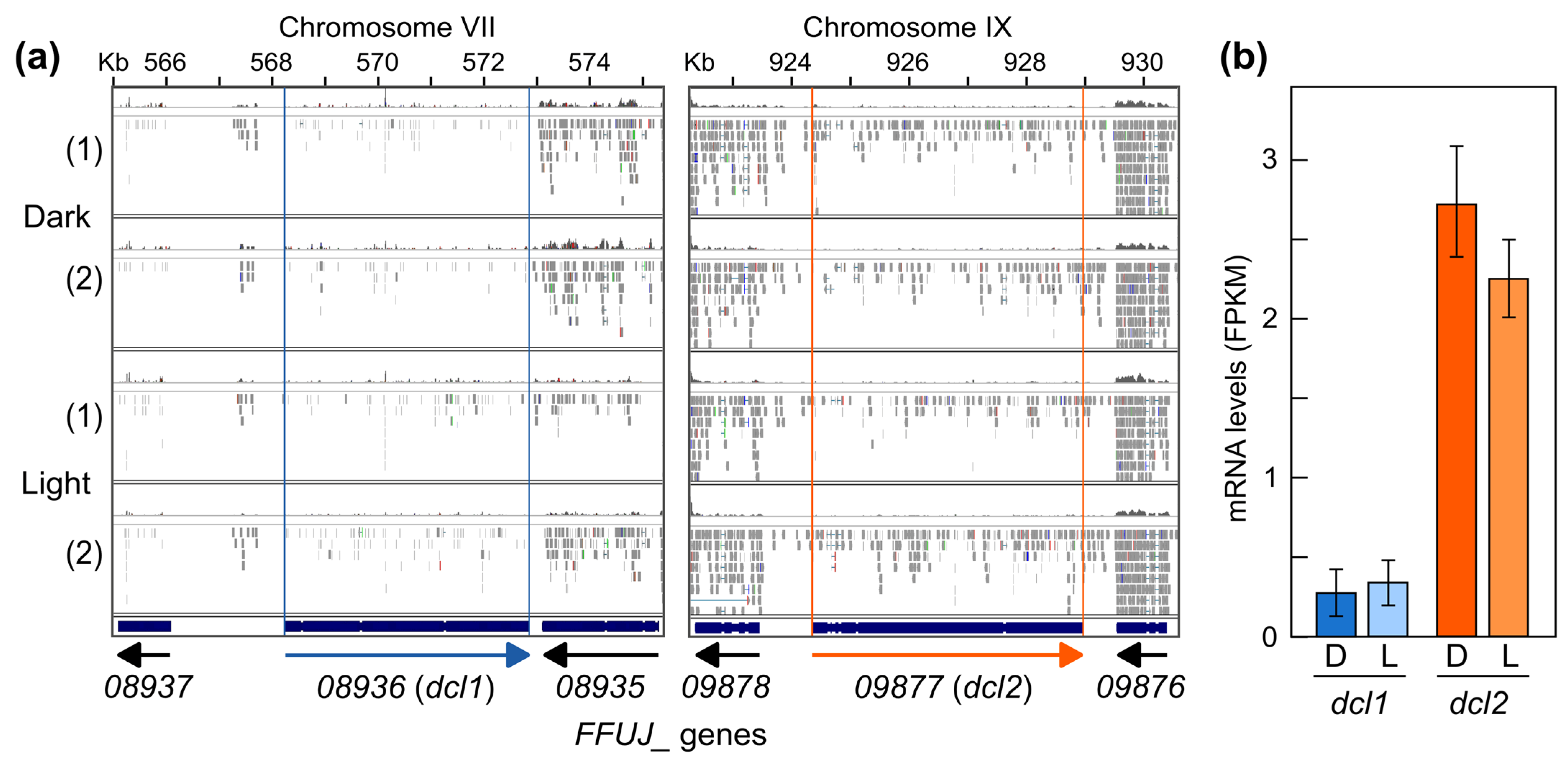
2.5. Differentially Expressed sRNA-Producing Loci under Dark and Light
2.6. Prediction of milRNAs by miRDeep2
2.7. Generation and Phenotypic Characterization of Δdcl2 Mutants
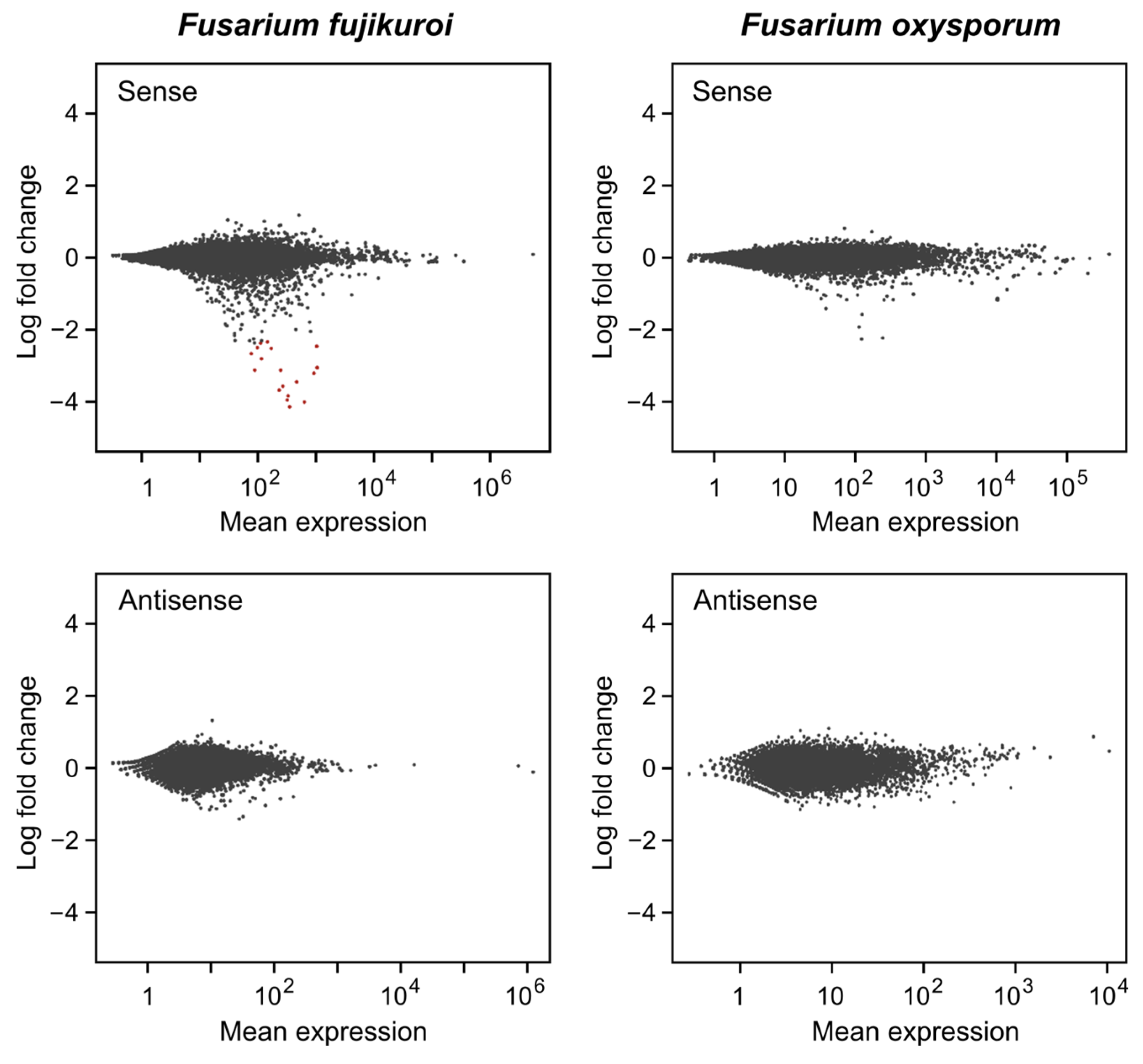
2.8. RNA-Seq Analysis of a Δdcl2 Mutant
3. Discussion
4. Materials and Methods
4.1. Strains and Culture Conditions
4.2. sRNA Sequencing and Analysis
4.3. Identification and Discarding of Redundant Sequences
4.4. Generation of ∆dcl2 Transformants
4.5. RNA-Seq Analyses of ∆dcl2 Mutant
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Torres-Martínez, S.; Ruiz-Vázquez, R.M. The RNAi Universe in Fungi: A Varied Landscape of Small RNAs and Biological Functions. Annu. Rev. Microbiol. 2017, 71, 371–391. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; Lemieux, C.; Jorgensen, R. Introduction of a Chimeric Chalcone Synthase Gene into Petunia Results in Reversible Co-Suppression of Homologous Genes in Trans. Plant Cell 1990, 2, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Cogoni, C.; Macino, G. Isolation of Quelling-Defective (qde) Mutants Impaired in Posttranscriptional Transgene-Induced Gene Silencing in Neurospora crassa. Proc. Natl. Acad. Sci. USA 1997, 94, 10233–10238. [Google Scholar] [CrossRef] [PubMed]
- Villalobos-Escobedo, J.M.; Herrera-Estrella, A.; Carreras-Villaseñor, N. The Interaction of Fungi with the Environment Orchestrated by RNAi. Mycologia 2016, 108, 556–571. [Google Scholar] [CrossRef] [PubMed]
- Nicolás, F.E.; Murcia, L.; Navarro, E.; Cánovas-Márquez, J.T.; Garre, V. Small RNAs in Fungi. In Genetics and Biotechnology; Benz, J.P., Schipper, K., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 105–122. ISBN 978-3-030-49923-5. [Google Scholar]
- Lax, C.; Tahiri, G.; Patiño-Medina, J.A.; Cánovas-Márquez, J.T.; Pérez-Ruiz, J.A.; Osorio-Concepción, M.; Navarro, E.; Calo, S. The Evolutionary Significance of RNAi in the Fungal Kingdom. Int. J. Mol. Sci. 2020, 21, 9348. [Google Scholar] [CrossRef] [PubMed]
