

Conceptualization and Investigation of Multicomponent Polymer Networks as Prospective Corticosteroid Carriers

 , and
, and
Abstract
1. Introduction
2. Results and Discussion
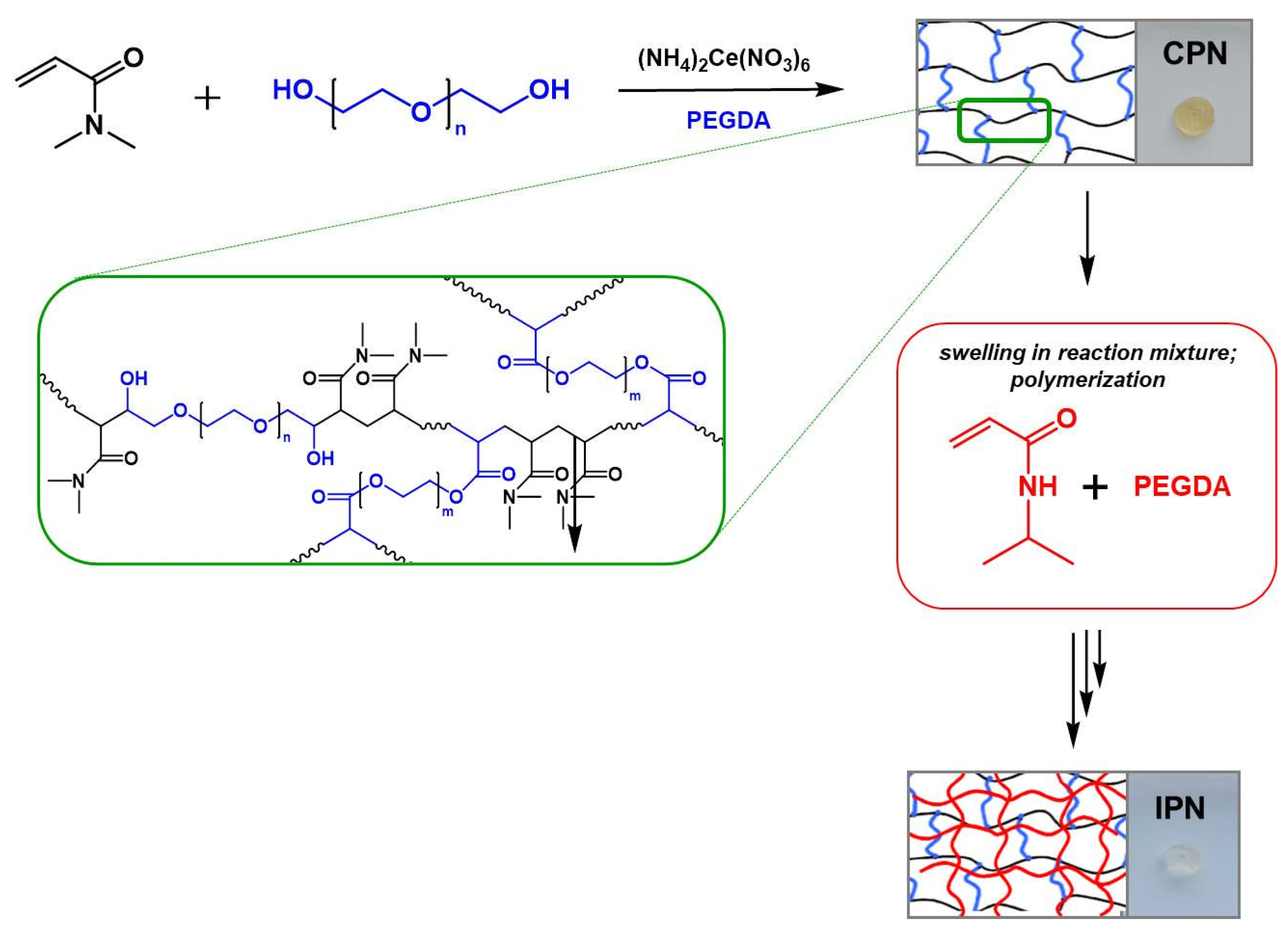
2.1. Synthesis of the Multicomponent Polymer Networks
