
Development, Characterization, and Evaluation of α-Mangostin-Loaded Polymeric Nanoparticle Gel for Topical Therapy in Skin Cancer

 , ,
, ,  , , , ,
, , , ,  and
and
Abstract
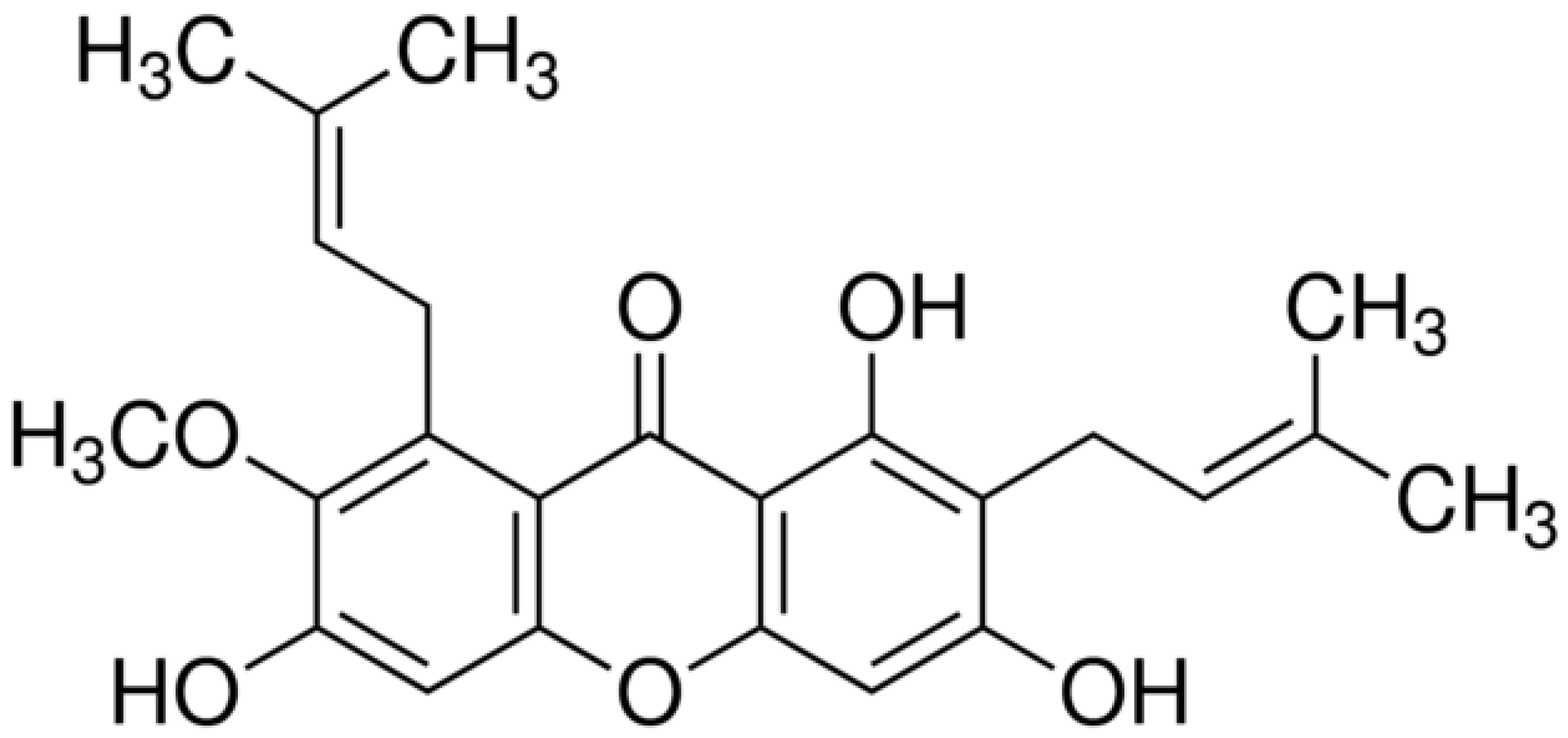
:1. Introduction
2. Results and Discussion
2.1. Optimization
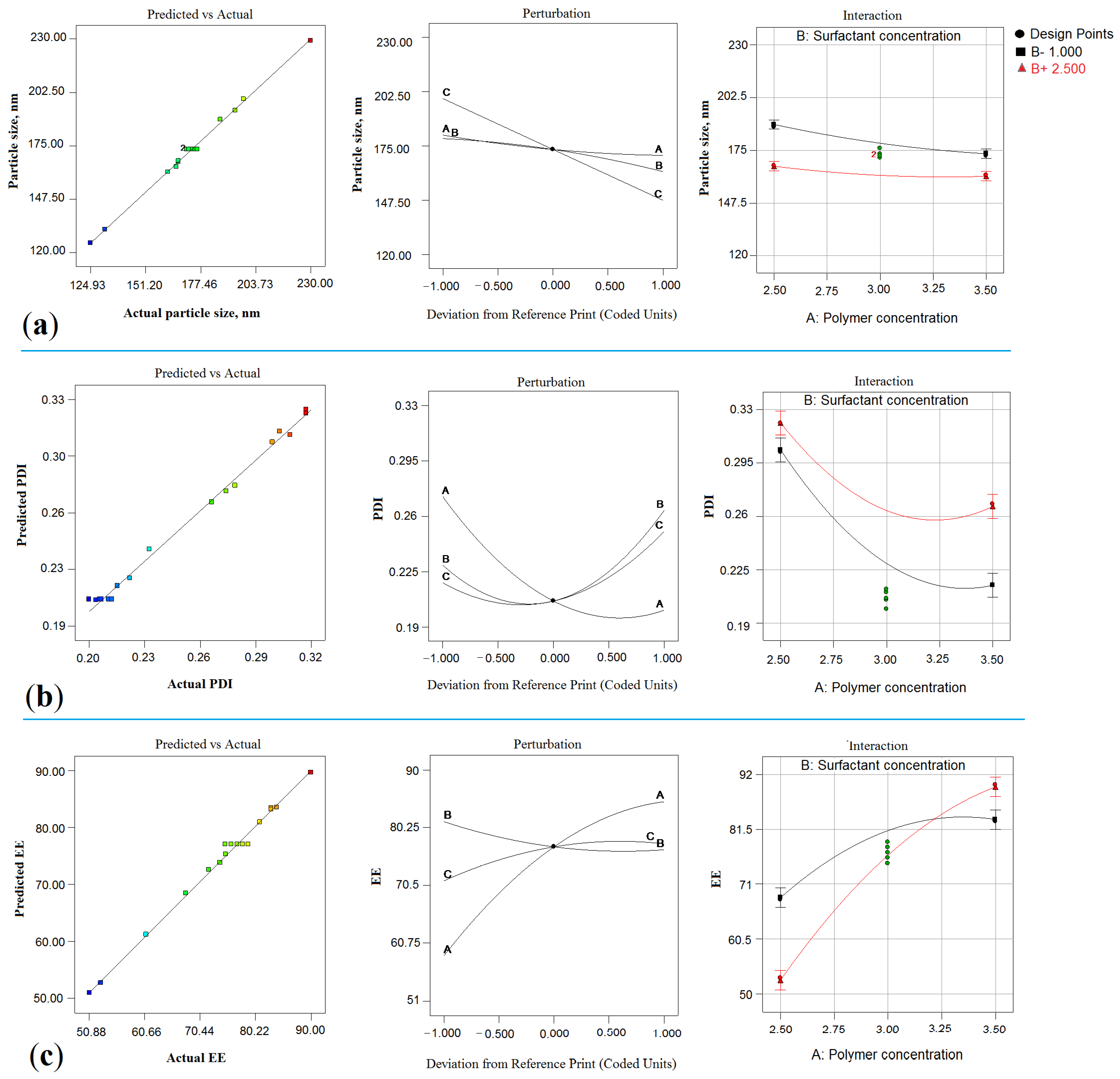
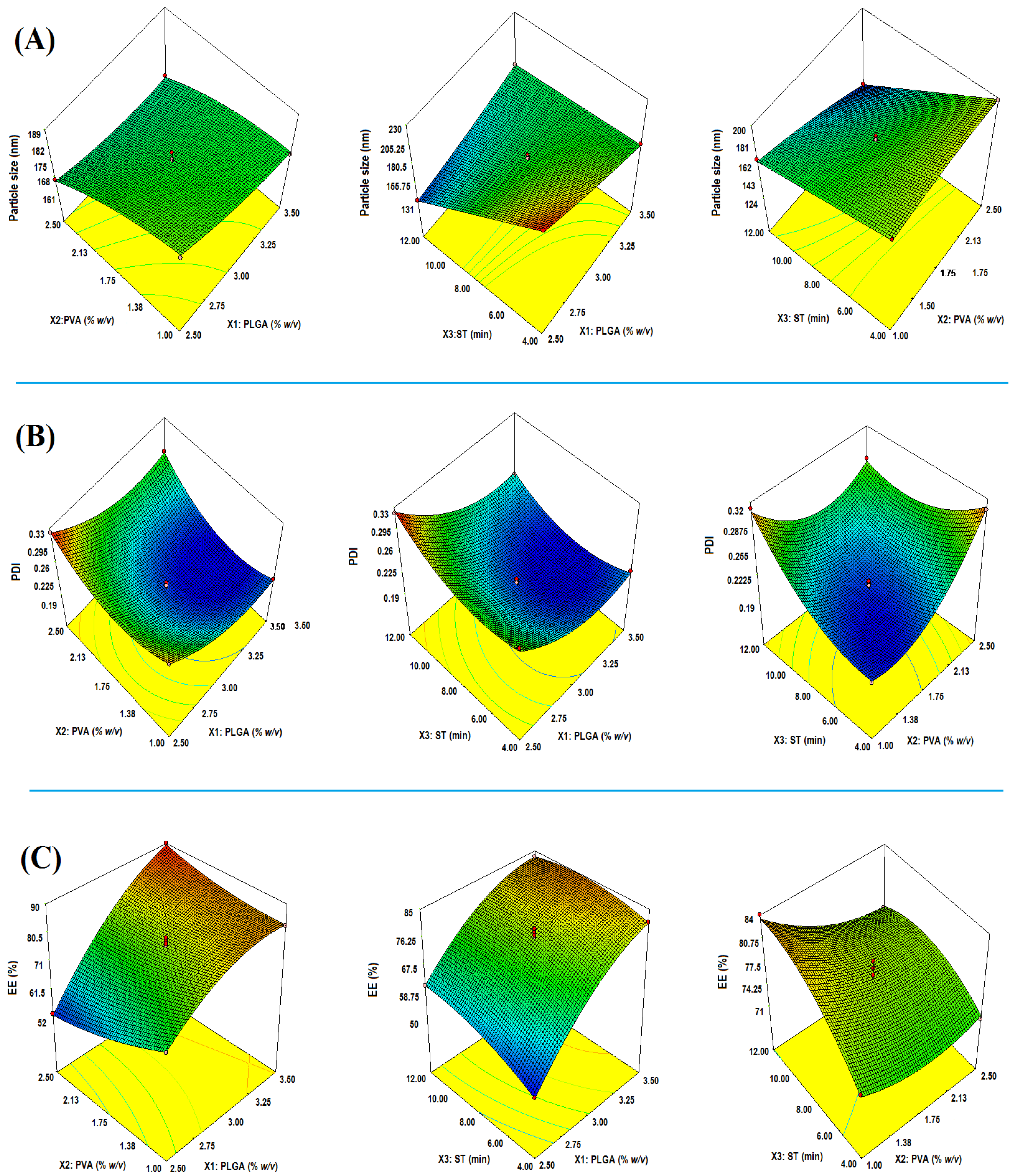
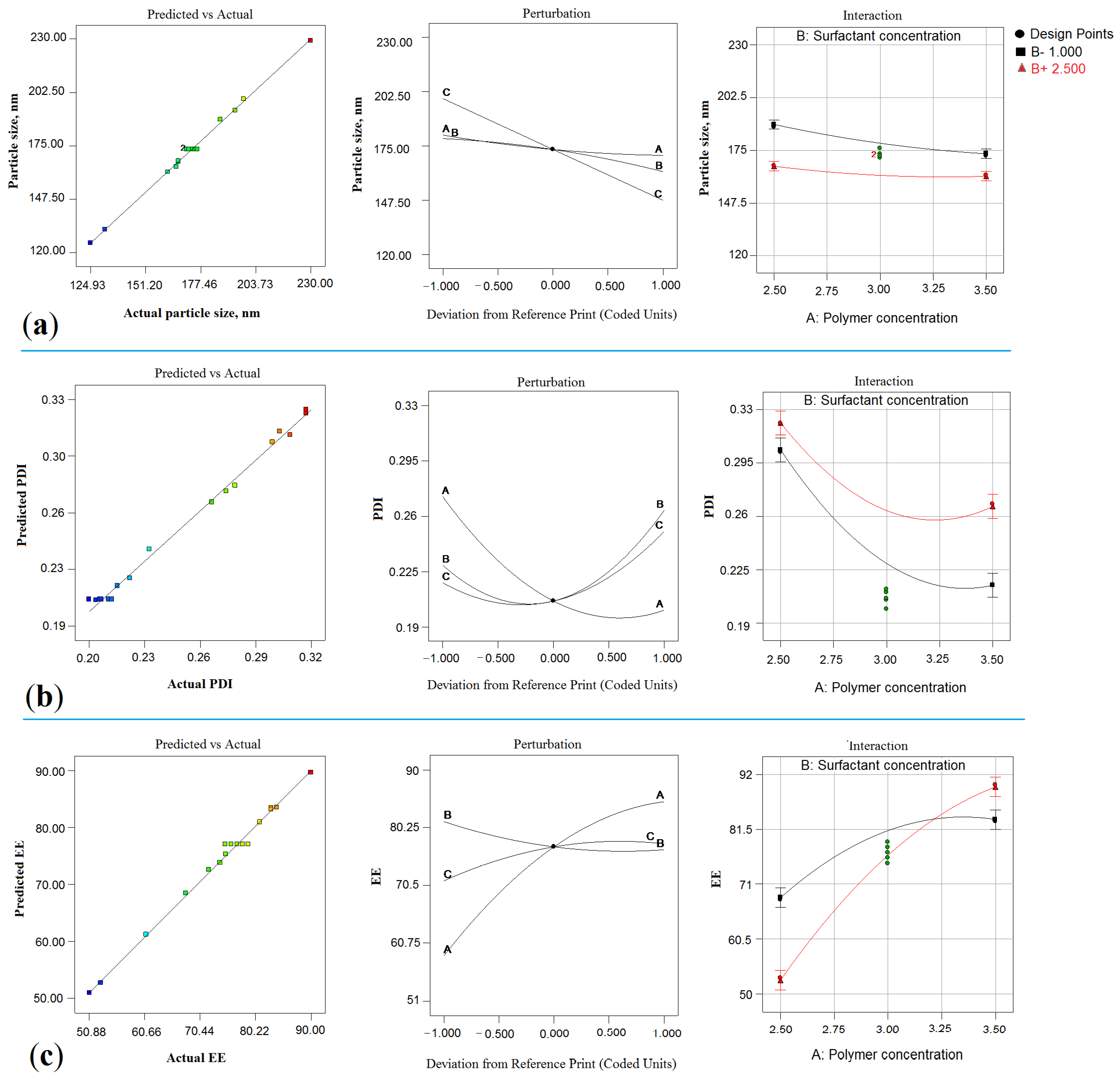
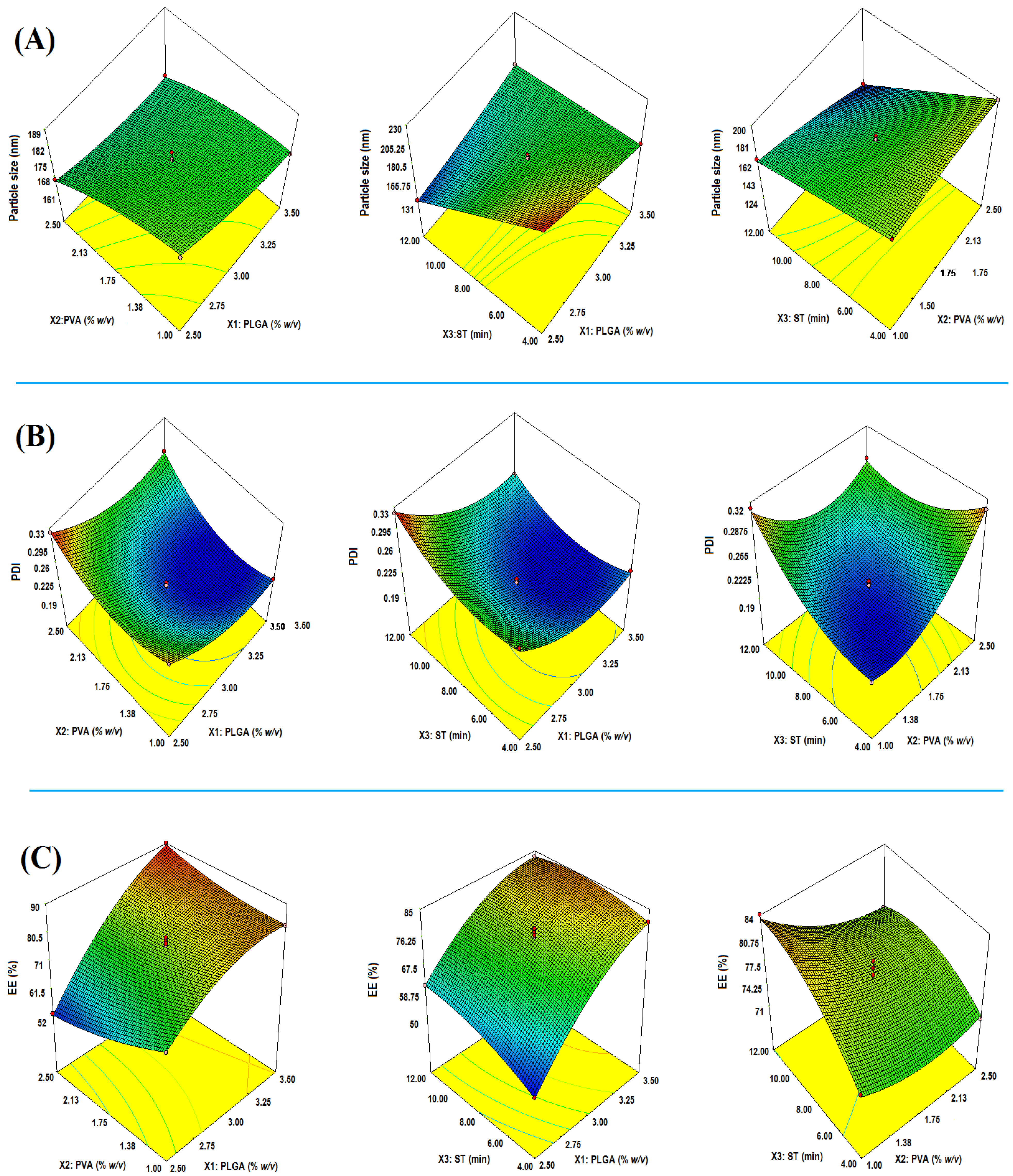
2.2. Effect on Particle Size (Y1)
2.2.1. Impact of PLGA
2.2.2. Impact of PVA
2.2.3. Impact of Sonication Time (ST)
2.3. Effect on PDI (Y2)
2.3.1. Impact of PLGA
2.3.2. Impact of PVA
2.3.3. Impact of ST
2.4. Y3: Effect of on EE
2.4.1. Impact of PLGA
2.4.2. Impact of PVA
2.4.3. Impact of ST
2.5. Numerical Optimization
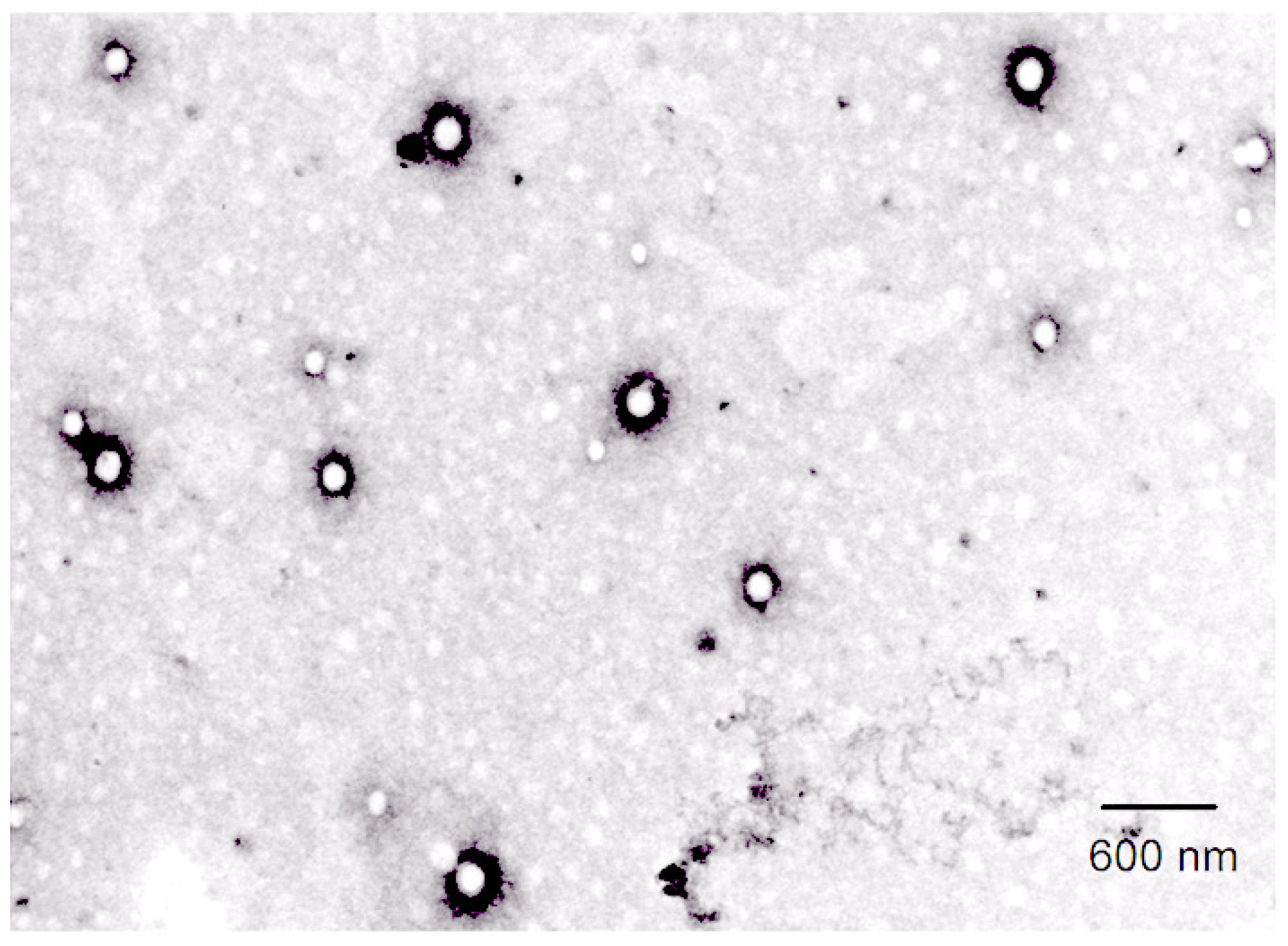
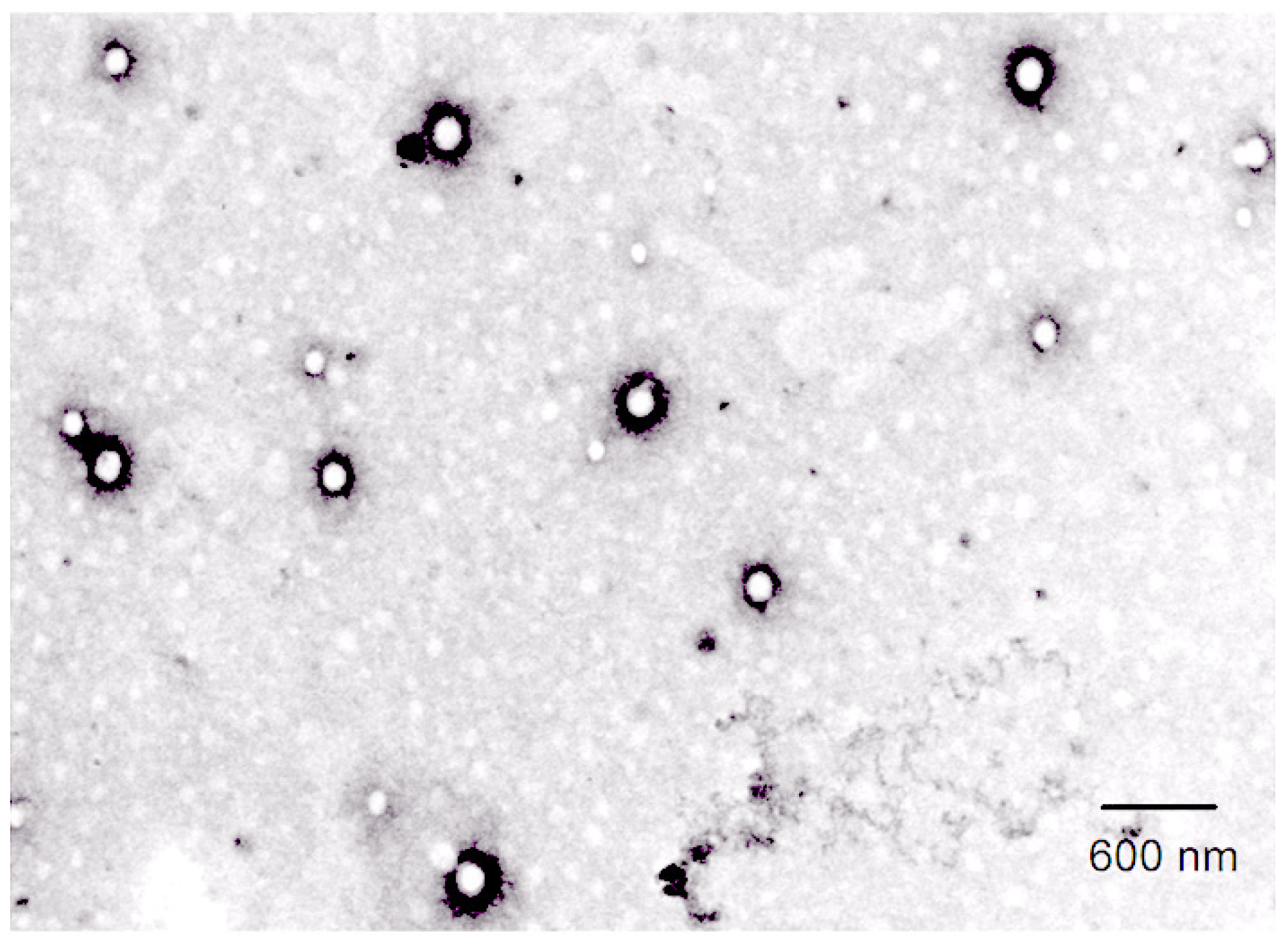
2.6. Characterization of α-MNG-PLGA NPs
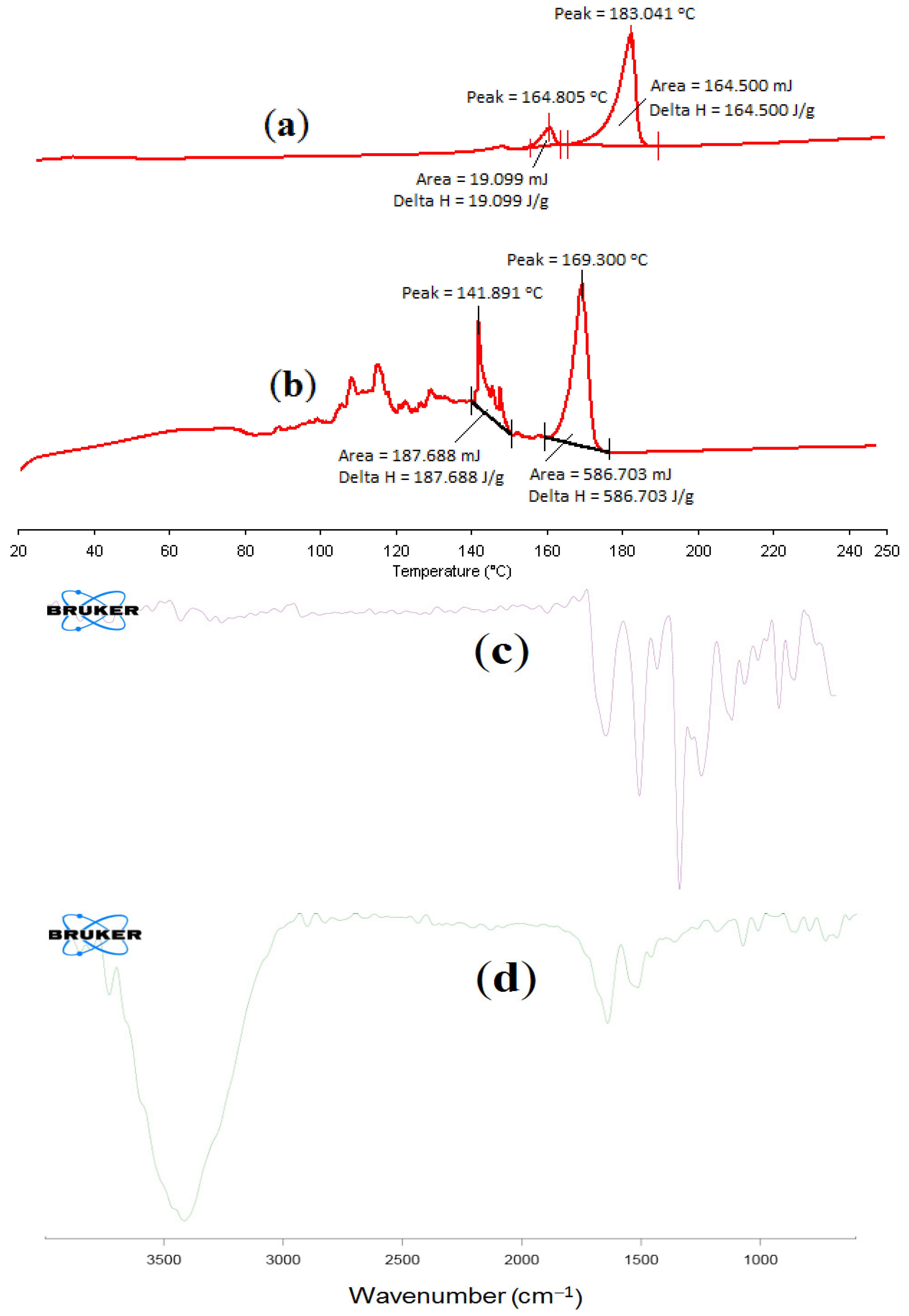
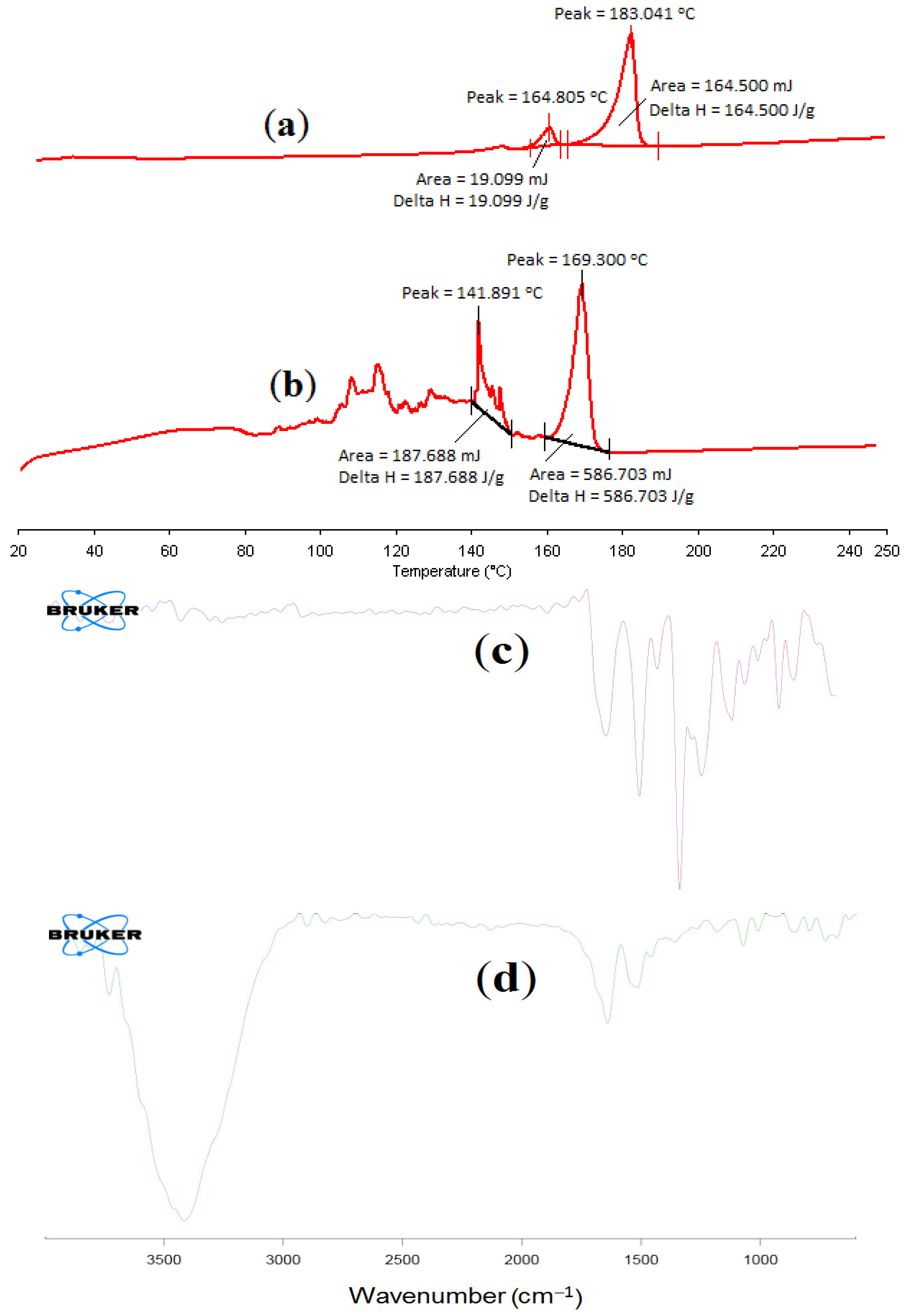
2.6.1. Differential Scanning Calorimetry of α-MNG NPs
2.6.2. FT-IR Spectral Analysis of α-MNG NPs
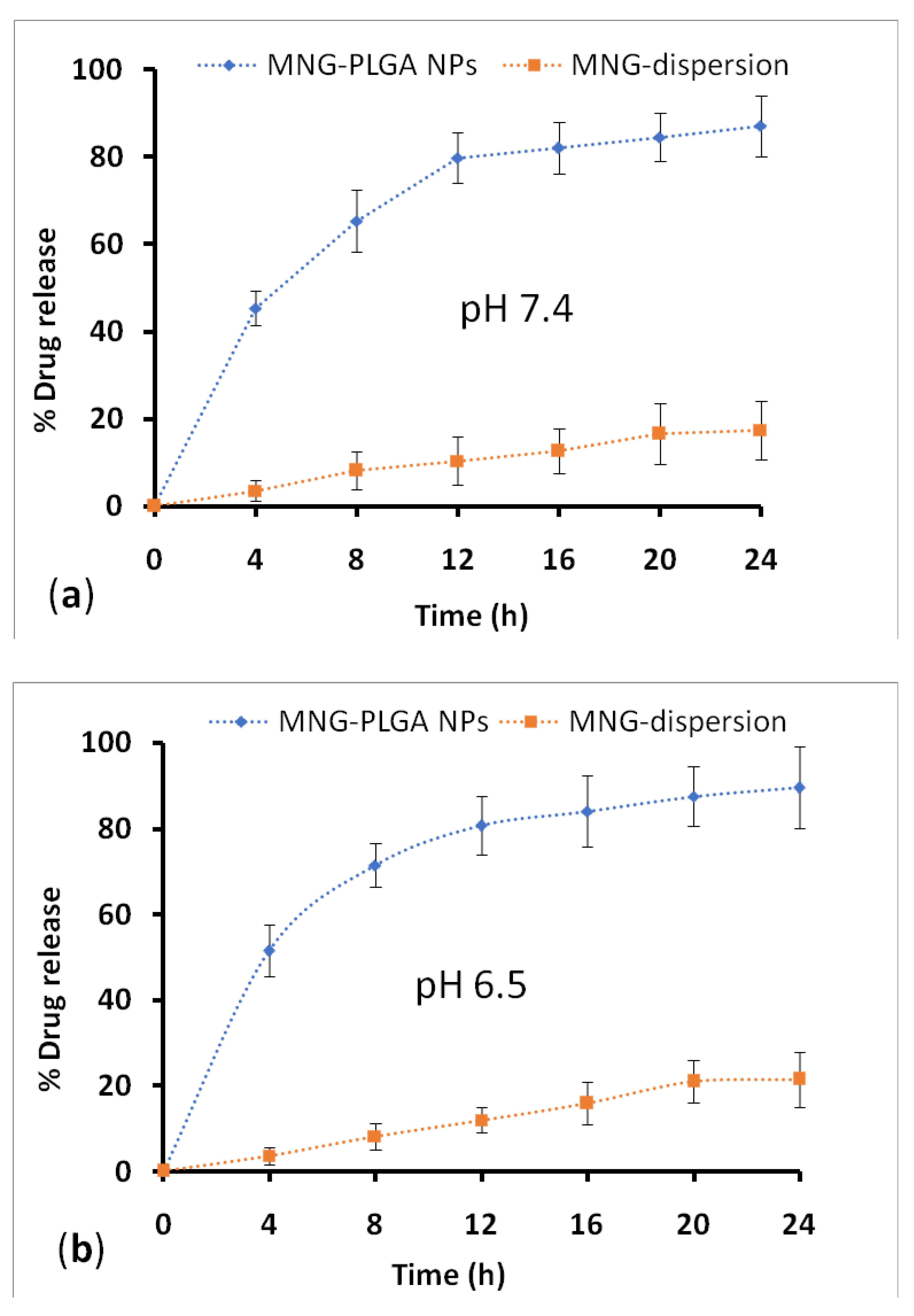
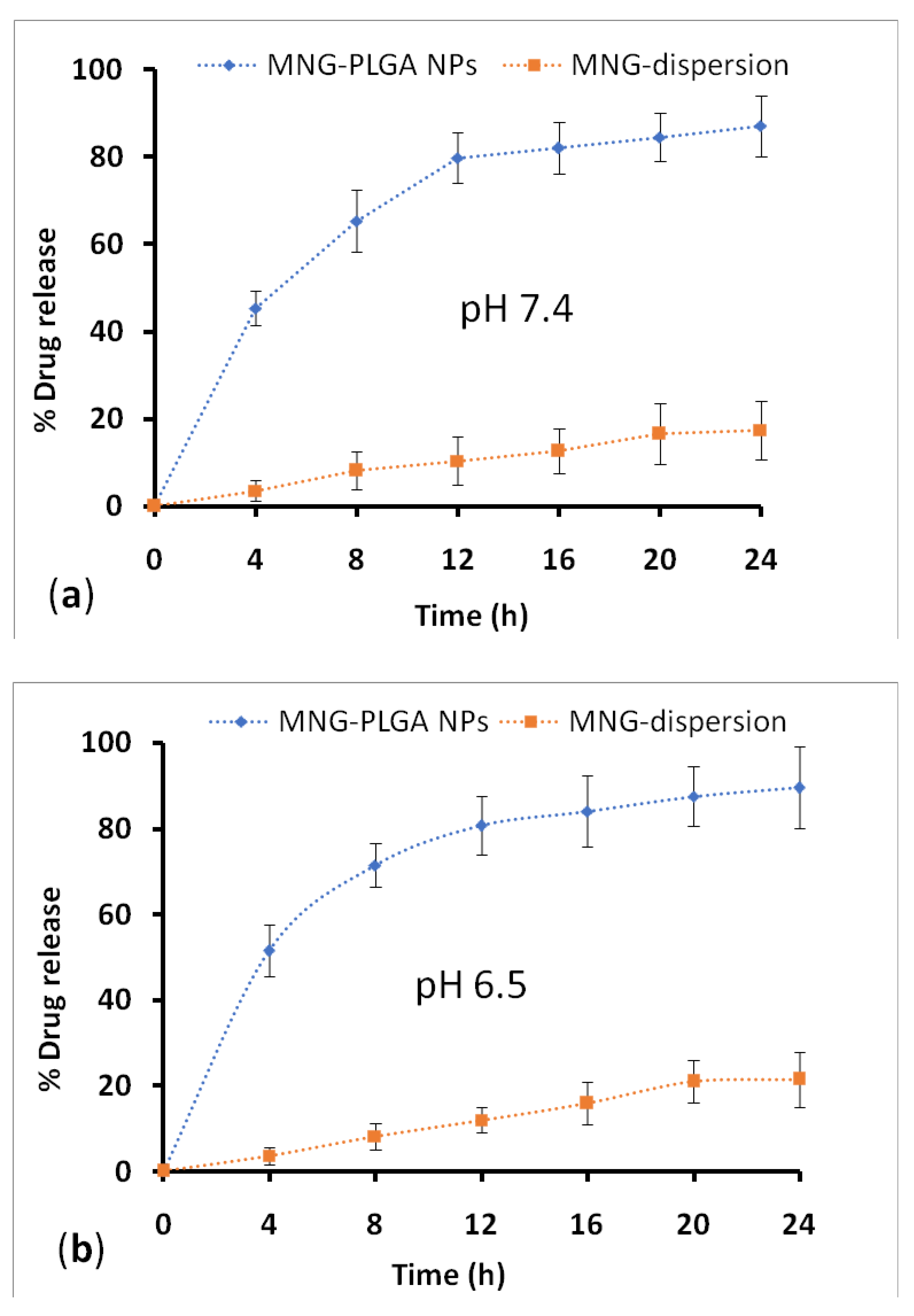