- Billmyre, R.B.; Calo, S.; Feretzaki, M.; Wang, X.; Heitman, J. RNAi Function, Diversity, and Loss in the Fungal Kingdom. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2013, 21, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Catalanotto, C.; Azzalin, G.; Macino, G.; Cogoni, C. Involvement of Small RNAs and Role of the qde Genes in the Gene Silencing Pathway in Neurospora. Genes Dev. 2002, 16, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Pickford, A.S.; Cogoni, C. RNA-Mediated Gene Silencing. Cell. Mol. Life Sci. CMLS 2003, 60, 871–882. [Google Scholar] [CrossRef]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA Biogenesis and Its Crosstalk with Other Cellular Pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef]
- Shiu, P.K.T.; Metzenberg, R.L. Meiotic Silencing by Unpaired DNA: Properties, Regulation and Suppression. Genetics 2002, 161, 1483–1495. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hsueh, Y.-P.; Li, W.; Floyd, A.; Skalsky, R.; Heitman, J. Sex-Induced Silencing Defends the Genome of Cryptococcus neoformans via RNAi. Genes Dev. 2010, 24, 2566–2582. [Google Scholar] [CrossRef] [PubMed]
- Son, H.; Min, K.; Lee, J.; Raju, N.B.; Lee, Y.-W. Meiotic Silencing in the Homothallic Fungus Gibberella zeae. Fungal Biol. 2011, 115, 1290–1302. [Google Scholar] [CrossRef]
- Yu, J.; Lee, K.-M.; Cho, W.K.; Park, J.Y.; Kim, K.-H. Differential Contribution of RNA Interference Components in Response to Distinct Fusarium graminearum Virus Infections. J. Virol. 2018, 92, e01756-17. [Google Scholar] [CrossRef]
- Halic, M.; Moazed, D. Dicer-Independent Primal RNAs Trigger RNAi and Heterochromatin Formation. Cell 2010, 140, 504–516. [Google Scholar] [CrossRef]
- Lee, H.-C.; Li, L.; Gu, W.; Xue, Z.; Crosthwaite, S.K.; Pertsemlidis, A.; Lewis, Z.A.; Freitag, M.; Selker, E.U.; Mello, C.C.; et al. Diverse Pathways Generate microRNA-like RNAs and Dicer-Independent Small Interfering RNAs in Fungi. Mol. Cell 2010, 38, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Nicolás, F.E.; Moxon, S.; de Haro, J.P.; Calo, S.; Grigoriev, I.V.; Torres-Martínez, S.; Moulton, V.; Ruiz-Vázquez, R.M.; Dalmay, T. Endogenous Short RNAs Generated by Dicer 2 and RNA-Dependent RNA Polymerase 1 Regulate mRNAs in the Basal Fungus Mucor circinelloides. Nucleic Acids Res. 2010, 38, 5535–5541. [Google Scholar] [CrossRef]
