


GastroPlus- and HSPiP-Oriented Predictive Parameters as the Basis of Valproic Acid-Loaded Mucoadhesive Cationic Nanoemulsion Gel for Improved Nose-to-Brain Delivery to Control Convulsion in Humans

 ,
,  ,
, 
Abstract
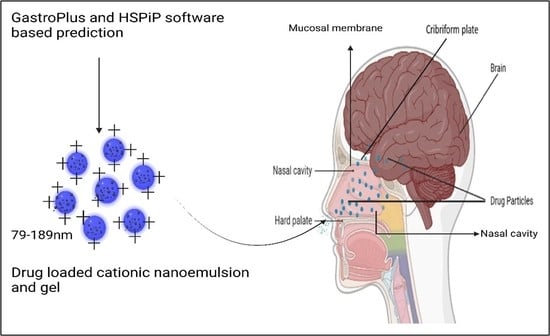
1. Introduction
2. Results and Discussion
2.1. Prediction and Simulation Study Using GastroPlus
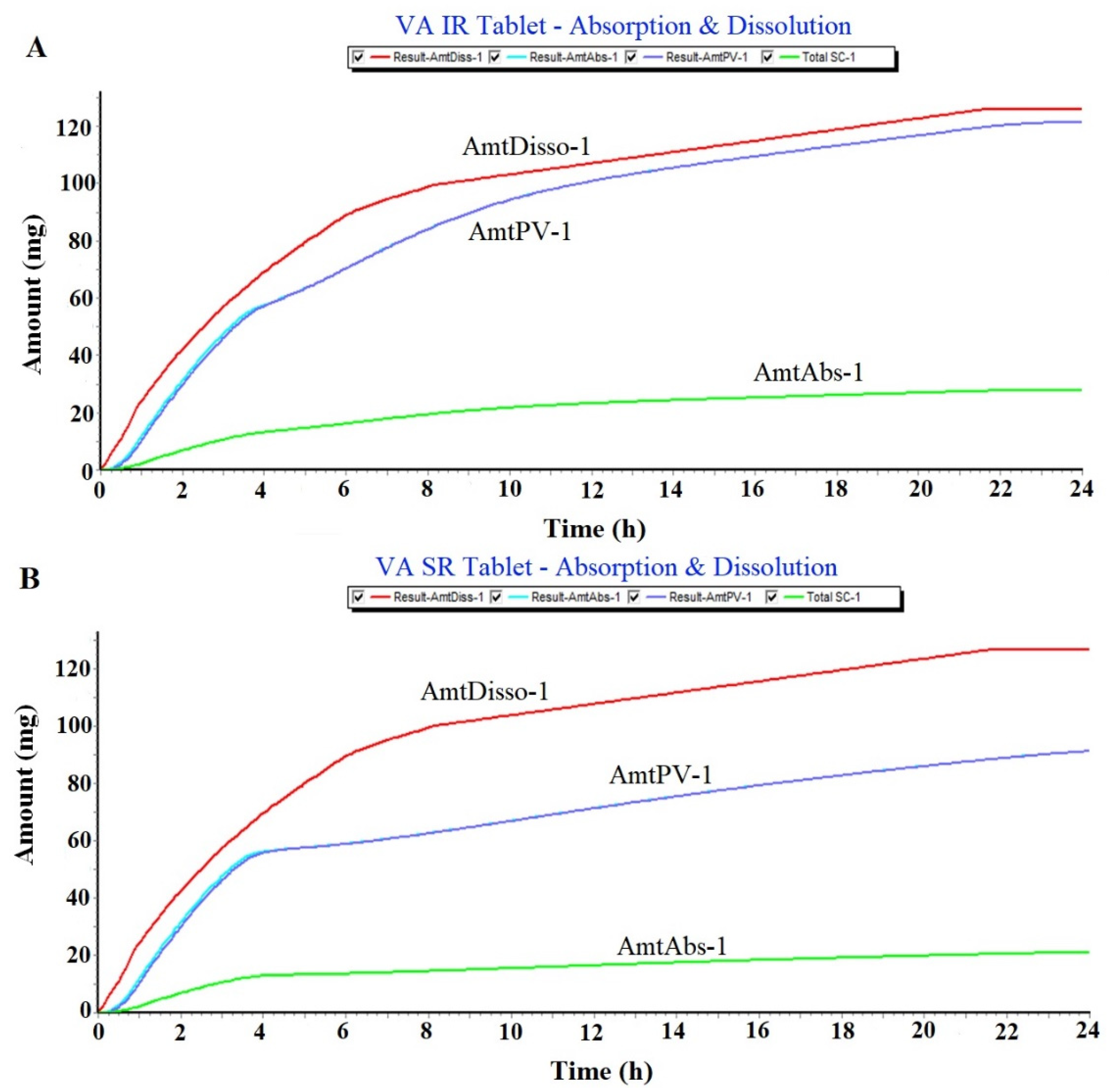
2.1.1. Prediction of Plasma Drug Concentration Time Profile
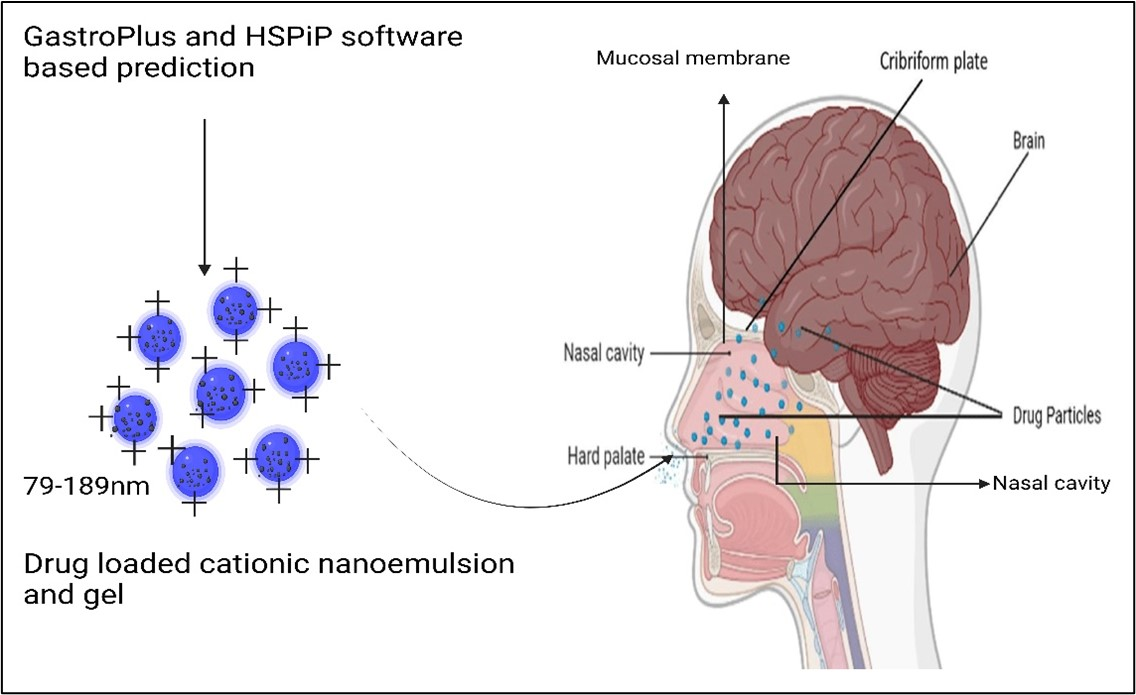
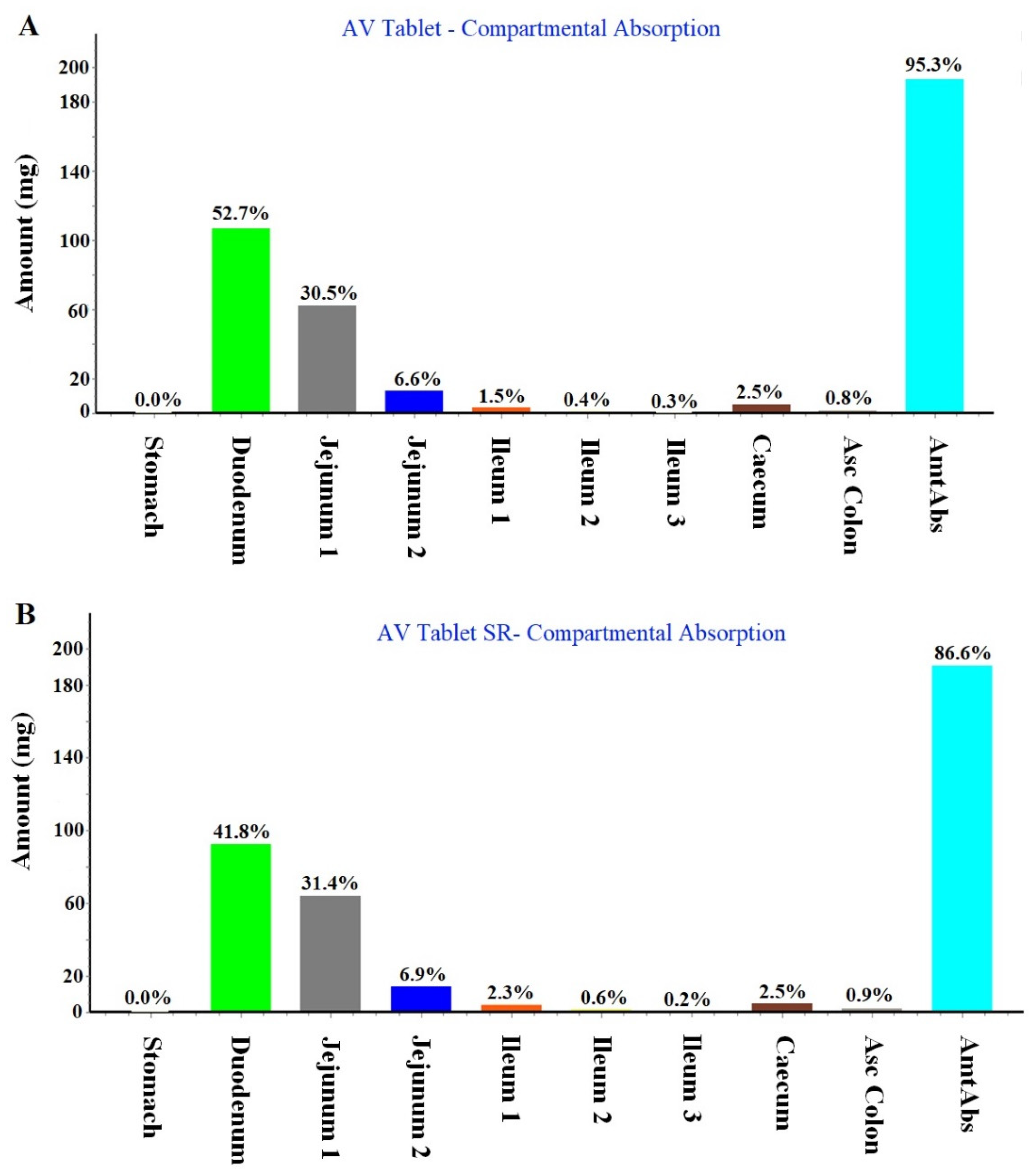
2.1.2. Regional Compartmental Absorption of Both Tablets
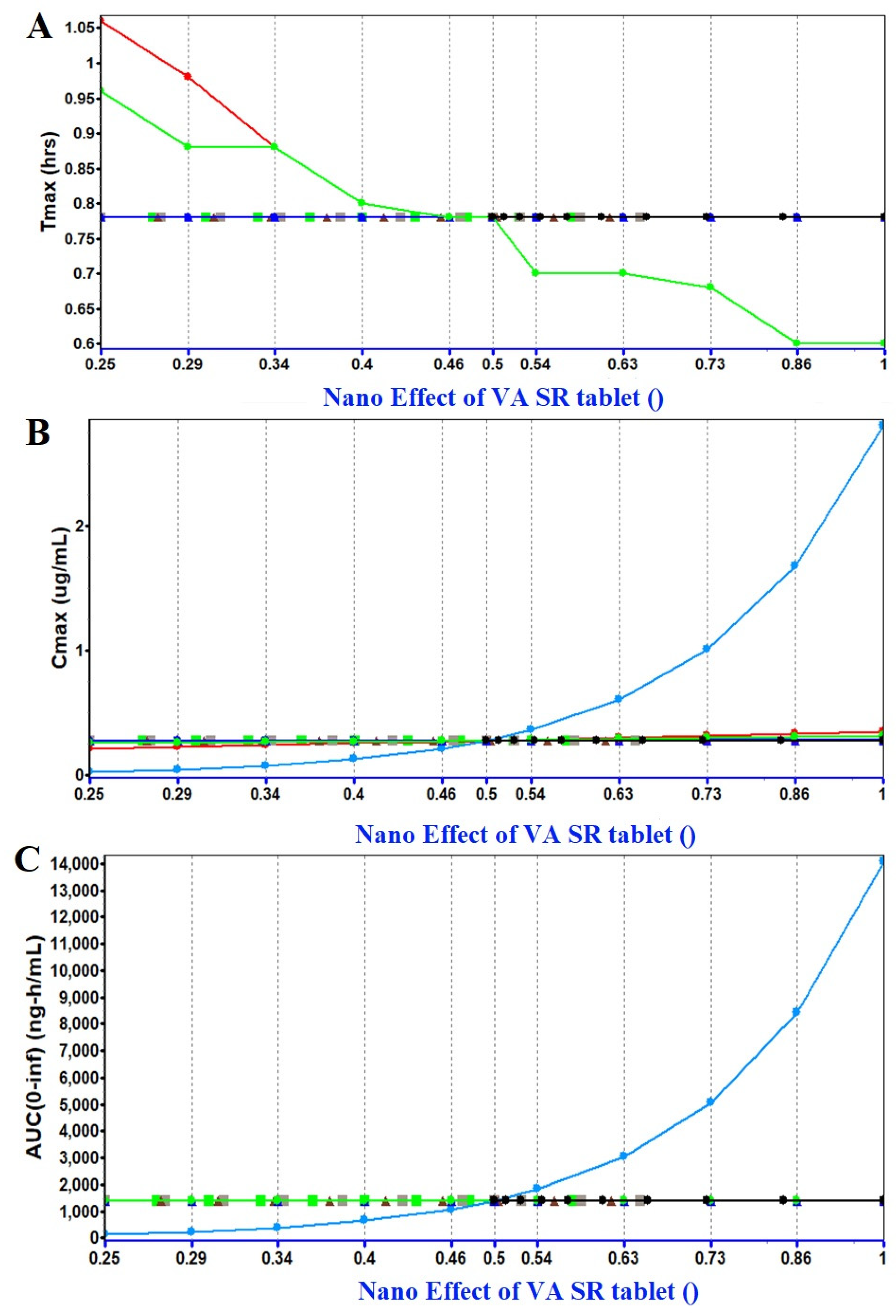
2.1.3. PSA (Parameter Sensitivity Analysis) Assessment