2.7. Drug Release and Kinetic Study
2.8. Gel Characteristics
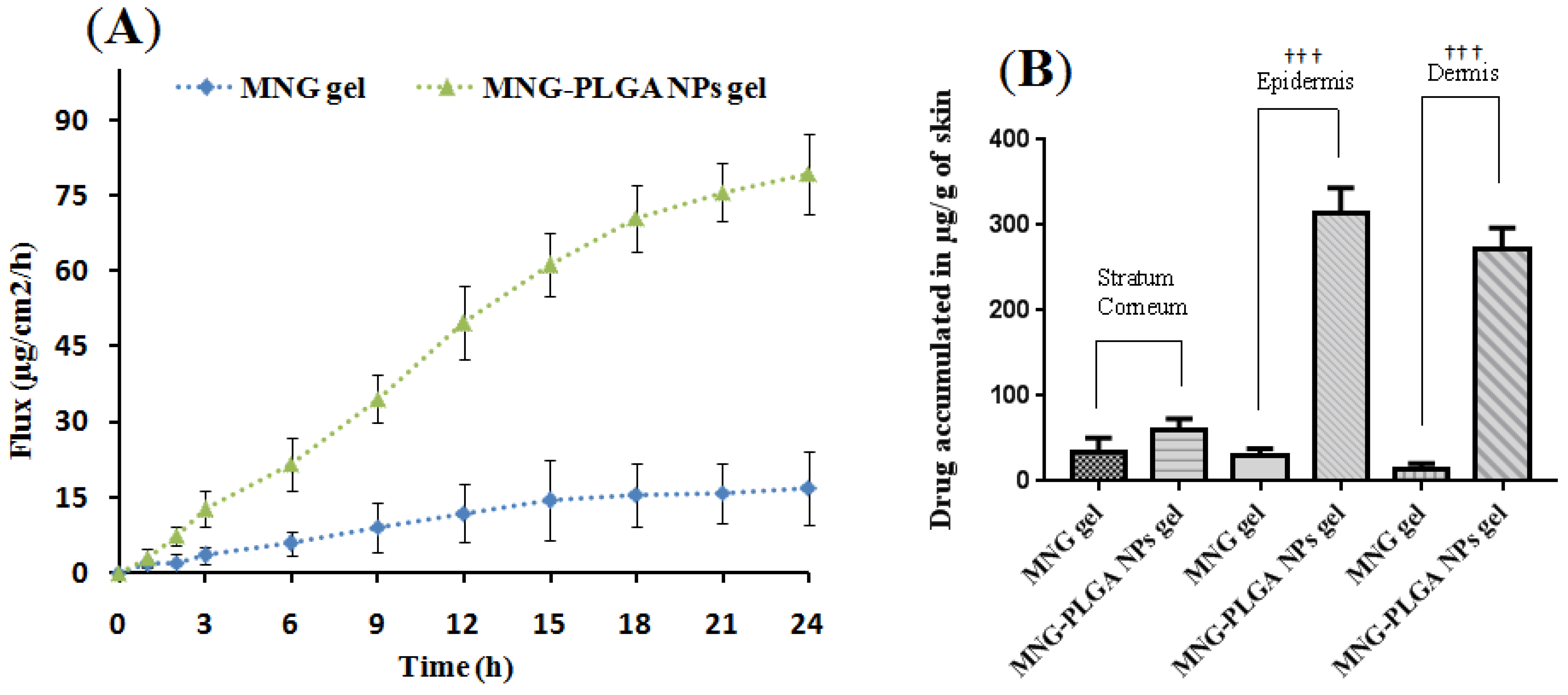
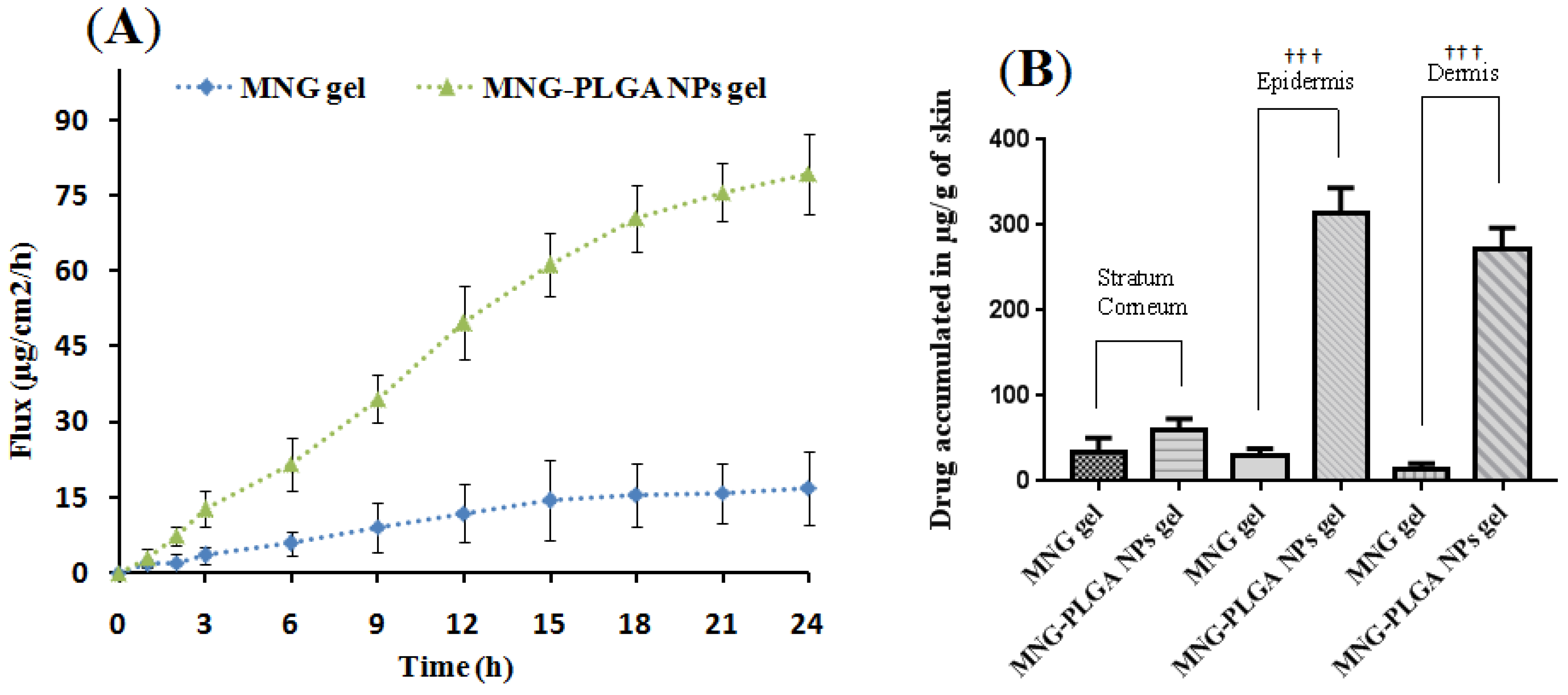
2.9. Ex Vivo Skin Permeation
Drug Estimation across Skin Layer
2.10. Confocal Laser Microscopy
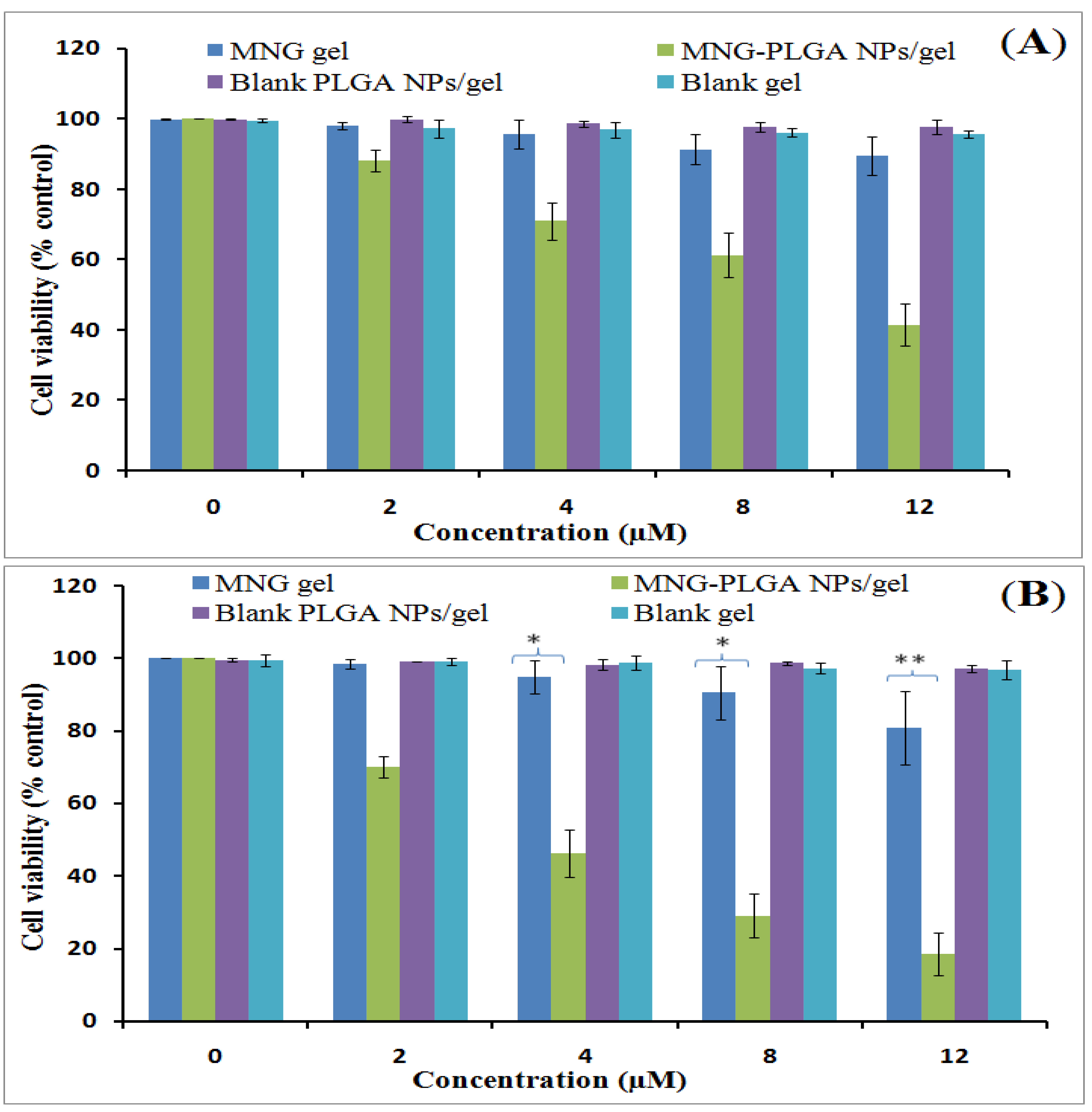
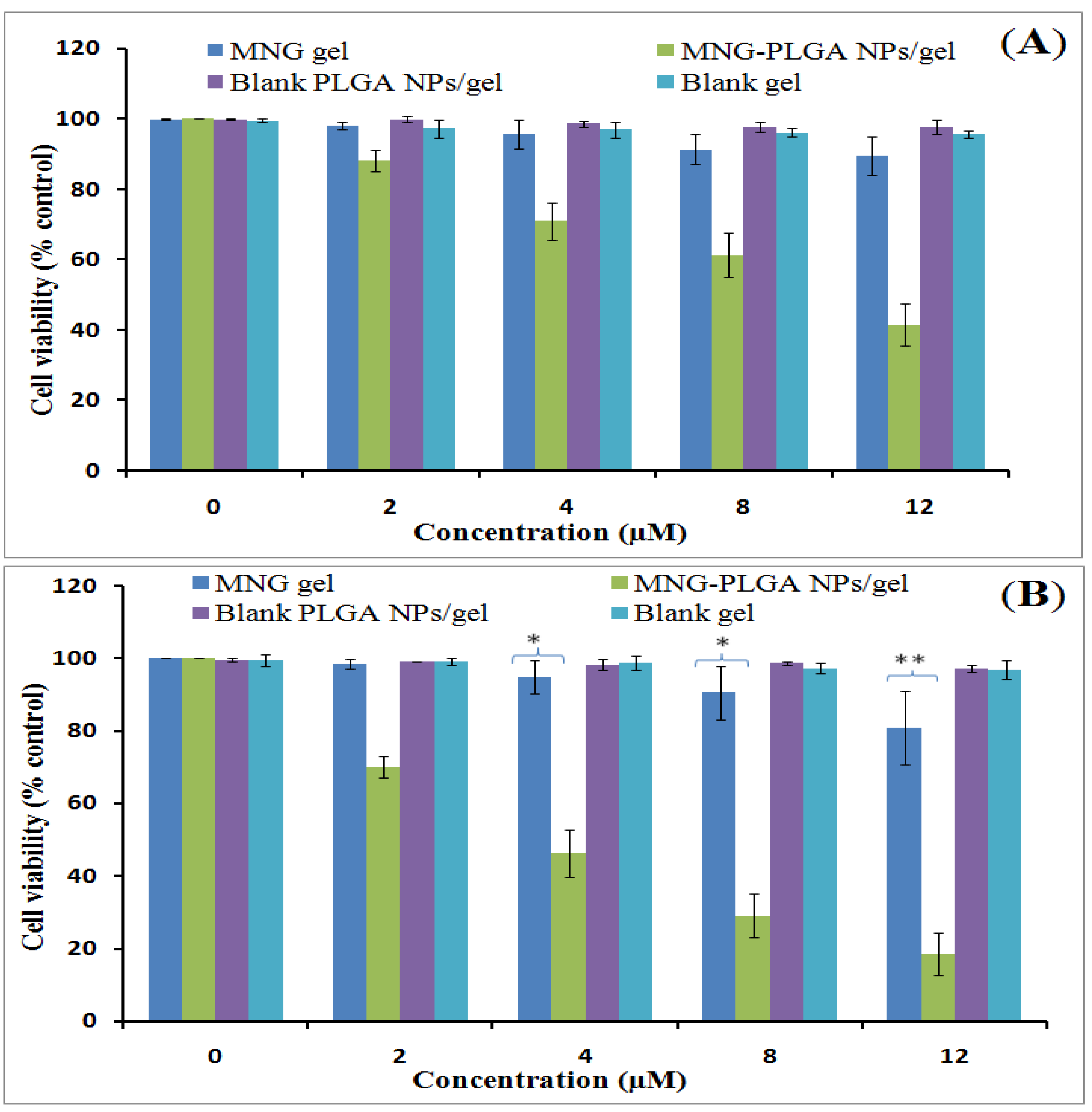
2.11. Cell Viability Assay
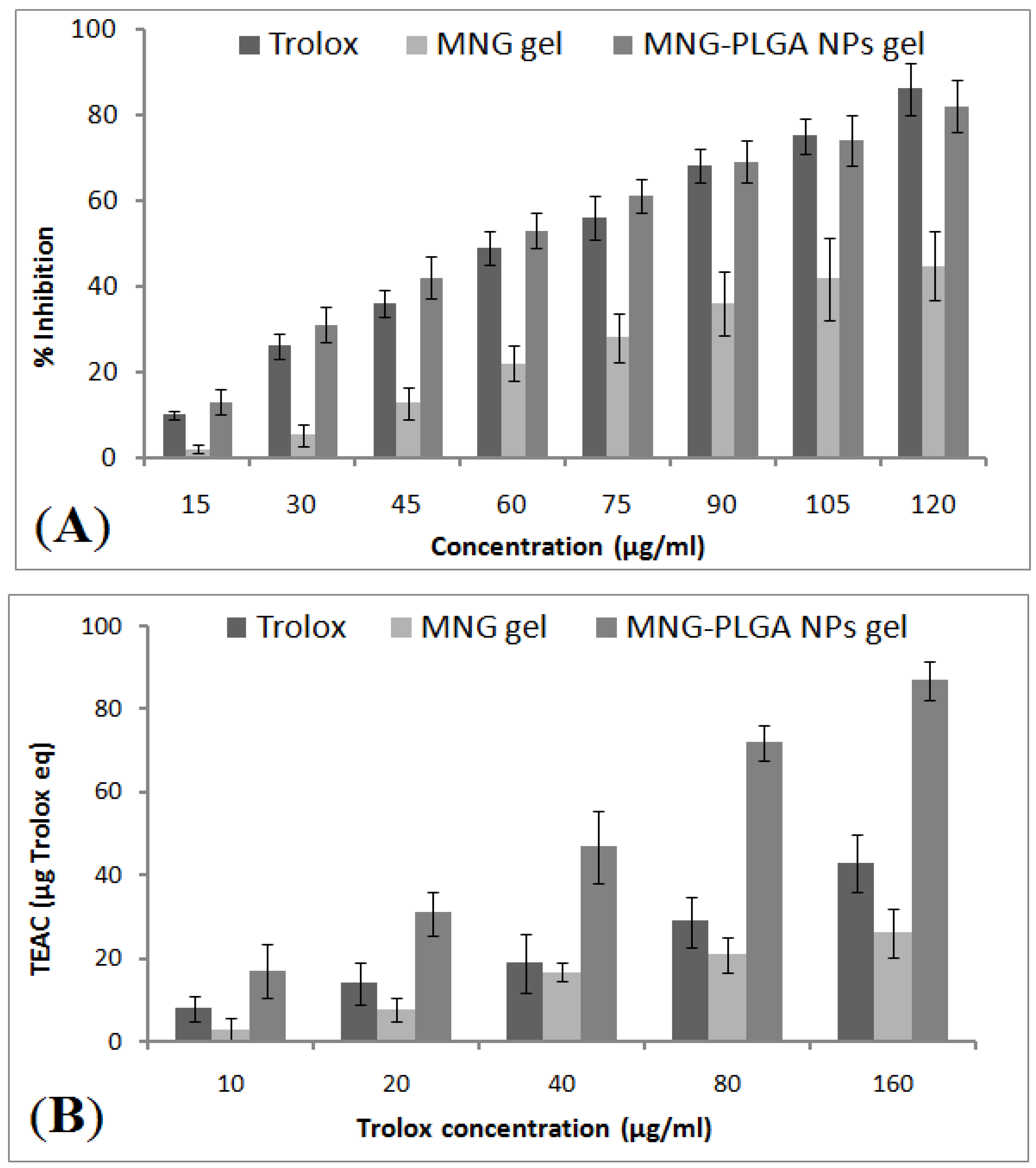
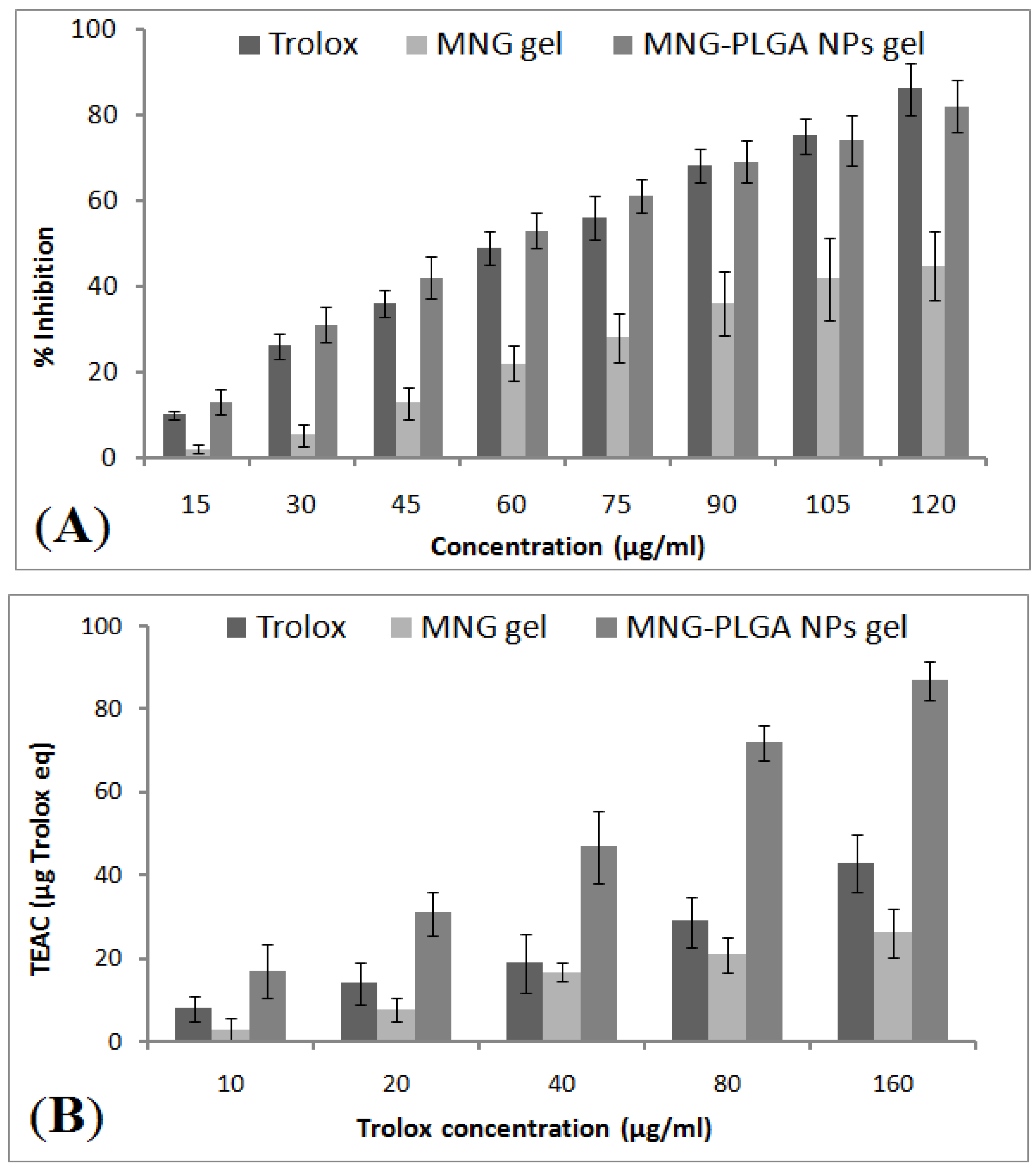
2.12. In Vitro Antioxidant Activity
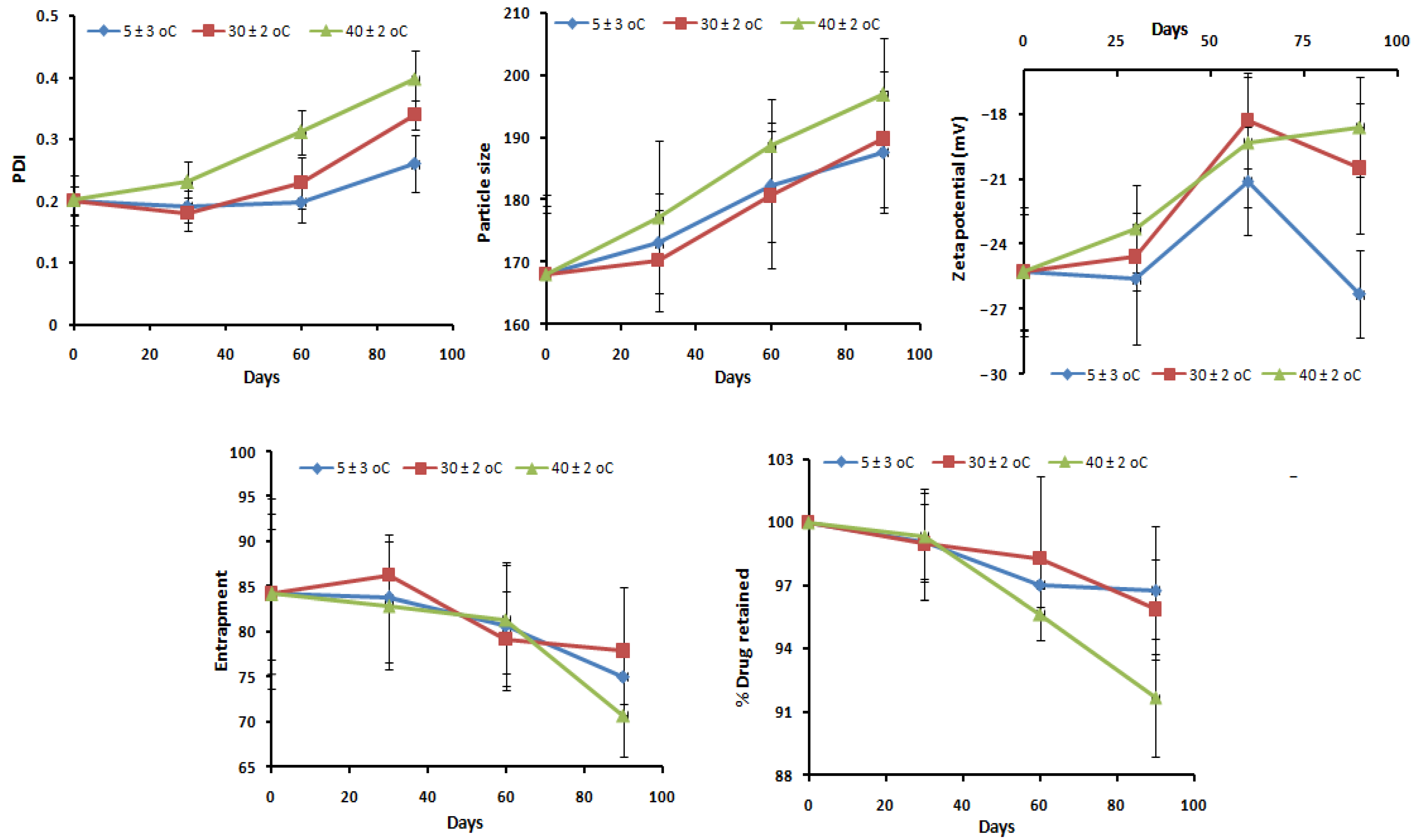
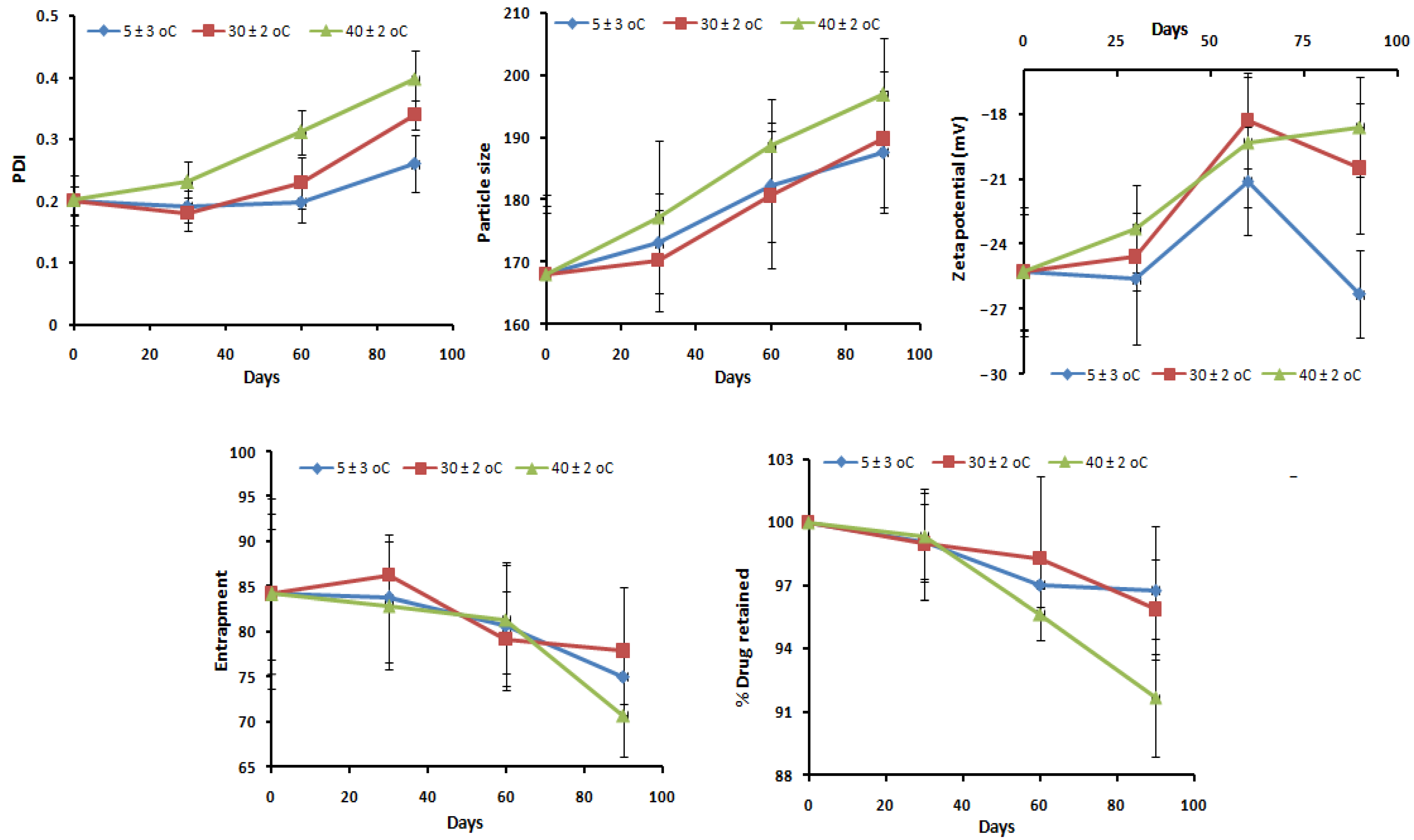
2.13. Stability Studies
3. Conclusions
4. Materials and Methods
4.1. Materials
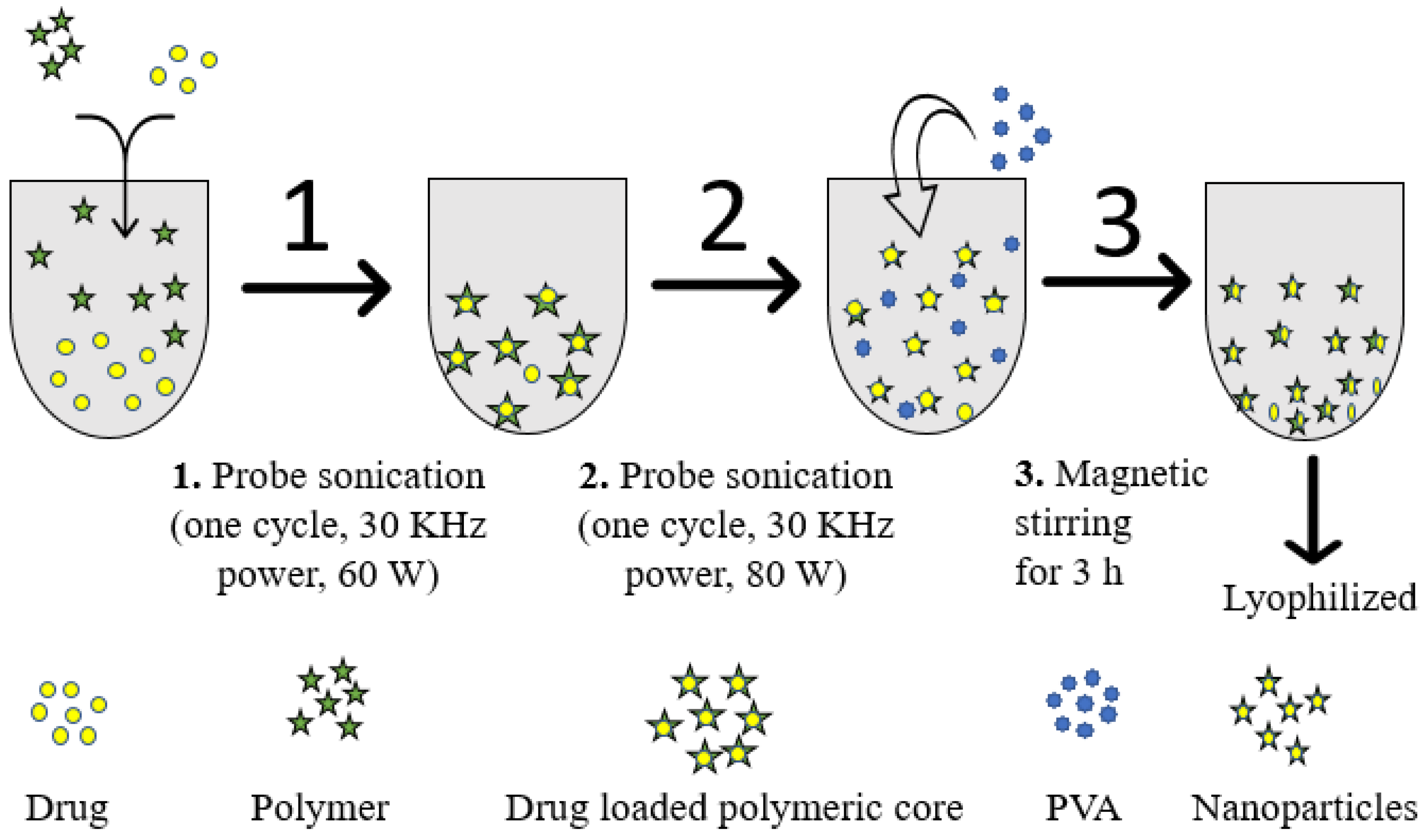
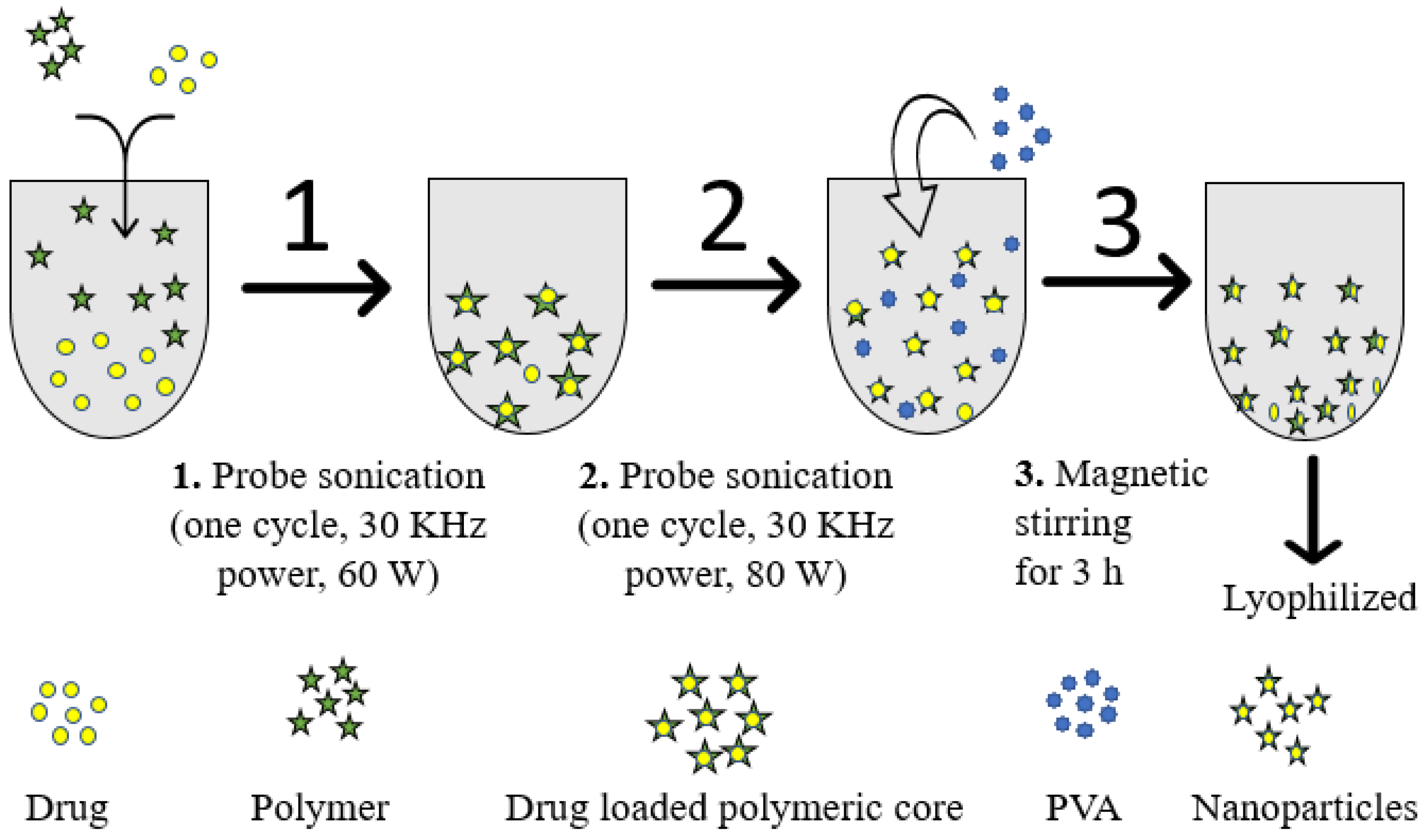
4.2. Fabrication of α-Mangostin-Loaded PLGA Nanoparticle
4.3. Analytical Method
4.4. Formulation Optimization
4.5. Nanoparticle Characterization
4.5.1. Particle Sizes and Their Distribution
4.5.2. Drug Entrapment and Loading
4.5.3. Transmission Electron Microscopy
4.5.4. Infrared Spectroscopy
4.5.5. Differential Scanning Calorimetry
4.5.6. Drug Release and Kinetics Studies
4.5.7. Preparation of Gel