- Yang, Q.; Li, L.; Xue, Z.; Ye, Q.; Zhang, L.; Li, S.; Liu, Y. Transcription of the Major Neurospora crassa microRNA-like Small RNAs Relies on RNA Polymerase III. PLoS Genet. 2013, 9, e1003227. [Google Scholar] [CrossRef]
- Trieu, T.A.; Navarro-Mendoza, M.I.; Pérez-Arques, C.; Sanchis, M.; Capilla, J.; Navarro-Rodriguez, P.; Lopez-Fernandez, L.; Torres-Martínez, S.; Garre, V.; Ruiz-Vázquez, R.M.; et al. RNAi-Based Functional Genomics Identifies New Virulence Determinants in Mucormycosis. PLoS Pathog. 2017, 13, e1006150. [Google Scholar] [CrossRef]
- Kang, K.; Zhong, J.; Jiang, L.; Liu, G.; Gou, C.Y.; Wu, Q.; Wang, Y.; Luo, J.; Gou, D. Identification of microRNA-Like RNAs in the Filamentous Fungus Trichoderma reesei by Solexa Sequencing. PLoS ONE 2013, 8, e76288. [Google Scholar] [CrossRef]
- Chen, R.; Jiang, N.; Jiang, Q.; Sun, X.; Wang, Y.; Zhang, H.; Hu, Z. Exploring microRNA-like Small RNAs in the Filamentous Fungus Fusarium oxysporum. PLoS ONE 2014, 9, e104956. [Google Scholar] [CrossRef] [PubMed]
- Son, H.; Park, A.R.; Lim, J.Y.; Shin, C.; Lee, Y.-W. Genome-Wide Exonic Small Interference RNA-Mediated Gene Silencing Regulates Sexual Reproduction in the Homothallic Fungus Fusarium graminearum. PLoS Genet. 2017, 13, e1006595. [Google Scholar] [CrossRef] [PubMed]
- Carreras-Villaseñor, N.; Esquivel-Naranjo, E.U.; Villalobos-Escobedo, J.M.; Abreu-Goodger, C.; Herrera-Estrella, A. The RNAi Machinery Regulates Growth and Development in the Filamentous Fungus Trichoderma atroviride. Mol. Microbiol. 2013, 89, 96–112. [Google Scholar] [CrossRef] [PubMed]
- Gaffar, F.Y.; Imani, J.; Karlovsky, P.; Koch, A.; Kogel, K.-H. Different Components of the RNA Interference Machinery Are Required for Conidiation, Ascosporogenesis, Virulence, Deoxynivalenol Production, and Fungal Inhibition by Exogenous Double-Stranded RNA in the Head Blight Pathogen Fusarium graminearum. Front. Microbiol. 2019, 10, 1662. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.-W.; Yang, P.; Zhang, J.-B.; Liu, G.; Yuan, Q.-S.; He, W.-J.; Nian, J.-N.; Yi, S.-Y.; Huang, T.; Liao, Y.-C. Expression of microRNA-like RNA-2 (Fgmil-2) and bioH1 from a Single Transcript in Fusarium graminearum Are Inversely Correlated to Regulate Biotin Synthesis during Vegetative Growth and Host Infection. Mol. Plant Pathol. 