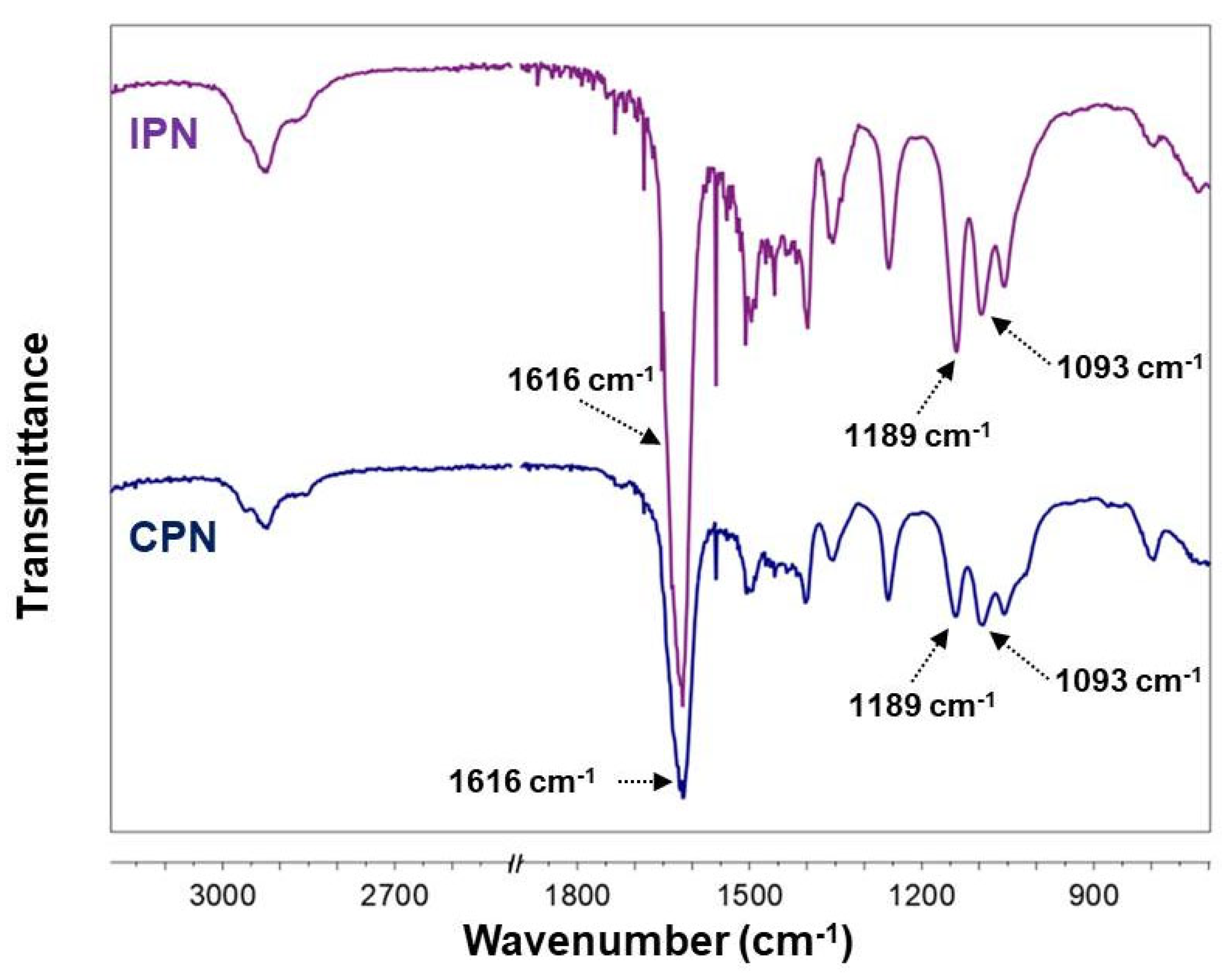
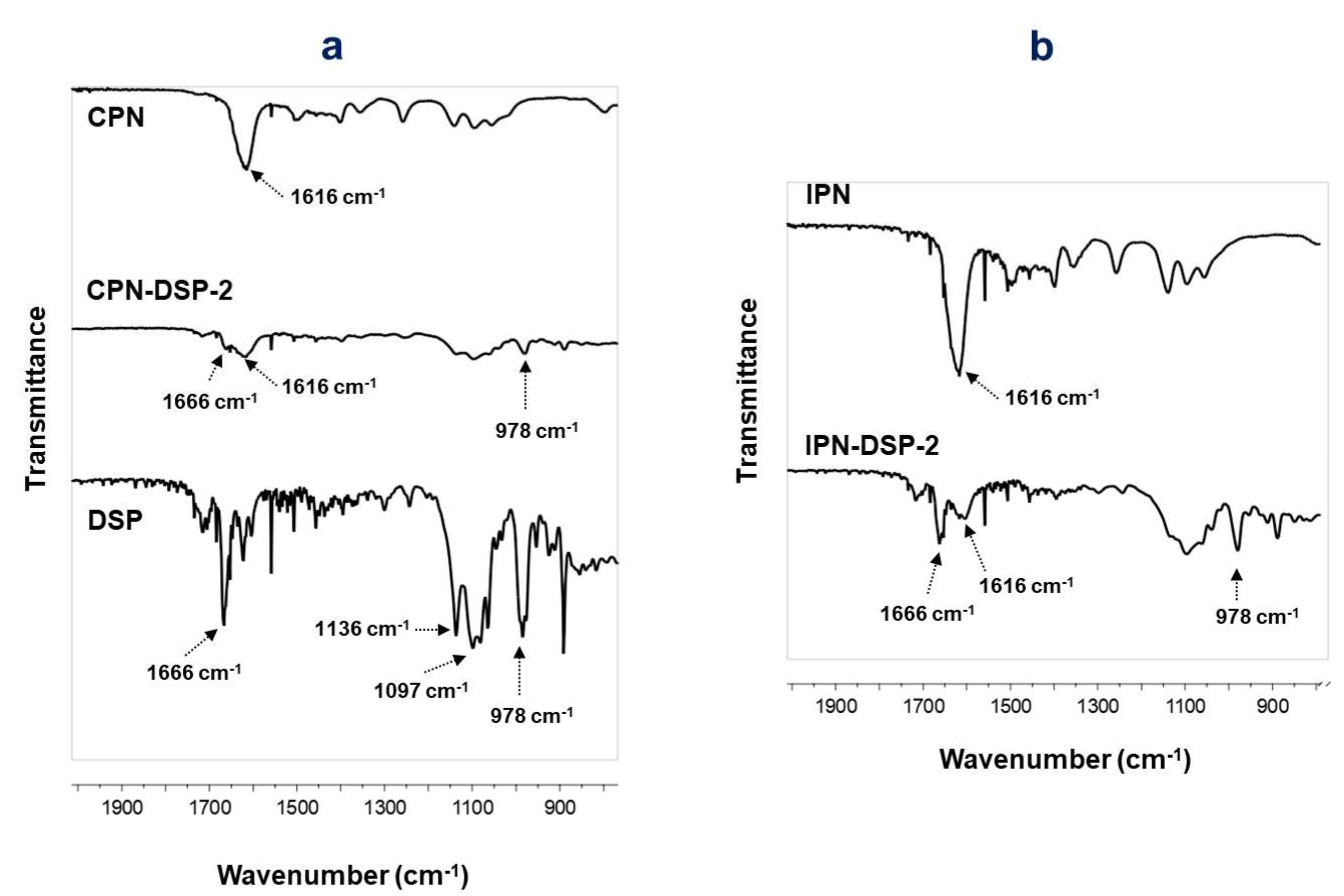
2.2. Fourier Transform Infrared (FTIR) Spectroscopy
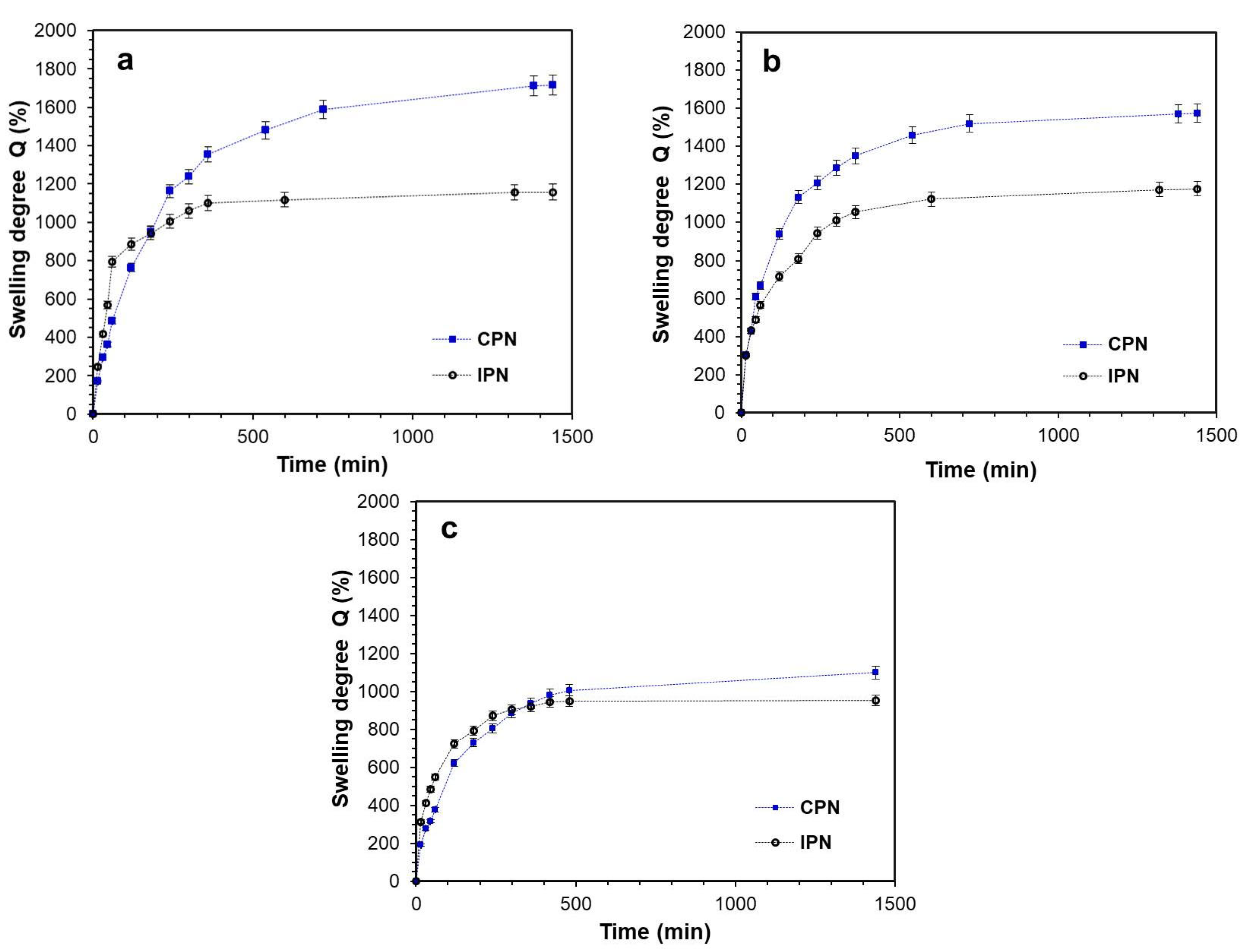
2.3. Swelling Experiments
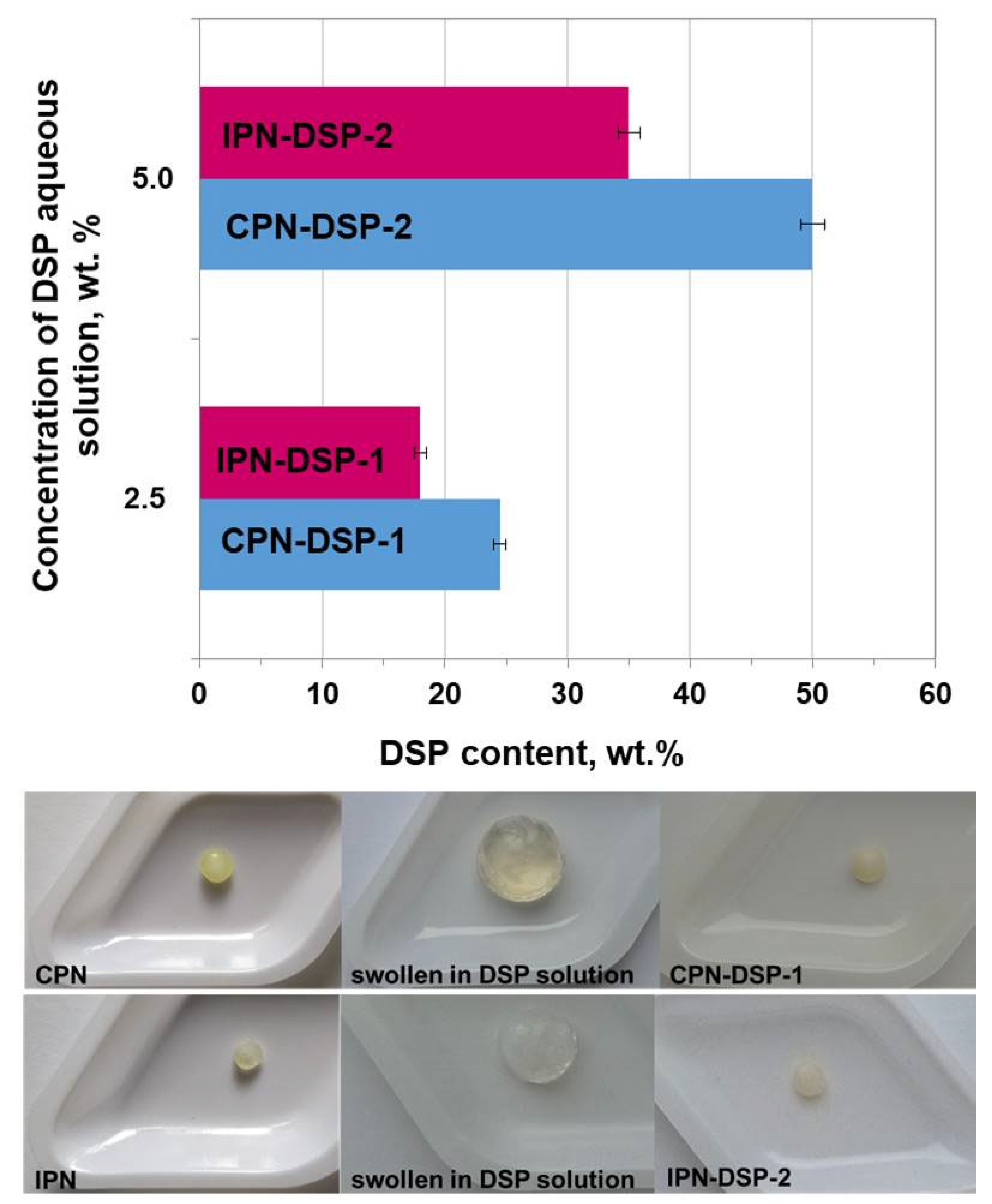