Gel Characterization
4.5.8. Ex Vivo Skin Permeation Studies
4.5.9. Drug Concentration Estimation in Skin Strata
4.5.10. Confocal Microscopy
4.5.11. Cell Viability Study
4.5.12. Free Radical Scavenging Activity
4.5.13. Stability Study
4.5.14. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dianzani, C.; Zara, G.P.; Maina, G.; Pettazzoni, P.; Pizzimenti, S.; Rossi, F.; Gigliotti, C.L.; Ciamporcero, E.S.; Daga, M.; Barrera, G. Drug Delivery Nanoparticles in Skin Cancers. BioMed Res. Int. 2014, 2014, 895986. [Google Scholar] [CrossRef]
- Omran, A.R. The Epidemiologic Transition: A Theory of the Epidemiology of Population Change. Milbank Q. 2005, 83, 731–757. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, B.K.; Kricker, A. The epidemiology of UV induced skin cancer. J. Photochem. Photobiol. B Biol. 2001, 63, 8–18. [Google Scholar] [CrossRef]
- Esteva, A.; Kuprel, B.; Novoa, R.A.; Ko, J.; Swetter, S.M.; Blau, H.M.; Thrun, S. Dermatologist-level classification of skin cancer with deep neural networks. Nature 2017, 542, 115–118. [Google Scholar] [CrossRef]