2019, 20, 1574–1581. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.-M.; Ayukawa, Y.; Yun, S.-H.; Komatsu, K.; Arie, T. A Putative RNA Silencing Component Protein FoQde-2 Is Involved in Virulence of the Tomato Wilt Fungus Fusarium oxysporum f. Sp. lycopersici. J. Gen. Plant Pathol. 2018, 84, 395–398. [Google Scholar] [CrossRef]
- Janevska, S.; Tudzynski, B. Secondary Metabolism in Fusarium Fujikuroi: Strategies to Unravel the Function of Biosynthetic Pathways. Appl. Microbiol. Biotechnol. 2018, 102, 615–630. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The Protein Families Database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef]
- Ruger-Herreros, M.; Parra-Rivero, O.; Pardo-Medina, J.; Romero-Campero, F.J.; Limón, M.C.; Avalos, J. Comparative Transcriptomic Analysis Unveils Interactions between the Regulatory CarS Protein and Light Response in Fusarium. BMC Genom. 2019, 20, 67. [Google Scholar] [CrossRef]
- Pardo-Medina, J.; Gutiérrez, G.; Limón, M.C.; Avalos, J. Impact of the White Collar Photoreceptor WcoA on the Fusarium fujikuroi Transcriptome. Front. Microbiol. 2021, 11, 619474. [Google Scholar] [CrossRef]
- Pardo-Medina, J.; Limón, M.C.; Avalos, J. Fusarium Photoreceptors. J. Fungi 2023, 9, 319. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Fu, Y.; Xie, J.; Li, B.; Jiang, D.; Li, G.; Cheng, J. Identification of microRNA-like RNAs in a Plant Pathogenic Fungus Sclerotinia sclerotiorum by High-Throughput Sequencing. Mol. Genet. Genom. MGG 2012, 287, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, T.A.; Kück, U. Dicer-Dependent Biogenesis of Small RNAs and Evidence for microRNA-like RNAs in the Penicillin Producing Fungus Penicillium chrysogenum. PLoS ONE 2015, 10, e0125989. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, Q.; Huang, M.; Liu, Y.; Liu, Z.; Liu, X.; Ma, Z. Characterization of RNA Silencing Components in the Plant Pathogenic Fungus Fusarium graminearum. Sci. Rep. 2015, 5, 12500. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Werner, B.T.; Koch, A.; Šečić, E.; Engelhardt, J.; Jelonek, L.; Steinbrenner, J.; Kogel, K.-H. Fusarium graminearum DICER-like-Dependent sRNAs Are Required for the Suppression of Host Immune Genes and Full Virulence. PLoS ONE 2021, 16, e0252365. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.-M.; Mao, H.-Y.; Li, S.-J.; Feng, T.; Zhang, Z.-Y.; Cheng, L.; Luo, S.-J.; Borkovich, K.A.; Ouyang, S.-Q. Fol-milR1, a Pathogenicity Factor of Fusarium oxysporum, Confers Tomato Wilt Disease Resistance by Impairing Host Immune Responses. New Phytol. 2021, 232, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xie, L.; Wang, M.; Lin, Y.; Zhong, J.; Zhang, Y.; Zeng, J.; Kong, G.; Xi, P.; Li, H.; et al. FoQDE2-Dependent milRNA Promotes Fusarium oxysporum f. Sp. Cubense Virulence by Silencing a Glycosyl Hydrolase Coding Gene Expression. PLoS Pathog. 2022, 18, e1010157. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-C.; Chang, S.-S.; Choudhary, S.; Aalto, A.P.; Maiti, M.; Bamford, D.H.; Liu, Y. qiRNA Is a New Type of Small Interfering RNA Induced by DNA Damage. Nature 2009, 459, 274–277. [Google Scholar] [CrossRef]
- Hammond, T.M.; Keller, N.P. RNA Silencing in Aspergillus nidulans Is Independent of RNA-Dependent RNA Polymerases. Genetics 2005, 169, 607–617. [Google Scholar] [CrossRef]
- Hu, Z.; Parekh, U.; Maruta, N.; Trusov, Y.; Botella, J.R. Down-Regulation of Fusarium oxysporum Endogenous Genes by Host-Delivered RNA Interference Enhances Disease Resistance. Front. Chem. 2015, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Wang, Z.; Wang, Y.; Zhu, H.; Huang, B. Dicer and Argonaute Genes Involved in RNA Interference in the Entomopathogenic Fungus Metarhizium robertsii. Appl. Environ. Microbiol. 2017, 83, e03230-16. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Weiberg, A.; Dellota, E.; Yamane, D.; Jin, H. Botrytis Small RNA Bc-siR37 Suppresses Plant Defense Genes by Cross-Kingdom RNAi. RNA Biol. 2017, 14, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; He, B.; Weiberg, A.; Buck, A.H.; Jin, H. Small RNAs and Extracellular Vesicles: New Mechanisms of Cross-Species Communication and Innovative Tools for Disease Control. PLoS Pathog. 2019, 15, e1008090. [Google Scholar] [CrossRef] [PubMed]