2.4. Drug Loading and DSP Content
2.5. Loading Efficiency
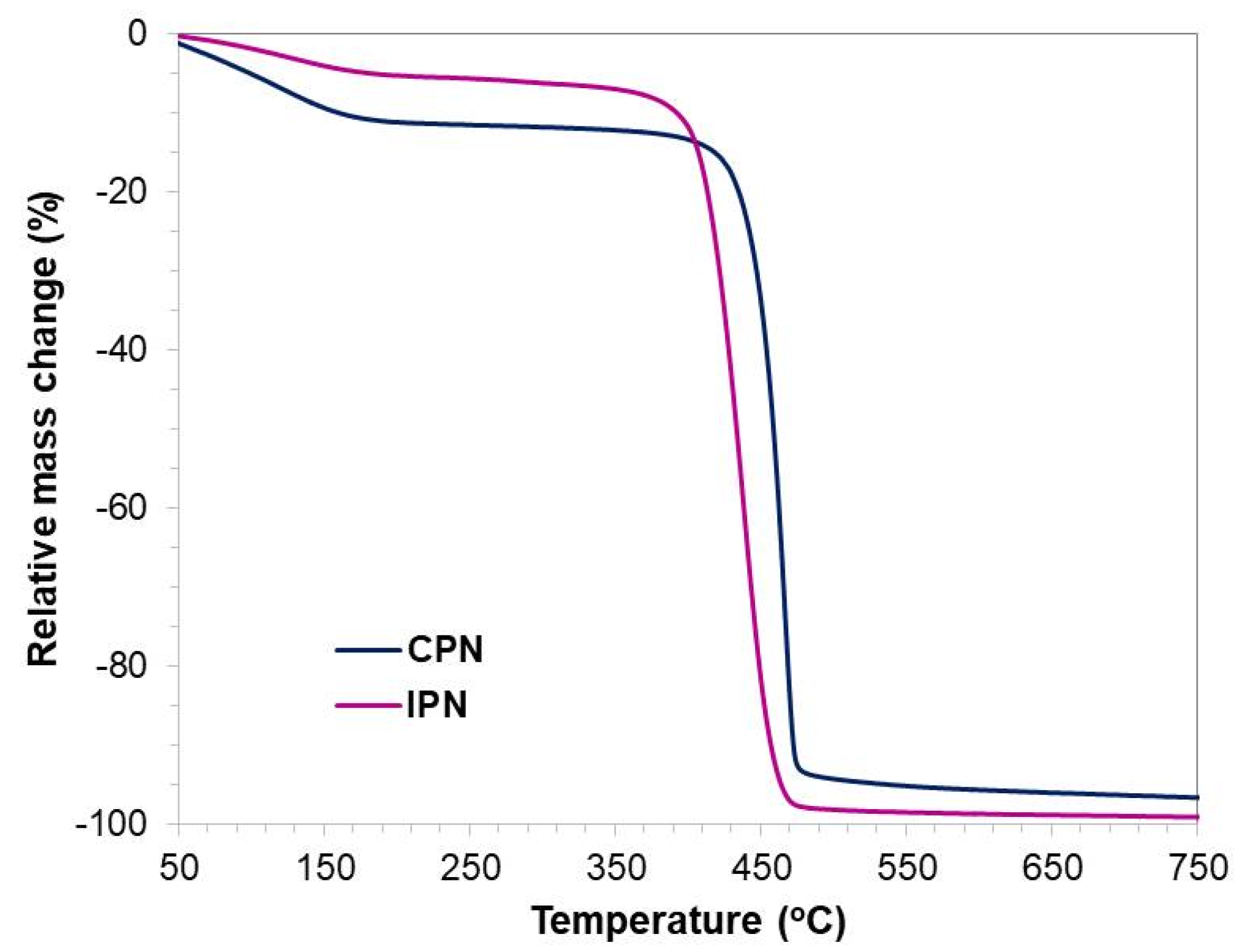
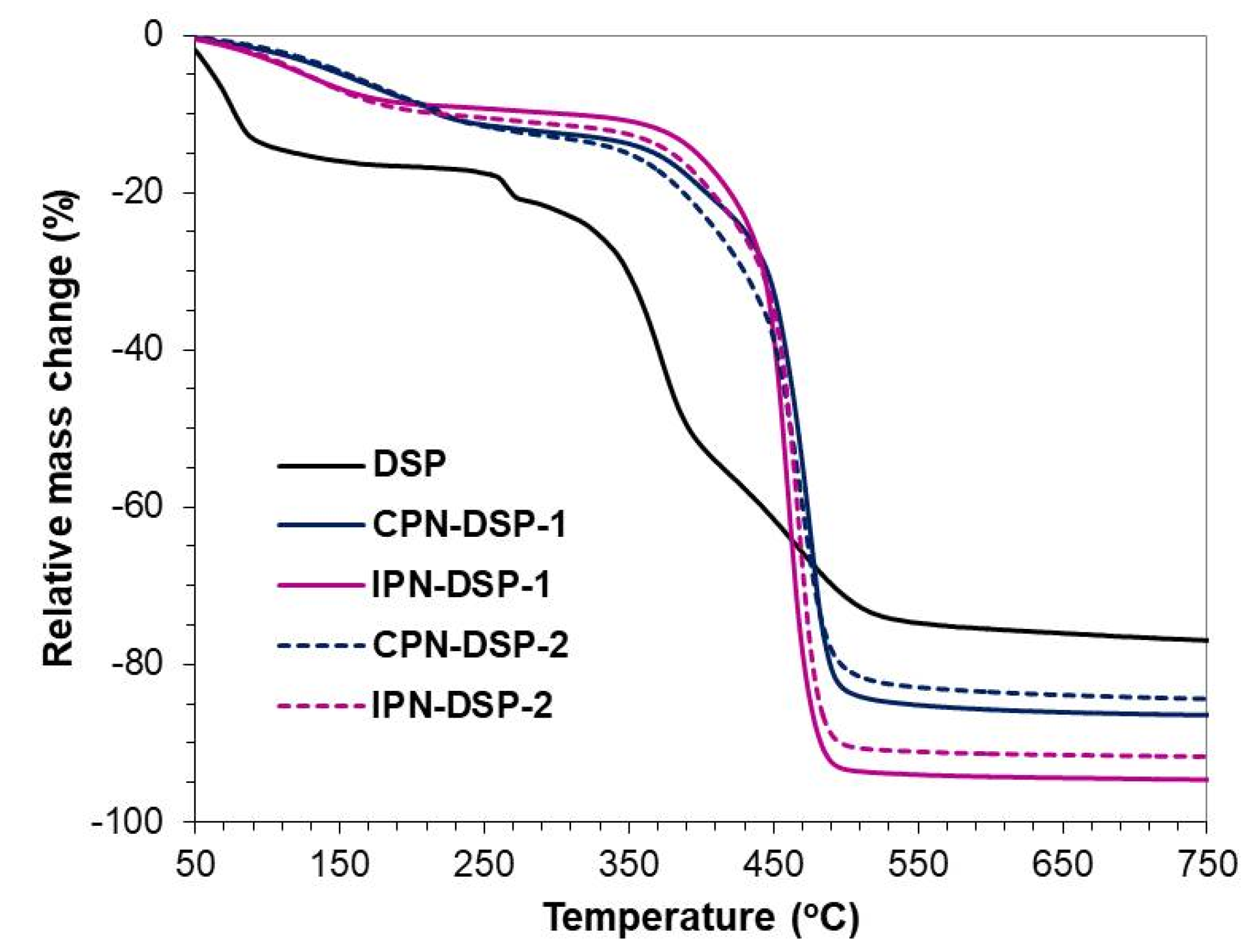
2.6. Thermogravimetric Analysis (TGA)
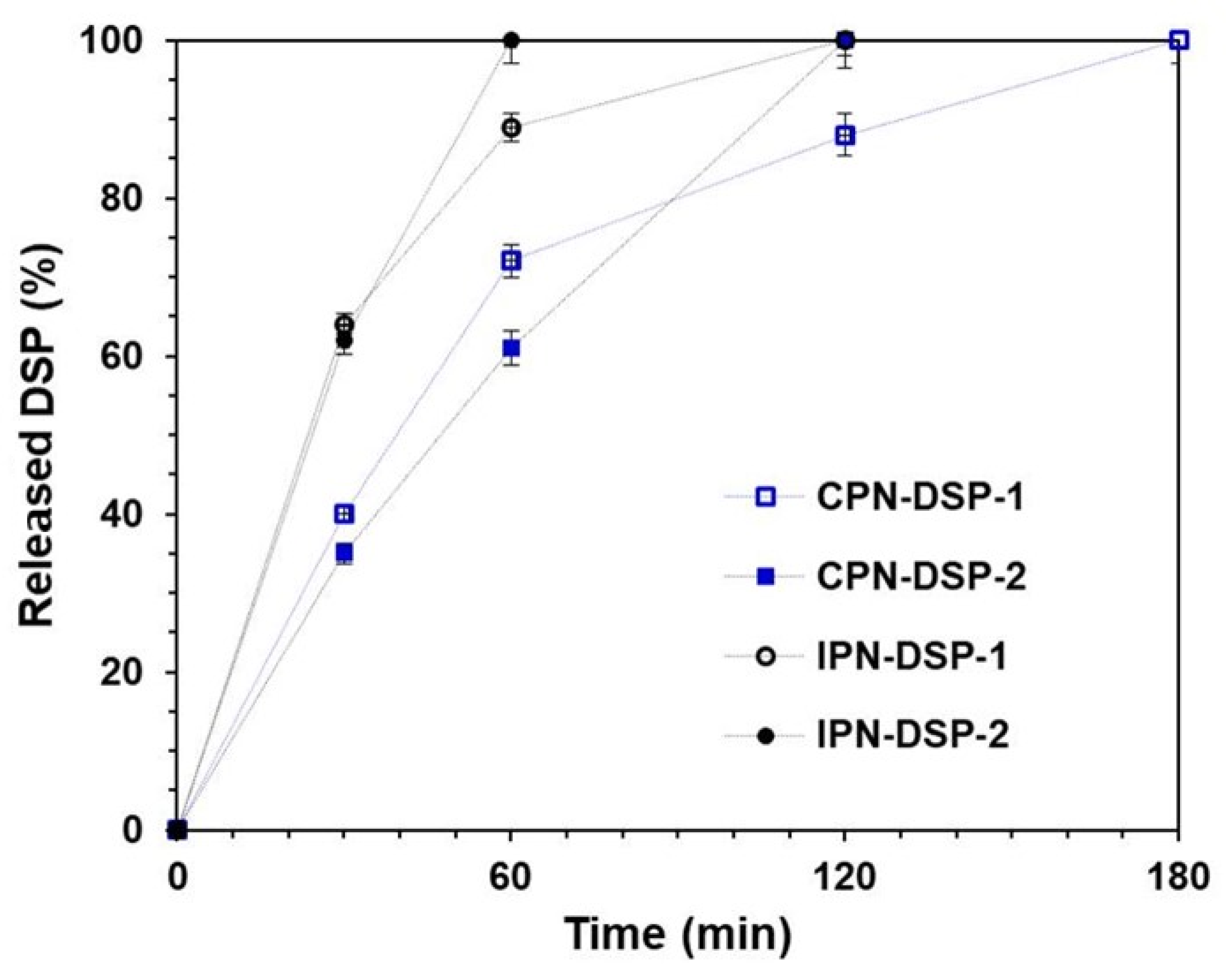
2.7. In Vitro Drug Release Study
2.8. Drug Release Kinetics
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Synthesis of Multicomponent Polymer Networks
4.2.2. Fourier Transform Infrared Spectroscopy (FTIR)
4.2.3. Swelling Experiments
4.2.4. Drug Loading and DSP Content
4.2.5. Loading Efficiency (%)
4.2.6. Thermogravimetric Analysis (TGA)
4.2.7. In Vitro Drug Release Study
4.2.8. Drug Release Kinetics
- Zero-order model—this model is applied on occasions when the drug release rate is not dependent on its concentration.