- Narayanan, D.L.; Saladi, R.N.; Fox, J.L. Review: Ultraviolet radiation and skin cancer. Int. J. Dermatol. 2010, 49, 978–986. [Google Scholar] [CrossRef]
- Akhter, M.H.; Amin, S. An Investigative Approach to Treatment Modalities for Squamous Cell Carcinoma of Skin. Curr. Drug Deliv. 2017, 14, 597–612. [Google Scholar] [CrossRef]
- Curran, S.; Vantangoli, M.M.; Boekelheide, K.; Morgan, J.R. Architecture of Chimeric Spheroids Controls Drug Transport. Cancer Microenviron. 2015, 8, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Li, H.; Ma, Y.; Gong, H.; Yang, S.; Fang, Q.; Hu, Z. Nanoparticle abraxane possesses impaired proliferation in A549 cells due to the underexpression of glucosamine 6-phosphate N-acetyltransferase 1 (GNPNAT1/GNA1). Int. J. Nanomed. 2017, 12, 1685–1697. [Google Scholar] [CrossRef] [Green Version]
- Sahibzada, M.U.K.; Sadiq, A.; Faidah, H.S.; Khurram, M.; Amin, M.U.; Haseeb, A.; Kakar, M. Berberine nanoparticles with enhanced in vitro bioavailability: Characterization and antimicrobial activity. Drug Des. Dev. Ther. 2018, 12, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Gokhale, J.P.; Mahajan, H.S.; Surana, S.J. Quercetin loaded nanoemulsion-based gel for rheumatoid arthritis: In vivo and in vitro studies. Biomed. Pharmacother. 2019, 112, 108622. [Google Scholar] [CrossRef]