2.1.4. Hansen Solubility Parameters for VA and Excipients
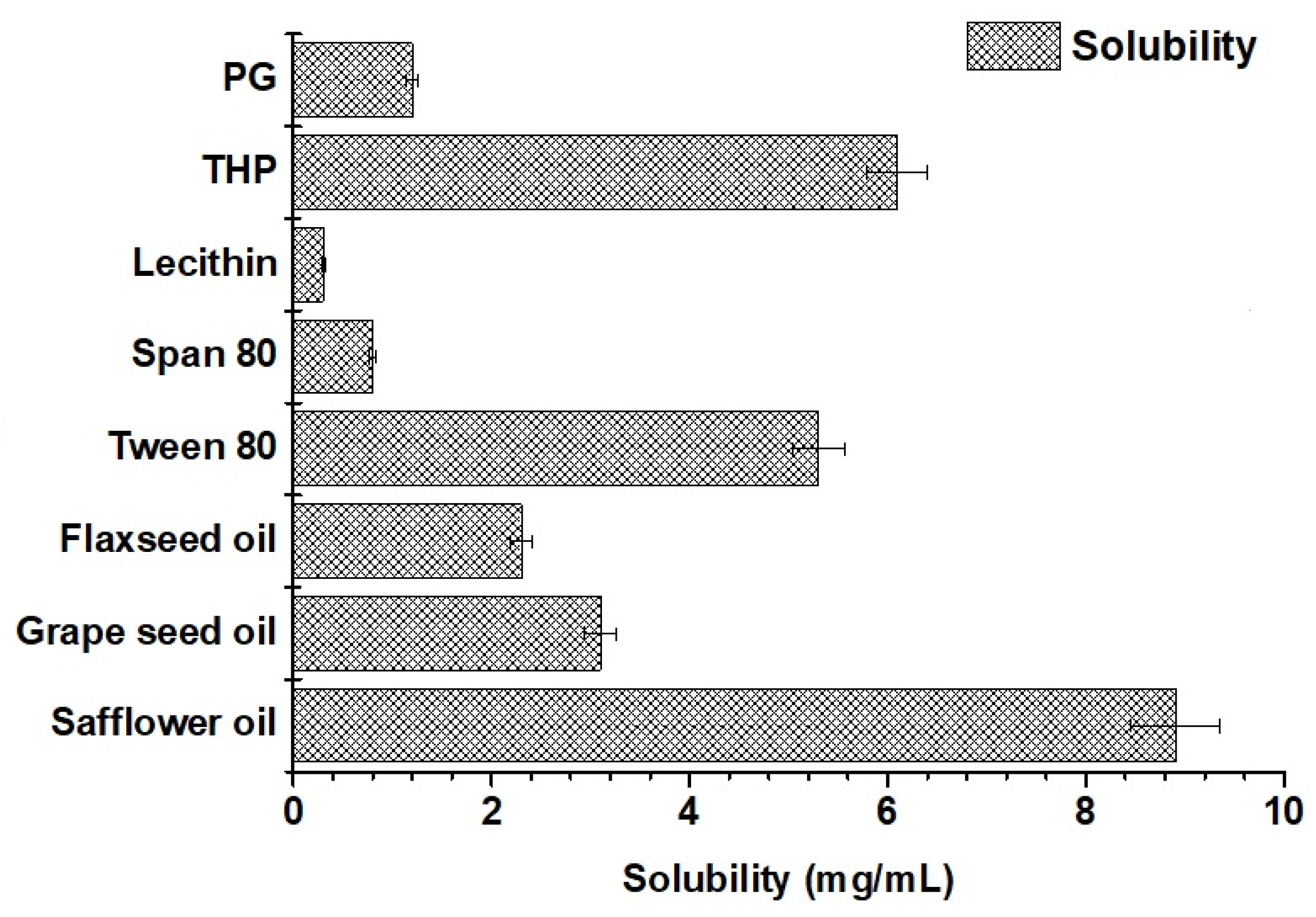
2.1.5. Solubility of Valproate (VA) in Various Excipients
2.1.6. VA Loaded Cationic Nanoemulsions Prepared
2.1.7. Freeze–Thaw Cycle and Ultracentrifugation of Nanoemulsions
2.2. Evaluation of Cationic and Anionic Nanoemulsions Gels
2.2.1. Morphological Evaluation of the Optimized Cationic Nanoemulsion and Respective Gel
2.2.2. Drug Content Estimation
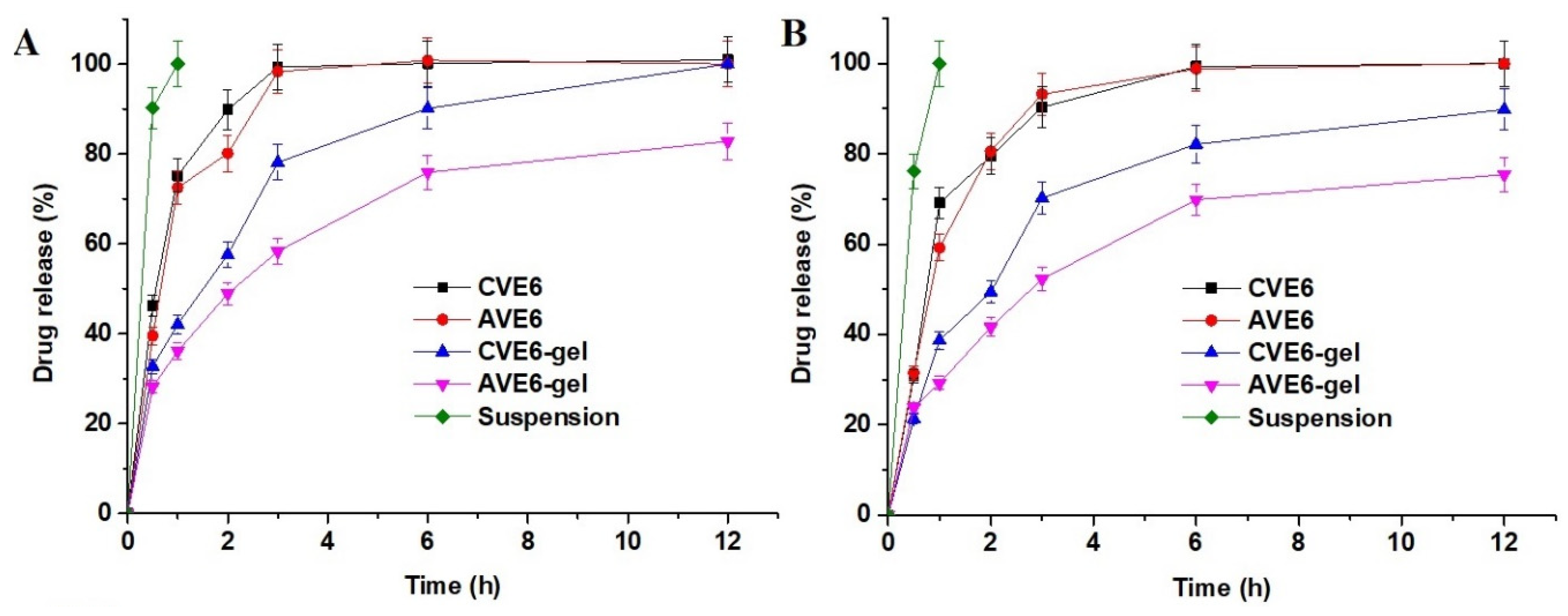
2.2.3. In Vitro Drug Release Profile
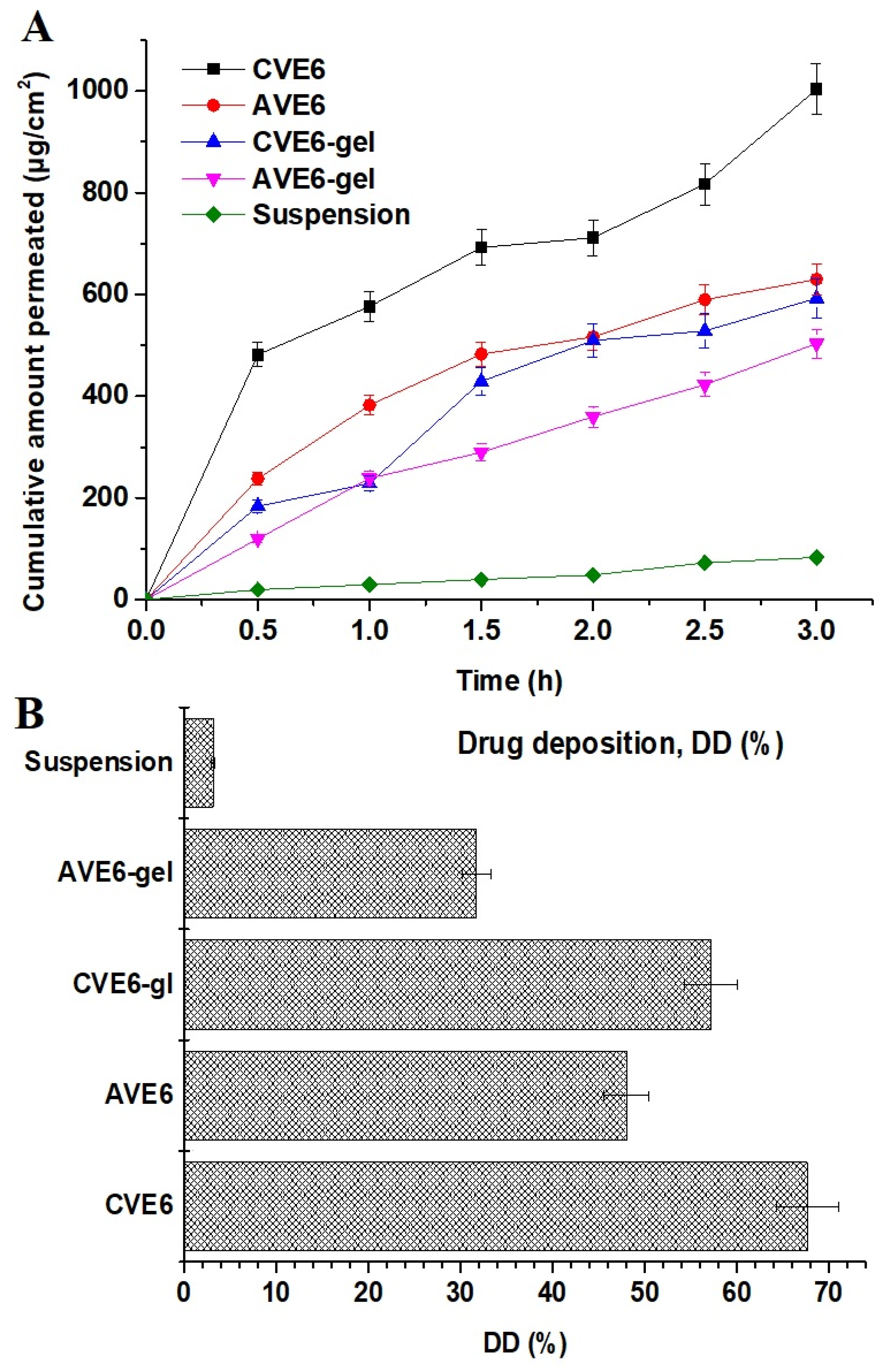
2.2.4. Ex Vivo Drug Permeation and Drug Deposition Using Goat Nasal Mucosal Tissue