- First-order model—according to this model, the drug release rate is directly proportional to the drug concentration. The first-order process is characterized by linear kinetics.
- Higuchi model—Higuchi suggested that two mechanisms are responsible for controlling the drug release rate: swelling and erosion/degradation.
- Korsmeyer–Peppas model—this model is used to describe the process when the release follows several kinetics mechanisms.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bunim, J.J.; Black, R.L.; Lutwak, L.; Peterson, R.E.; Whedon, G.D. Studies on dexamethasone, a new synthetic steroid, in Rheu rheumatoid arthritis: A preliminary report; adrenal cortical, metabolic and early clinical effects. Arthritis Rheum. 1958, 1, 313–331. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Goyal, A.; Sonthalia, S. Corticosteroid Adverse Effects. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- European Medicines Agency. Assessment Report of Dexamethasone Alapis; European Medicines Agency: Amsterdam, The Netherlands, 2011.
- Yoon, J.J.; Kim, J.H.; Park, T.G. Dexamethasone-releasing biodegradable polymer scaffolds fabricated by a gas-foaming/salt-leaching method. Biomaterials 2003, 24, 2323–2329. [Google Scholar] [CrossRef] [PubMed]
- Shoji, J.; Sakimoto, T.; Muromoto, K.; Inada, N.; Mitsuru, R. Comparison of Topical Dexamethasone and Topical FK506 Treatment for the Experimental Allergic Conjunctivitis Model in Balb/c Mice. Jpn. J. Ophthalmol. 2005, 49, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Fernandez, S.; Martin-Mola, E. Uveitis. Best Pract. Res. Clin. Rheumatol. 2006, 20, 487–505. [Google Scholar] [CrossRef]
- Yülek, F.; Ozdek, S.; Gurelik, G.; Hasanreisoglu, B. Effect of topical steroids on corneal epithelial healing after vitreoretinal surgery. Acta Ophthalmol. Scand. 2006, 84, 319–322. [Google Scholar] [CrossRef]
- Matsumoto, K.; Mizukoshi, K.; Oyobikawa, M.; Ohshima, H.; Sakai, Y.; Tagami, H. Objective Evaluation of the Efficacy of Daily Topical Applications of Cosmetics Bases Using the Hairless Mouse Model of Atopic Dermatitis. Skin. Res. Technol. 2005, 11, 209–217. [Google Scholar] [CrossRef]
- Trautmann, A.; Akdis, M.; Schmid-Grendelmeier, P.; Disch, R.; Bröcker, E.B.; Blaser, K.; Akdis, A.C. Targeting keratinocyte apoptosis in the treatment of atopic dermatitis and allergic contact dermatitis. J. Allergy Clin. Immunol. 2001, 108, 839–846. [Google Scholar] [CrossRef]
- Zampieri, N.; Corroppolo, M.; Zuin, V.; Bianchi, S.; Camoglio, F.S. Phimosis and topical steroids: New clinical findings. Pediatr. Surg. Int. 2007, 23, 331–335. [Google Scholar] [CrossRef]
- Valencia, I.C.; Kerdel, F.A. Chapter 216. Topical Corticosteroids. In Fitzpatrick’s Dermatology in General Medicine, 8th ed.; Goldsmith, L.A., Katz, S.I., Gilchrest, B.A., Paller, A.S., Leffel, D.J., Wolff, K., Eds.; McGraw Hill: New York, NY, USA, 2012. [Google Scholar]
- Gómez-Gaete, C.; Tsapis, N.; Besnard, M.; Bochot, A.; Fattal, E. Encapsulation of dexamethasone into biodegradable polymeric nanoparticles. Int. J. Pharm. 2007, 331, 153–159. [Google Scholar] [CrossRef]
- Kim, D.H.; Martin, D.C. Sustained release of dexamethasone from hydrophilic matrices using PLGA nanoparticles for neural drug delivery. Biomaterials 2006, 27, 3031–3037. [Google Scholar] [CrossRef]
- Friedrich, R.B.; Fontana, M.C.; Beck, R.C.R.; Pohlmann, A.R.; Guterres, S.S. Development and physicochemical characterization of dexamethasone-loaded polymeric nanocapsule suspensions. Quim. Nova 2008, 31, 1131–1136. [Google Scholar] [CrossRef]
- Beck, R.C.R.; Guterres, S.S.; Freddo, R.J.; Michalowski, C.B.; Barcellos, I.; Funck, J.A.B. Nanoparticles containing dexamethasone: Physicochemical properties and anti-inflammatory activity. Acta Farm. Bonaerense 2003, 22, 11–15. [Google Scholar]
- Zhang, Z.; Grijpma, D.W.; Feijen, J. Poly(trimethylene carbonate) and monomethoxy poly(ethylene glycol)-block-poly(trimethylene carbonate) nanoparticles for the controlled release of dexamethasone. J. Control. Release 2006, 111, 263–270. [Google Scholar] [CrossRef]
- Rençber, S.; Köse, F.A.; Karavana, S.Y. Dexamethasone loaded PLGA nanoparticles for potential local treatment of oral precancerous lesions. Pharm. Dev. Technol. 2020, 25, 149–158. [Google Scholar] [CrossRef]
- Simón-Vázquez, R.; Tsapis, N.; Lorscheider, M.; Rodríguez, A.; Calleja, P.; Mousnier, L.; González-Fernández, Á.; Fattal, E. Improving dexamethasone drug loading and efficacy in treating arthritis through a lipophilic prodrug entrapped into PLGA-PEG nanoparticles. Drug. Deliv. Transl. Res. 2022, 12, 1270–1284. [Google Scholar] [CrossRef]
- Sankar, V.; Harikrishnan, V.; Madhura Keerthi, L.; Prasanth, K.G.; Sundar, D.; Ruckmani, K. In vivo evaluation of dexamethasone sodium phosphate nanoparticles for post cataract treatment. Pharmacologyonline 2009, 3, 822–828. [Google Scholar]