- Akhter, M.H.; Rizwanullah, M.; Ahmad, J.; Amin, S.; Ahmad, M.Z.; Minhaj, A.; Mujtaba, A.; Ali, J. Molecular Targets and Nanoparticulate Systems Designed for the Improved Therapeutic Intervention in Glioblastoma Multiforme. Drug Res. 2021, 71, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Akhter, M.H.; Beg, S.; Tarique, M.; Malik, A.; Afaq, S.; Choudhry, H.; Hosawi, S. Receptor-based targeting of engineered nanocarrier against solid tumors: Recent progress and challenges ahead. Biochim. Biophys. ActaBBA-Gen. Subj. 2021, 1865, 129777. [Google Scholar] [CrossRef] [PubMed]
- Barua, S.; Mitragotri, S. Challenges associated with penetration of nanoparticles across cell and tissue barriers: A review of current status and future prospects. Nano Today 2014, 9, 223–243. [Google Scholar] [CrossRef]
- Akhter, M.H.; Khalilullah, H.; Gupta, M.; Alfaleh, M.A.; Alhakamy, N.A.; Riadi, Y.; Shadab, M. Impact of Protein Corona on the Biological Identity of Nanomedicine: Understanding the Fate of Nanomaterials in the Biological Milieu. Biomedicines 2021, 9, 1496. [Google Scholar] [CrossRef]