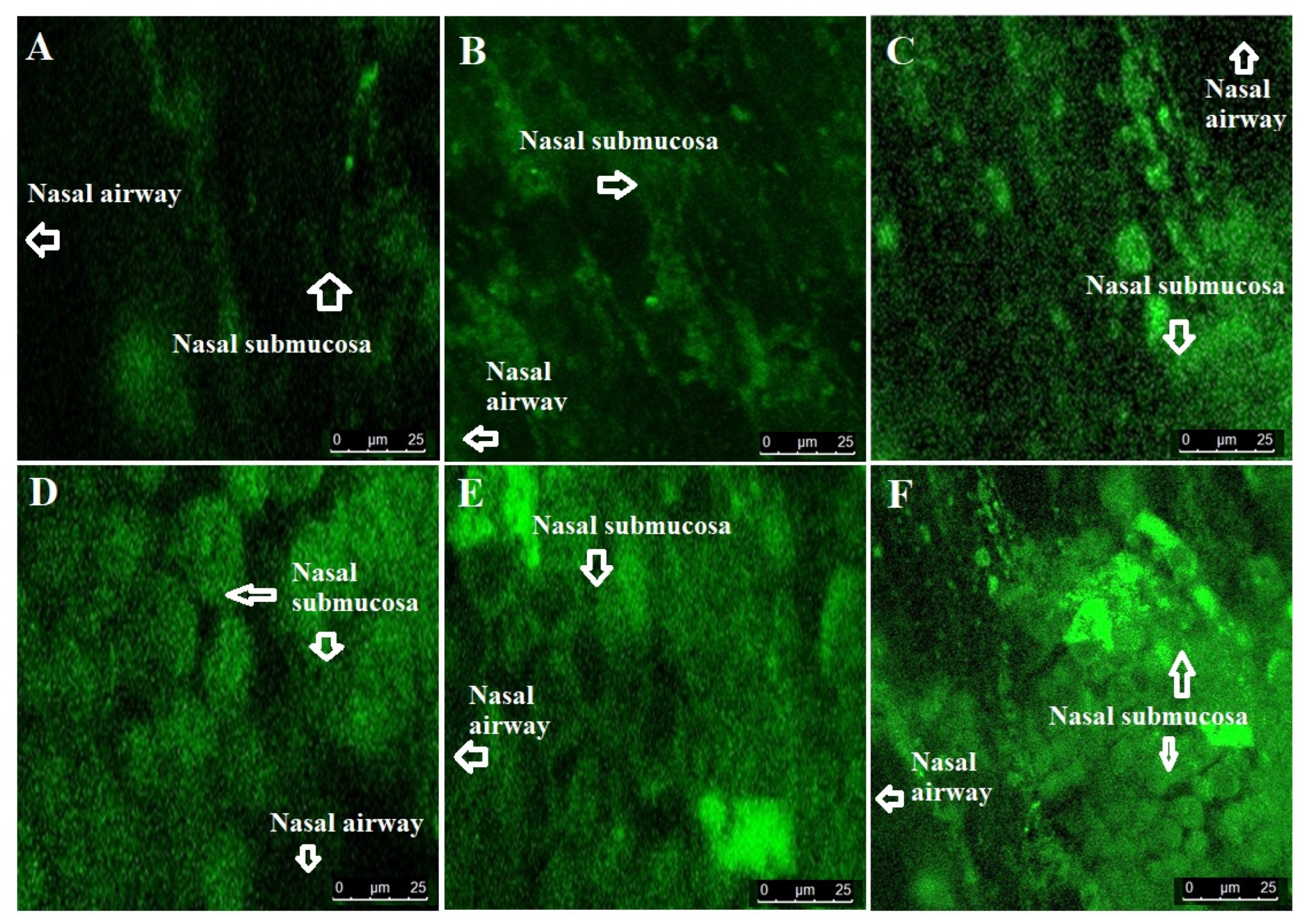
2.2.5. Confocal Laser Scanning Microscopy (CLSM)
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Prediction and Simulation Study Using GastroPlus for Oral Tablet
Hansen Solubility Parameters for VA and Excipients
Solubility of Valproate Sodium in Various Excipients
Pseudo Ternary Phase Diagram, Cationic Nanoemulsions, and Nanoemulsion Gel
Thermodynamic Stability of Cationic Nanoemulsion: Freeze–Thaw Cycle and Ultracentrifugation
4.2.2. Evaluation of Cationic Nanoemulsions and Gels
Morphological Evaluation of the Optimized Cationic Nanoemulsion and Respective Gel
Drug Content Estimation
4.2.3. In Vitro Drug Release Profile