- Dustgani, A.; Farahani, E.V.; Imani, M. Preparation of chitosan nanoparticles loaded by dexamethasone sodium phosphate. Iran. J. Pharm. Sci. 2008, 4, 111–114. [Google Scholar]
- Puri, S.; Kallinteri, P.; Higgins, S.; Hutcheon, G.A.; Garnett, M.C. Drug incorporation and release of water soluble drugs from novel functionalized poly(glycerol adipate) nanoparticles. J. Control. Release 2008, 125, 59–67. [Google Scholar] [CrossRef]
- Thote, A.J.; Gupta, R.B. Formation of nanoparticles of a hydrophilic drug using supercritical carbon dioxide and microencapsulation for sustained release. Nanomedicine 2005, 1, 85–90. [Google Scholar] [CrossRef]
- Yang, K.; Weng, L.; Cheng, Y.; Zhang, H.; Zhang, J.; Wu, Q.; Xu, T. Host-guest chemistry of dendrimer-drug complexes. 6. Fully acetylated dendrimers as biocompatible drug vehicles using dexamethasone 21-phosphate as a model drug. J. Phys. Chem. B 2011, 115, 2185–2195. [Google Scholar] [CrossRef]
- Wang, B.; Tang, Y.; Oh, Y.; Lamb, N.W.; Xia, S.; Ding, Z.; Chen, B.; Suarez, M.J.; Meng, T.; Kulkarni, V.; et al. Controlled release of dexamethasone sodium phosphate with biodegradable nanoparticles for preventing experimental corneal neovascularization. Nanomed. Nanotechnol. Biol. Med. 2019, 17, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Boroojen, F.R.; Mashayekhan, S.; Abbaszadeh, H.A. The Controlled Release of Dexamethasone Sodium Phosphate from Bioactive Electrospun PCL/Gelatin Nanofiber Scaffold. Iran. J. Pharm. Res. 2019, 18, 111–124. [Google Scholar]
- Georgieva, D.; Kostova, B.; Ivanova, S.; Rachev, D.; Tzankova, V.; Kondeva-Burdina, M.; Christova, D. pH-Sensitive cationic copolymers of different macromolecular architecture as potential dexamethasone sodium phosphate delivery systems. J. Pharm. Sci. 2014, 103, 2406–2413. [Google Scholar] [CrossRef] [PubMed]
- Yahia, L.; Chirani, N.; Gritsch, L.; Motta, F.L. History and Applications of Hydrogels. J. Biomed. Sci. 2015, 4, 1–23. [Google Scholar] [CrossRef]
- Peppas, N.A. Hydrogels and drug delivery. Curr. Opin. Colloid Interface Sci. 1997, 2, 531–537. [Google Scholar] [CrossRef]
- Singh, T.R.R.; Laverty, G.; Donnelly, R. Hydrogels: Design, Synthesis and Application in Drug Delivery and Regenerative Medicine; CRC Press: Boca Raton, FL, USA; Taylor and Francis Group: Abingdon, UK, 2018. [Google Scholar] [CrossRef]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef]
- Ahmed, E.M. Hydrogel: Preparation, characterization, and applications: A review. J. Adv. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef]
- Das, N. Preparation methods and properties of hydrogel: A review. Int. J. Pharm. Pharm. Sci. 2013, 5, 112–117. [Google Scholar]
- Bashir, S.; Hina, M.; Iqbal, J.; Rajpar, A.H.; Mujtaba, M.A.; Alghamdi, N.A.; Wageh, S.; Ramesh, K.; Ramesh, S. Fundamental Concepts of Hydrogels: Synthesis, Properties, and Their Applications. Polymers 2020, 12, 2702. [Google Scholar] [CrossRef]
- Bashir, S.; Teo, Y.Y.; Ramesh, S.; Ramesh, K.; Mushtaq, M.W. Rheological behavior of biodegradable N-succinyl chitosan-g-poly (acrylic acid) hydrogels and their applications as drug carrier and in vitro theophylline release. Int. J. Biol. Macromol. 2018, 117, 454–466. [Google Scholar] [CrossRef]
- Bhattarai, N.; Gunn, J.; Zhang, M. Chitosan-based hydrogels for controlled, localized drug delivery. Adv. Drug Deliv. Rev. 2010, 62, 83–99. [Google Scholar] [CrossRef]
- Liu, J.; Ni, R.; Chau, Y. A self-assembled peptidic nanomillipede to fabricate a tuneable hybrid hydrogel. Chem. Commun. 2019, 55, 7093–7096. [Google Scholar] [CrossRef]
- Kostova, B.; Ivanova-Mileva, K.; Rachev, D.; Christova, D. Study of the potential of amphiphilic conetworks based on poly(2-ethyl-2-oxazoline) as new platforms for delivery of drugs with limited solubility. AAPS PharmSciTech 2013, 14, 352–359. [Google Scholar] [CrossRef]
- Kostova, B.; Ivanova, S.; Balashev, K.; Rachev, D.; Christova, D. Evaluation of poly(2-ethyl-2-oxazoline) containing copolymer networks of varied composition as sustained metoprolol tartrate delivery systems. AAPS PharmSciTech 2014, 15, 939–946. [Google Scholar] [CrossRef]
- Kostova, B.; Georgieva, D.; Dundarova, M.; Ivanova, S.; Ivanova-Mileva, K.; Tzankova, V.; Christova, D. Design and study of the potential of crosslinked cationic polymers as drug delivery systems for dermal application. J. Appl. Polym. Sci. 2018, 135, 46524. [Google Scholar] [CrossRef]
- Zhang, J.T.; Huang, S.W.; Cheng, S.X.; Zhuo, R.X. Preparation and properties of poly(N-isopropylacrylamide)/poly(N-isopropylacrylamide) interpenetrating polymer networks for drug delivery. J. Polym. Sci. A Polym. Chem. 2004, 42, 1249–1254. [Google Scholar] [CrossRef]