- Jian, J.; Liang, X. One Small RNA of Fusarium graminearum Targets and Silences CEBiP Gene in Common Wheat. Microorganisms 2019, 7, 425. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Liu, N.; Su, Q.; Liu, X.; Jia, H.; Liu, Y.; Sun, M.; Cao, Z.; Dong, J. MicroRNAs Involved in the Trans-Kingdom Gene Regulation in the Interaction of Maize Kernels and Fusarium verticillioides. Int. J. Biol. Macromol. 2023, 242, 125046. [Google Scholar] [CrossRef]
- Marente, J.; Ortega, P.; Pardo-Medina, J.; Avalos, J.; Limón, M.C. Modulation of Activity of a Carotenoid Pathway through the Use of the TET-on Regulatory System: Application in the Fungus Fusarium fujikuroi. In Plant and Food Carotenoids: Methods and Protocols; Rodríguez-Concepción, M., Welsch, R., Eds.; Methods in Molecular Biology; Springer US: New York, NY, USA, 2020; pp. 343–360. ISBN 978-1-4939-9952-1. [Google Scholar]
- Colot, H.V.; Park, G.; Turner, G.E.; Ringelberg, C.; Crew, C.M.; Litvinkova, L.; Weiss, R.L.; Borkovich, K.A.; Dunlap, J.C. A High-Throughput Gene Knockout Procedure for Neurospora Reveals Functions for Multiple Transcription Factors. Proc. Natl. Acad. Sci. USA 2006, 103, 10352–10357. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Metzker, M.L. Sequencing Technologies—The next Generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transposable Element | F. fujikuroi | F. oxysporum | ||||
|---|---|---|---|---|---|---|
| Gene ID | Identity (%) | E-Value | Gene ID | Identity (%) | E-Value | |
| HobS | FFUJ_04535 | 100 | 0 | FOXG_15494 | 39.09 | 9 × 10−142 |
| FFUJ_02576 | 100 | 0 | FOXG_12541 | 25.94 | 6 × 10−65 | |
| FFUJ_04521 | 96.24 | 0 | FOXG_15105 | 26.89 | 4 × 10−57 | |
| FFUJ_05678 | 96.24 | 0 | FOXG_06872 | 43.48 | 8 × 10−49 | |
| FFUJ_05881 | 100 | 0 | FOXG_16138 | 43.48 | 1 ×10−48 | |
| Impala | FFUJ_07491 | 87.35 | 0 | chr14: 1236756-1237727 1 | 89.56 | 0 |
| chrV: 2117840-2118331 1 | 29.09 | 7 × 10−13 | chr14: 1117562-1116699 1 | 87.84 | 0 | |
| - | - | - | chr14: 121747-121423 1 | 87.80 | 1 × 10−103 | |
| - | - | - | chr1: 6134797-6136077 1 | 82.54 | 0.0 | |
| - | - | - | chr15: 1546451-1545171 1 | 82.54 | 0.0 | |
| Ty1/copia | FFUJ_08062 | 31.08 | 7 × 10−66 | chr3: 342220-343926 1 | 98.42 | 0.0 |
| FFUJ_13355 2 | 31.08 | 7 × 10−66 | chr3: 415150-416856 1 | 98.42 | 0.0 | |
| FFUJ_00408 | 31.08 | 1 × 10−65 | chr3: 982302-984008 1 | 98.42 | 0.0 | |