4.2.4. Ex Vivo Drug Permeation and Drug Deposition Using a Goat Nasal Mucosa
4.2.5. Confocal Laser Scanning Microscopy (CLSM)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization Epilepsy. Available online: https://www.who.int/news-room/fact-sheets/detail/epilepsy (accessed on 20 December 2021).
- Available online: https://www.ninds.nih.gov/current-research/focus-disorders/focus-epilepsy-research/curing-epilepsies-promise-research (accessed on 20 December 2021).
- Al Rajeh, S.; Awada, A.; Bademosi, O.; Gunniyi, A. The prevalence of epilepsy and other seizure disorders in an Arab population: A community-based study. Seizure 2001, 10, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Ishizue, N.; Niwano, S.; Saito, M.; Fukaya, H.; Nakamura, H.; Igarashi, T.; Fujiishi, T.; Yoshizawa, T.; Oikawa, J.; Satoh, A.; et al. Polytherapy with Sodium Channel-Blocking Antiepileptic Drugs Is Associated with Arrhythmogenic ST-T Abnormality in Patients with Epilepsy. Seizure 2016, 40, 81–87. [Google Scholar] [CrossRef][Green Version]
- Hammond, E.J.; Perchalski, R.J.; Villarreal, H.J.; Wilder, B.J. In vivo uptake of valproic acid into brain. Brain Res. 1982, 240, 195–198. [Google Scholar] [CrossRef] [PubMed]
- US FDA (US Food and Drug Administration). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/018081s065_018082s048lbl.pdf (accessed on 1 July 2023).