- Georgieva, D.; Ivanova-Mileva, K.; Ivanova, S.; Kostova, B.; Rachev, D.; Christova, D. Thermoresponsive poly(N-isopropylacrylamide) copolymer networks for galantamine hydrobromide delivery. Colloid Polym. Sci. 2020, 298, 377–384. [Google Scholar] [CrossRef]
- Gupta, A.; Upadhyay, N.K.; Parthasarathy, S.; Rajagopal, C.; Roy, P.K. Nitrofurazone-loaded PVA–PEG semi-IPN for application as hydrogel dressing for normal and burn wounds. J. Appl. Polym. Sci. 2013, 128, 4031–4039. [Google Scholar] [CrossRef]
- Saghazadeh, S.; Rinoldi, C.; Schot, M.; Kashaf, S.S.; Sharifi, F.; Jalilian, E.; Nuutila, K.; Giatsidis, G.; Mostafalu, P.; Derakhshandeh, H.; et al. Drug delivery systems and materials for wound healing applications. Adv. Drug Deliv. Rev. 2018, 1, 138–166. [Google Scholar] [CrossRef]
- Gurney, A.B.; Wascher, D.C. Absorption of dexamethasone sodium phosphate in human connective tissue using iontophoresis. Am. J. Sports Med. 2008, 36, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Nirschl, R.P.; Rodin, D.M.; Ochiai, D.H.; Maartmann-Moe, C. DEX-AHE-01-99 Study Group. Iontophoretic administration of dexamethasone sodium phosphate for acute epicondylitis. A randomized, double-blinded, placebo-controlled study. Am. J. Sports Med. 2003, 31, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Rigby, J.H.; Draper, D.O.; Johnson, A.W.; Myrer, J.W.; Eggett, D.L.; Mack, G.W. The Time Course of Dexamethasone Delivery Using Iontophoresis Through Human Skin, Measured via Microdialysis. J. Orthop. Sport. Phys. Ther. 2015, 45, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Ajaz, N.; Khan, I.U.; Khalid, I.; Khan, R.U.; Khan, H.A.; Asghar, S.; Khalid, S.H.; Shahzad, Y.; Yousaf, A.M.; Hussain, T.; et al. In vitro and toxicological assessment of dexamethasone sodium phosphate loaded pH sensitive Pectin-g-poly(AA)/PVP semi interpenetrating network. Mater. Today Commun. 2020, 25, 101325. [Google Scholar] [CrossRef]
- Ajaz, N.; Khan, I.U.; Irfan, M.; Khalid, S.H.; Asghar, S.; Mehmood, Y.; Asif, M.; Usra; Hussain, G.; Shahzad, Y.; et al. In Vitro and Biological Characterization of Dexamethasone Sodium Phosphate Laden pH-Sensitive and Mucoadhesive Hydroxy Propyl β-Cyclodextrin-g-poly(acrylic acid)/Gelatin Semi-Interpenetrating Networks. Gels 2022, 8, 290. [Google Scholar] [CrossRef]
- Lazarova, K.; Vasileva, M.; Ivanova, S.; Novakov, C.; Christova, D.; Babeva, T. Influence of macromolecular architecture on the optical and humidity sensing properties of poly(N,N-dimethylacrylamide)-based block copolymers. Polymers 2018, 10, 769. [Google Scholar] [CrossRef]
- Topp, M.D.C.; Dijkstra, P.J.; Talsma, H.; Feijen, J. Thermosensitive micelle-forming block copolymers of poly(ethylene glycol) and poly(N-isopropylacrylamide). Macromolecules 1997, 30, 8518–8520. [Google Scholar] [CrossRef]
- Chowdhury, P.; Pal, C.M. Graft copolymerization of methyl acrylate onto polyvinyl alcohol using Ce(IV) initiator. Eur. Polym. J. 1999, 35, 2207–2213. [Google Scholar] [CrossRef]
- Wang, W.R.; Li, A.; Mei, W.; Zhu, R.R.; Li, K.; Sun, X.Y.; Qian, Y.C.; Wang, S.L. Dexamethasone sodium phosphate intercalated layered double hydroxides and their therapeutic efficacy in a murine asthma model. RSC Adv. 2015, 5, 23826–23834. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinetic Model | CPN-DSP-1 | CPN-DSP-2 | IPN-DSP-1 | IPN-DSP-2 |
|---|---|---|---|---|
| Zero-order | k0 = 3.9223 R2 = 0.8368 | k0 = 4.2645 R2 = 0.8584 | k0 = 2.3058 R2 = 0.6393 | k0 = 7.2929 R2 = 0.9815 |
| First-order | k1 = 2.6353 R2 = 0.7764 | k1 = 3.3339 R2 = 0.9175 | k1 = 2.0223 R2 = 0.8035 | k1 = 1.2990 R2 = 0.4576 |
| Higuchi | kH = 18.1229 R2 = 0.9692 | kH = 15.3779 R2 = 0.9971 | kH = 23.0216 R2 = 0.9513 | kH = 20.0597 R2 = 1 |
| Korsmeyer–Peppas | n = 1.1519 R2 = 0.8507 | n = 1.3031 R2 = 0.9898 | n = 2.8795 R2 = 0.9305 | n = 1.4469 R2 = 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Georgieva, D.; Alexandrova, M.; Ivanova, S.; Christova, D.; Kostova, B. Conceptualization and Investigation of Multicomponent Polymer Networks as Prospective Corticosteroid Carriers. Gels 2023, 9, 470. https://doi.org/10.3390/gels9060470
Georgieva D, Alexandrova M, Ivanova S, Christova D, Kostova B. Conceptualization and Investigation of Multicomponent Polymer Networks as Prospective Corticosteroid Carriers. Gels. 2023; 9(6):470. https://doi.org/10.3390/gels9060470
Chicago/Turabian StyleGeorgieva, Dilyana, Mariela Alexandrova, Sijka Ivanova, Darinka Christova, and Bistra Kostova. 2023. "Conceptualization and Investigation of Multicomponent Polymer Networks as Prospective Corticosteroid Carriers" Gels 9, no. 6: 470. https://doi.org/10.3390/gels9060470
APA StyleGeorgieva, D., Alexandrova, M., Ivanova, S., Christova, D., & Kostova, B. (2023). Conceptualization and Investigation of Multicomponent Polymer Networks as Prospective Corticosteroid Carriers. Gels, 9(6), 470. https://doi.org/10.3390/gels9060470