- Li, C.; Obireddy, S.R.; Lai, W.-F. Preparation and use of nanogels as carriers of drugs. Drug Deliv. 2021, 28, 1594–1602. [Google Scholar] [CrossRef]
- Boinpelly, V.C.; Verma, R.K.; Srivastav, S.; Srivastava, R.K.; Shankar, S. α-Mangostin-encapsulated PLGA na-noparticles inhibit colorectal cancer growth by inhibiting Notch pathway. J. Cell. Mol. Med. 2020, 24, 11343–11354. [Google Scholar] [CrossRef]
- Rezvantalab, S.; Drude, N.; Moraveji, M.K.; Güvener, N.; Koons, E.K.; Shi, Y.; Lammers, T.; Kiessling, F. PLGA-Based Nanoparticles in Cancer Treatment. Front. Pharmacol. 2018, 9, 1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, A.T.C.R.; Cardoso, B.C.O.; e Silva, M.E.S.R.; Freitas, R.F.S.; Sousa, R.G. Synthesis, Characterization, and Study of PLGA Copolymer in Vitro Degradation. J. Biomater. Nanobiotechnol. 2015, 6, 8–19. [Google Scholar] [CrossRef] [Green Version]
- Rodeiro, I.; Delgado, R.; Garrido, G. Effects of a Mangiferaindica L. stem bark extract and mangiferin on radiation-induced DNA damage in human lymphocytes and lymphoblastoid cells. Cell Prolif. 2013, 47, 48–55. [Google Scholar] [CrossRef]