- Eskandari, S.; Varshosaz, J.; Minaiyan, M.; Tabbakhian, M. Brain delivery of valproic acid via intranasal administration of nanostructured lipid carriers: In vivo pharmacodynamic studies using rat electroshock model. Int. J. Nanomed. 2011, 6, 363–371. [Google Scholar]
- Wang, H.; Huang, Q.; Chang, H.; Xiao, J.; Cheng, Y. Stimuli-responsive dendrimers in drug delivery. Biomater. Sci. 2016, 4, 375–390. [Google Scholar] [CrossRef]
- Lopez, T. Biocompatible Titania Microtubes Formed by Nanoparticles and its Application in the Drug Delivery of Valproic Acid. Opt. Mater. 2006, 29, 70–74. [Google Scholar] [CrossRef]
- Kirby, T.B.P.; Stanslas, J.; Basri, H.B. Characterization, in-vitro and in-vivo evaluation of valproic acid-loaded nanoemulsion for improved brain bioavailability. J. Pharm. Pharmacol. 2017, 69, 1447–1457. [Google Scholar]
- Mori, N.; Ohta, S. Comparison of anticonvulsant effects of valproic acid entrapped in positively and negatively charged liposomes in amygdaloid-kindled rats. Brain Res. 1992, 593, 329–331. [Google Scholar] [CrossRef]
- Tan, S.F.; Masoumi, H.R.F.; Karjiban, R.A.; Stanslas, J.; Kirby, B.P.; Basri, M.; Basri, H.B. Ultrasonic emulsification of parenteral valproic acid-loaded nanoemulsion with response surface methodology and evaluation of its stability. Ultrason. Sonochemistry 2016, 29, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Gandham, S.K.; Panicucci, R.; Amiji, M.M. Intranasal brain delivery of cationic nanoemulsion-encapsulated TNFα siRNA in prevention of experimental neuroinflammation. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 987–1002. [Google Scholar] [CrossRef]
- Azambuja, J.H.; Schuh, R.S.; Michels, L.R.; Gelsleichter, N.E.; Beckenkamp, L.R.; Iser, I.C.; Lenz, G.S.; de Oliveira, F.H.; Venturin, G.; Greggio, S.; et al. Nasal Administration of Cationic Nanoemulsions as CD73-siRNA Delivery System for Glioblastoma Treatment: A New Therapeutical Approach. Mol. Neurobiol. 2020, 57, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Samad, A.; Singh, S.K.; Ahsan, M.N.; Haque, M.W.; Faruk, A.; Ahmed, F.J. Nanoemulsion gel-based topical delivery of an antifungal drug: In vitro activity and in vivo evaluation. Drug Deliv. 2016, 23, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Teixeira-da-Silva, P.; Pérez-Blanco, J.S.; Santos-Buelga, D.; Otero, M.J.; García, M.J. Population pharmacokinetics of valproic acid in pediatric and adult caucasian patients. Pharmaceutics 2022, 14, 811. [Google Scholar] [CrossRef] [PubMed]
- Zaccara, G.; Messori, A.; Moroni, F. Clinical Pharmacokinetics of Valproic Acid—1988. Clin. Pharmacokinet. 1988, 15, 367–389. [Google Scholar] [CrossRef]
- Johannessen, C.U.; Johannessen, S.I. Valproate: Past, present, and future. CNS Drug Rev. 2003, 9, 199–216. [Google Scholar] [CrossRef]
- Winter, M.E. Basic Clinical Pharmacokinetics, 5th ed.; Lippincott Williams & Wilkins Health: Philadelphia, PA, USA, 2010. [Google Scholar]
- Loscher, W. Serum protein binding and pharmacokinetics of valproate in man, dog, rat and mouse. J. Pharmacol. Exp. Ther. 1978, 204, 255–261. [Google Scholar]
- Dickinson, R.G.; Harland, R.C.; Ilias, A.M.; Rodgers, R.M.; Kaufman, S.N.; Lynn, R.K.; Gerber, N. Disposition of valproic acid in the rat: Dose dependent metabolism, distribution, enterohepatic recirculation and choleretic effect. J. Pharmacol. Exp. Ther. 1979, 211, 583–595. [Google Scholar] [PubMed]
- Hansen, S.; Lehr, C.-M.; Schaefer, U.F. Improved input parameters for diffusion models of skin absorption. Adv. Drug Deliv. Rev. 2013, 65, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Methaneethorn, J. A systematic review of population pharmacokinetics of valproic acid. Br. J. Clin. Pharmacol. 2018, 84, 816–834. [Google Scholar] [CrossRef] [PubMed]
- Gugler, R.; von Unruh, G.E. Clinical Pharmacokinetics of Valproic Acid1. Clin. Pharmacokinet. 1980, 5, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Alshehri, S.; Ramzan, M.; Afzal, O.; Altamimi, A.S.A.; Alossaimi, M.A. Biocompatible solvent selection based on thermodynamic and computational solubility models, in-silico GastroPlus prediction, and cellular studies of ketoconazole for subcutaneous delivery. J. Drug Deliv. Sci. Technol. 2021, 65, 102699. [Google Scholar] [CrossRef]
- Alsarra, I.A.; Al-Omar, M.; Belal, F. Valproic Acid and Sodium Valproate: Comprehensive Profile. Profiles of Drug Substances. Excip. Relat. Methodol. 2005, 32, 209–240. [Google Scholar] [CrossRef]
- Vay, K.; Scheler, S.; Frie, W. Application of Hansen solubility parameters for understanding and prediction of drug distribution in microspheres. Int. J. Pharm. 2011, 416, 202–209. [Google Scholar] [CrossRef] [PubMed]
- De La Peña-Gil, A.; Toro-Vazquez, J.F.; Rogers, M.A. Simplifying Hansen Solubility Parameters for Complex Edible Fats and Oils. Food Biophys. 2016, 11, 283–291. [Google Scholar] [CrossRef]
- Hamilton, J.A.; Brunaldi, K. A model for fatty acid transport into the brain. J. Mol. Neurosci. 2007, 33, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Altamimi, M.A.; Afzal, O.; Altamimi, A.S.A.; Ali, A.; Ali, A.; Martinez, F.; Siddique, M.U.M.; Acree, W.E., Jr.; Jouyban, A. Preferential solvation study of the synthesized aldose reductase inhibitor (SE415) in the {PEG400 (1) + Water (2)} cosolvent mixture and GastroPlus-based prediction. ACS Omega 2022, 7, 1197–1210. [Google Scholar] [CrossRef]