| FFUJ_14407 | 30.59 | 3 × 10−65 | chr3: 3405691-3403985 1 | 98.42 | 0.0 | |
| - | - | - | FOXG_14142 | 43.42 | 6 × 10−62 | |
| Skippy | chrIV: 3140654-3142846 1 | 53.08 | 0 | chr5: 4489311-4493195 1,2 | 97.99 | 0 |
| chrVI: 3662701-3660476 1 | 52.96 | 0 | DS231747: 67362-63478 1 | 97.99 | 0 | |
| chrIII: 4890426-4892675 1 | 49.47 | 0 | chr14: 1418859-1422395 1 | 97.71 | 0 | |
| chrX: 887601-889802 1 | 52.04 | 0 | FOXG_17761 | 36.73 | 2 × 10−137 | |
| FFUJ_00160 | 25.77 | 6 × 10−68 | FOXG_17760 | 40.37 | 9 × 10−133 | |
| MAGGY | chrX: 4392-6848 | 37.61 | 4 × 10−134 | FOXG_17760 | 68.33 | 0.0 |
| FFUJ_00160 | 26.67 | 3 × 10−78 | FOXG_17761 | 66.33 | 0.0 | |
| ID | Score | Read Count | Mature milRNA 5′-3′ Sequence | Physical Location 1 | Genetic Location | Gene Function |
|---|---|---|---|---|---|---|
| #1 | 1.3 × 101 | 34 | UGGGACGAGGACAAGGCUGAA | Chr VII 1735744 | FFUJ_08557 ORF Antisense orientation | Uncharacterized protein |
| #2 | 3.8 | 14 | UCACCGUUAGACCAUUACAG | Chr VII 2455572 | FFUJ_08366 promoter 451 pb upstream SC 2 Sense orientation | Uncharacterized protein |
| #3 | 2.7 | 5 | GUCCUGGAGGCACUUGA | Chr VII 1512406 | FFUJ_08631 ORF Sense orientation | Related to hydrolase |
| #4 | 2.3 | 2 | GGCGCGAGAAGAGAUCGAGGAUC | Chr VII 868943 | FFUJ_02416 ORF Sense orientation | Related to nitrate reductase |
| #5 | 2.1 | 3 | AGCCCAAUCCUUGUGCCACU | Chr II 1257164 | FFUJ_04898 ORF Antisense orientation | SRP40-Suppressor of mutant AC40 of RNA polymerase I and III |
| #6 | 1.9 | 3 | AGAGGAAUCGACGAUGUGACU | Chr I 3119036 | FFUJ_01140 promoter 652 bp upstream from SC 2 Antisense orientation | Uncharacterized protein |
| #7 | 1.5 | 68 | UGCAGAGCUUAUUCUAUCCC | Chr III 2168980 | FFUJ_02787 promoter 2117 bp upstream from SC 2 Sense orientation | Uncharacterized protein |
| #8 | 1.5 | 330 | UUAGGGUUAGGGUUAGGGUUA | Chr X 2543622 | Subtelomeric region | No gene |
| #9 | 1.4 | 2 | UCCGAGCGCCAUGGUUGAUGAGA | Chr VII 2396236 | FFUJ_08382 ORF Antisense orientation | Uncharacterized protein |
| #10 | 0.6 | 25 | UUCCACUACCUAUGGUCGUAU | Chr II 2372098 | EFFFUG00000000016 ORF Antisense orientation | |
| #11 | 0 | 5 | UCGACAACCUCGUCUGCCUC | Chr IV 1640658 | FFUJ_13515 ORF Antisense orientation | Probable acetolactate synthase small subunit precursor |