- Han, X.; Cheng, L.; Zhang, R.; Bi, J. Extraction of safflower seed oil by supercritical CO2. J. Food Eng. 2009, 92, 370–376. [Google Scholar] [CrossRef]
- Afzal, O.; Alshammari, H.A.; Altamimi, M.A.; Hussain, A.; Almohaywi, B.; Altamimia, A.S. Hansen solubility parameters and green nanocarrier based removal of trimethoprim from contaminated aqueous solution. J. Mol. Liq. 2022, 361, 119657. [Google Scholar] [CrossRef]
- Abdullah, G.Z.; Abdulkarim, M.F.; Mallikarjun, C.; Mahdi, E.S.; Basri, M.; Sattar, M.A.; Noor, A.M. Carbopol 934, 940 and Ultrez 10 as viscosity modifiers of palm olein esters based nano-scaled emulsion containing ibuprofen. Pak. J. Pharm. Sci. 2013, 26, 75–83. [Google Scholar] [PubMed]
- Jones, M.N.; Song, Y.-H.; Kaszuba, M.; Reboiras, M.D. The Interaction of Phospholipid Liposomes with Bacteria and Their Use in the Delivery of Bactericides. J. Drug Target. 1997, 5, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Altamimi, M.A.; Hussain, A.; Alshehri, S.; Imam, S.S.; Alnemer, U.A. Development and evaluations of transdermally delivered luteolin loaded cationic nanoemulsion: In vitro and ex vivo evaluations. Pharmaceutics 2021, 13, 1218. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.M.; Chuong, M.C.; Le, H.; Pham, L.; Bendas, E. Hydrocortisone diffusion through synthetic membrane, mouse skin, and Epiderm™ cultured skin. Arch. Drug Inf. 2011, 4, 10–21. [Google Scholar] [CrossRef]
- Yuwanda, A.; Surini, S.; Harahap, Y.; Jufri, M. Study of valproic acid liposomes for delivery into the brain through an intranasal route. Heliyon 2022, 8, e09030. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Maity, S. Preparation and Characterisation of Mucoadhesive Nasal Gel of Venlafaxine Hydrochloride for Treatment of Anxiety Disorders. Indian J. Pharm. Sci. 2012, 74, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Khuroo, T.; Khuroo, A.; Hussain, A.; Mirza, M.A.; Panda, A.K.; Iqbal, Z. Qbd based and Box-Behnken design assisted oral delivery of stable lactone (active) form of topotecan as polymeric nanoformulation: Cytotoxicity, pharmacokinetic, in vitro, and ex vivo gut permeation studies. J. Drug Deliv. Sci. Technol. 2022, 77, 103850. [Google Scholar] [CrossRef]
- Trenkel, M.; Scherlie, R. Nasal Powder Formulations: In-Vitro Characterisation of the Impact of Powders on Nasal Residence Time and Sensory Effects. Pharmaceutics 2021, 13, 385. [Google Scholar] [CrossRef] [PubMed]
- Edmond, J. Essential Polyunsaturated Fatty Acids and the Barrier to the Brain: The Components of a Model for Transport. J. Mol. Neurosci. 2001, 16, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Garg, T.; Rath, G.; Goyal, A.K. In situ nasal gel drug delivery: A novel approach for brain targeting through the mucosal membrane. Artif. Cells Nanomed. Biotechnol. 2015, 44, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Shilpa, P.; Vibhavari, C.; Chatur, M. Development of Valproic Acid Niosomal in situ Nasal Gel Formulation for Epilepsy. Indian J. Pharm. Educ. Res. 2013, 47, 31–41. [Google Scholar]
- Ehrick, J.D.; Shah, S.A.; Shaw, C.; Kulkarni, V.S.; Coowanitwong, I.; De, S.; Suman, J.D. Considerations for the Development of Nasal Dosage Forms. AAPS Adv. Pharm. Sci. Ser. 2013, 6, 99–144. [Google Scholar]
- Vidgren, M.T.; Kublik, H. Nasal delivery systems and their effect on deposition and absorption. Adv. Drug Deliv. Rev. 1998, 29, 157–177. [Google Scholar] [PubMed]
- Dhuria, S.V.; Hanson, L.R.; Frey, W.H. Intranasal delivery to the central nervous system: Mechanisms and experimental considerations. J. Pharm. Sci. 2010, 99, 1654–1673. [Google Scholar] [CrossRef]
- Kumar, S.; Wong, H.; Yeung, S.A.; Riggs, K.W.; Abbott, F.S.; Rurak, D.W. Disposition of valproic acid in maternal, fetal, and newborn sheep II: Metabolism and renal elimination. Drug Metab. Dispos. 2000, 28, 845–856. [Google Scholar] [PubMed]
- Tang, W.; Borel, A.G.; Fujimia, T.; Abbott, F.S. Fluorinated analogs as mechanistic probes in valproic acid hepatotoxicity: Hepatic microvesicular steatosis and glutathione status. Chem. Res. Toxicol. 1995, 8, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.A. Targeting anti-epileptic drug therapy without collateral damage: Nanocarrier-based drug delivery. Epilepsy Curr. 2012, 12, 199–200. [Google Scholar] [CrossRef] [PubMed]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef]
- Andersen, I.; Proctor, D.F. Measurement of nasal mucociliary clearance. Eur. J. Respir. Dis. Suppl. 1983, 127, 37–40. [Google Scholar] [PubMed]
- Patel, R.B.; Patel, M.R.; Bhatt, K.K.; Patel, B.G. Formulation consideration and characterization of microemulsion drug delivery system for transnasal administration of carbamazepine. Bull. Fac. Pharm. Cairo Univ. 2013, 51, 243–253. [Google Scholar] [CrossRef]
- Yang, Y.; Jing, Y.; Yang, J.; Yang, Q. Effects of intranasal administration with Bacillus subtilis on immune cells in the nasal mucosa and tonsils of piglets. Exp. Ther. Med. 2018, 15, 5189–5198. [Google Scholar] [PubMed]
- Ezati, N.; Roberts, M.S.; Zhang, Q.; Moghimi, H.R. Measurement of Hansen Solubility Parameters of Human Stratum Corneum. Iran J. Pharm. Res. 2020, 19, 572–578. [Google Scholar]
- Hansen, C.M.; Andersen, B. The affinities of organic solvents in biological systems. Am. Ind. Hyg. Assoc. 1988, 49, 301–308. [Google Scholar] [CrossRef]
- Hussain, A.; Singh, S.K. Evidences for anti-mycobacterium activities of lipids and surfactants. World J. Microbiol. Biotechnol. 2015, 32, 7. [Google Scholar] [CrossRef] [PubMed]
- Wolf, L.; Hoffmann, H.; Talmon, Y.; Teshigawara, T.; Watanabe, K. Cryo-TEM imaging of a novel microemulsion system of silicone oil with an anionic/nonionic surfactant mixture. Soft Matter 2010, 6, 5367. [Google Scholar] [CrossRef]
- Baghdasaryan, A.; Wang, F.; Ren, F.; Ma, Z.; Li, J.; Zhou, X.; Grigoryan, L.; Xu, C.; Dai, H. Phosphorylcholine-conjugated gold-molecular clusters improve signal for Lymph Node NIR-II fluorescence imaging in preclinical cancer models. Nat. Commun. 2022, 13, 5613. [Google Scholar] [CrossRef]
- Hansen, C.M. The significance of the surface condition in solutions to the diffusion equation: Explaining “anomalous” sigmoidal, Case II, and Super Case II absorption behavior. Eur. Polym. J. 2010, 46, 651–662. [Google Scholar] [CrossRef]
- Nava, G.; Piñon, E.; Mendoza, L.; Mendoza, N.; Quintanar, D.; Ganem, A. Formulation and in vitro, ex vivo and in vivo evaluation of elastic liposomes for transdermal delivery of ketorolac tromethamine. Pharm. Times 2011, 3, 954–970. [Google Scholar]
- Chen, H.; Chang, X.; Du, D.; Liu, W.; Liu, J.; Weng, T.; Yang, Y.; Xu, H.; Yang, X. Podophyllotoxin-loaded solid lipid nanoparticles for epidermal targeting. J. Control. Release 2006, 110, 296–306. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Values |
|---|---|
| Molecular formula | C8H15NaO2 |
| Molecular weight (g/mol) | 166.19 |
| Melting point (°C) | 300 |
| Aqueous solubility (mg/mL) at 25 °C | <1 |
| Density (g/mL) | 0.9 |
| Pka | 5.14 |
| Log p | 3.08 |
| Apparent permeability coefficient (cm/h) across hCMEC/D3 and CC-2565 of in vitro blood brain barrier | 0.625 |
| Dose (mg) | 200 |
| Body weight (kg) | 70 |
| Dosing volume (mL) | 1 |
| Mean precipitation time (s) | 30 |
| AUC (µg. h/mL) | 10–160 |
| Cmax (mg/L) | ~120 |
| Tmax (h) (mean) | 5 |
| Elimination half-life (h) | 8–15 |
| Clearance (L/h) | 0.206–1.154 |
| Plasma protein binding (%) | 90–95 |
| Vd (L) | 8.4–23.3 |
| pH for reference solubility | 7.0 |
| Simulation time (h) | 24 |
| Drug and Excipient | δd (MPa1/2) | δp (MPa1/2) | δh (MPa1/2) |
|---|---|---|---|
| AV | 16.1 | 4.3 | 9.0 |
| Safflower seed oil (87%) * | 14.5 | 2.7 | 5.3 |
| Grape seed oil (70%) * | 11.69 | 2.17 | 4.27 |
| Flaxseed oil (60%) * | 10.02 | 1.86 | 3.66 |
| Tween 80 | 16.6 | 5.3 | 7.5 |
| Span 80 | 16.7 | 6.1 | 12.4 |
| Lecithin (PC as 20%) * | 3.2 | 0.54 | 0.64 |
| Linoleic acid * | 16.7 | 3.1 | 6.1 |
| Transcutol HP | 16.0 | 2.8 | 6.2 |
| PG ϕ | 16.8 | 10.4 | 21.3 |
| PC * | 16 | 2.7 | 3.2 |
| Code | SO (%) | Smix (%) | Water (%) | Smix Ratio | ST (%) | Size (nm) | PDI | ZP (mV) | %T | Product Strength (% w/w) |
|---|---|---|---|---|---|---|---|---|---|---|
| CVE1 | 16.46 | 30.21 | 48.59 | 1:2 | 0.04 | 162 | 0.27 | +24.7 | 98.5 | 0.4 |
| CVE2 | 20.75 | 23.5 | 50.22 | 1:3 | 0.05 | 189 | 0.32 | +26.8 | 96.8 | 0.5 |
| CVE3 | 14.72 | 21.67 | 57.12 | 1:2 | 0.06 | 185 | 0.31 | +31.6 | 97.2 | 0.6 |
| CVE4 | 19.88 | 43.51 | 32.16 | 1:2 | 0.04 | 148 | 0.18 | +23.9 | 96.9 | 0.4 |
| CVE5 | 9.8 | 21.84 | 63.04 | 2:1 | 0.05 | 79 | 0.11 | +27.1 | 95.3 | 0.5 |
| CVE6 | 14.46 | 17.15 | 60.99 | 3:1 | 0.07 | 113 | 0.26 | +34.7 | 97.8 | 0.7 |
| AVE6 | 14.46 | 17.15 | 60.92 | 3:1 | 0.0 | 126 | 0.29 | −22.8 | 95.6 | 0.7 |
| Nanoemulsion gel (0.5% w/w) composition (VA strength) | Evaluated parameters | |||||||||
| 0.5% VE gel | NE (g) | Gel-blank (g) | Triethanolamine (g) | CVE6:gel ratio | Size (nm) | PDI | ZP (mV) | Viscosity (cP) | pH | |
| CVE6 gel (0.35%) | 1 | 0.95 | 0.05 g | 1:1 | 129 | 0.24 | +21.9 | 1837.3 | 6.8 | |
| AVE6 gel (0.35%) | 1 | 0.95 | 0.05 g | 1:1 | 142 | 0.31 | −26.5 | 1907.1 | 7.1 | |
| Formulations | Freezing (−21 °C) | Room Temperature | Thaw (40 °C) | Centrifugation | Inference * |
|---|---|---|---|---|---|
| CVE1 | ✓ | ✓ | ✓ | ✓ | Stable |
| CVE2 | ✓ | ✓ | ✓ | ✓ | Stable |
| CVE3 | ✓ | ✓ | ✓ | ✓ | Stable |
| CVE4 | ✓ | ✓ | ✓ | ✓ | Stable |
| CVE5 | ✓ | ✓ | ✓ | ✓ | Stable |
| CVE6 | ✓ | ✓ | ✓ | ✓ | Stable |
| AVE6 | ✓ | ✓ | ✓ | ✓ | Stable |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, A.; Altamimi, M.A.; Ramzan, M.; Mirza, M.A.; Khuroo, T. GastroPlus- and HSPiP-Oriented Predictive Parameters as the Basis of Valproic Acid-Loaded Mucoadhesive Cationic Nanoemulsion Gel for Improved Nose-to-Brain Delivery to Control Convulsion in Humans. Gels 2023, 9, 603. https://doi.org/10.3390/gels9080603
Hussain A, Altamimi MA, Ramzan M, Mirza MA, Khuroo T. GastroPlus- and HSPiP-Oriented Predictive Parameters as the Basis of Valproic Acid-Loaded Mucoadhesive Cationic Nanoemulsion Gel for Improved Nose-to-Brain Delivery to Control Convulsion in Humans. Gels. 2023; 9(8):603. https://doi.org/10.3390/gels9080603
Chicago/Turabian StyleHussain, Afzal, Mohammad A. Altamimi, Mohhammad Ramzan, Mohd Aamir Mirza, and Tahir Khuroo. 2023. "GastroPlus- and HSPiP-Oriented Predictive Parameters as the Basis of Valproic Acid-Loaded Mucoadhesive Cationic Nanoemulsion Gel for Improved Nose-to-Brain Delivery to Control Convulsion in Humans" Gels 9, no. 8: 603. https://doi.org/10.3390/gels9080603
APA StyleHussain, A., Altamimi, M. A., Ramzan, M., Mirza, M. A., & Khuroo, T. (2023). GastroPlus- and HSPiP-Oriented Predictive Parameters as the Basis of Valproic Acid-Loaded Mucoadhesive Cationic Nanoemulsion Gel for Improved Nose-to-Brain Delivery to Control Convulsion in Humans. Gels, 9(8), 603. https://doi.org/10.3390/gels9080603