| ID | E-Value | Identity Sequence 1 | Target Gene | Putative Function |
|---|---|---|---|---|
| #1 | 0.36 | CGAGGACAAGGCTGA (AS) | FFUJ_01736 | Related to plasma membrane phosphatase required for sodium stress response |
| #1 | 0.36 | GAGGACAAGGCTGAA (S) | FFUJ_09223 | Related to alpha-L-rhamnosidase A |
| #4 | 0.42 | CGAGAAGAGATCGAG (S) | FFUJ_02865 | Probable D-xylose reductase |
| #5 | 0.079 | CCAATCCTTGTGCCAC (S) | FFUJ_02776 | Related to cysteine-rich protein NFX-1 |
| #7 | 0.079 | TGCAGAGCTTAT[C]CTATCCC 2 (AS) | FFUJ_13355 FFUJ_14407 FFUJ_00408 | Retrotransposon HobS hobase (FFUJ_13355 and FFUJ_14407) TY2B-TY2B protein (FFUJ_00408) |
| #8 | - | UUAGGGUUAGGGUUAGGGUUA | None | Telomeric DNA |
| #9 | 0.42 | CGCCATGGTTGATGA (AS) | FFUJ_01768 | Uncharacterized protein |
| #10 | 0.001 | TTCCACTACCTATGGTCGT (AS) | rDNA loci | 8 S ribosomal RNA (116 bases) |
| #11 | 0.31 | AACCTCGTCTGCCTC (AS) | FFUJ_13704 | Related to COG5-conserved oligomeric Golgi complex |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pardo-Medina, J.; Dahlmann, T.A.; Nowrousian, M.; Limón, M.C.; Avalos, J. The RNAi Machinery in the Fungus Fusarium fujikuroi Is Not Very Active in Synthetic Medium and Is Related to Transposable Elements. Non-Coding RNA 2024, 10, 31. https://doi.org/10.3390/ncrna10030031
Pardo-Medina J, Dahlmann TA, Nowrousian M, Limón MC, Avalos J. The RNAi Machinery in the Fungus Fusarium fujikuroi Is Not Very Active in Synthetic Medium and Is Related to Transposable Elements. Non-Coding RNA. 2024; 10(3):31. https://doi.org/10.3390/ncrna10030031
Chicago/Turabian StylePardo-Medina, Javier, Tim A. Dahlmann, Minou Nowrousian, M. Carmen Limón, and Javier Avalos. 2024. "The RNAi Machinery in the Fungus Fusarium fujikuroi Is Not Very Active in Synthetic Medium and Is Related to Transposable Elements" Non-Coding RNA 10, no. 3: 31. https://doi.org/10.3390/ncrna10030031
APA StylePardo-Medina, J., Dahlmann, T. A., Nowrousian, M., Limón, M. C., & Avalos, J. (2024). The RNAi Machinery in the Fungus Fusarium fujikuroi Is Not Very Active in Synthetic Medium and Is Related to Transposable Elements. Non-Coding RNA, 10(3), 31. https://doi.org/10.3390/ncrna10030031