- Xiao, J.; Liu, L.; Zhong, Z.; Xiao, C.; Zhang, J. Mangiferin regulates proliferation and apoptosis in glioma cells by induction of microRNA-15b and inhibition of MMP-9 expression. Oncol. Rep. 2015, 33, 2815–2820. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Yao, Y.; Yang, J.; Tang, Y.; Huang, X. Mangiferin induces cell cycle arrest at G2/M phase through ATR-Chk1 pathway in HL-60 leukemia cells. Genet. Mol. Res. 2015, 14, 4989–5002. [Google Scholar] [CrossRef] [PubMed]
- Quan, X.; Wang, Y.; Ma, X.; Liang, Y.; Tian, W.; Ma, Q.; Jiang, H.; Zhao, Y. α-Mangostin Induces Apoptosis and Suppresses Differentiation of 3T3-L1 Cells via Inhibiting Fatty Acid Synthase. PLoS ONE 2012, 7, e33376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhter, M.H.; Rizwanullah, M.; Ahmad, J.; Ahsan, M.J.; Mujtaba, A.; Amin, S. Nanocarriers in advanced drug targeting: Setting novel paradigm in cancer therapeutics. Artif. Cells Nanomed. Biotechnol. 2018, 46, 873–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhter, M.H.; Madhav, N.S.; Ahmad, J. Epidermal growth factor receptor based active targeting: A paradigm shift towards advance tumor therapy. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1188–1198. [Google Scholar] [CrossRef] [Green Version]
- Pongsumpun, P.; Iwamoto, S.; Siripatrawan, U. Response surface methodology for optimization of cinnamon essential oil nanoemulsion with improved stability and antifungal activity. Ultrason. Sonochem. 2020, 60, 104604. [Google Scholar] [CrossRef] [PubMed]
- Kumbhar, S.A.; Kokare, C.R.; Shrivastava, B.; Gorain, B.; Choudhury, H. Preparation, characterization, and optimization of asenapine maleate mucoadhesivenanoemulsion using Box-Behnken design: In vitro and in vivo studies for brain targeting. Int. J. Pharm. 2020, 586, 119499. [Google Scholar] [CrossRef]
- Venugopal, V.; Kumar, K.J.; Muralidharan, S.; Parasuraman, S.; Raj, P.V. Optimization and in-vivo evaluation of isradipine nanoparticles using Box-Behnken design surface response methodology. OpenNano 2016, 1, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kausar, H.; Mujeeb, M.; Ahad, A.; Moolakkadath, T.; Aqil, M.; Ahmad, A.; Akhter, M.H. Optimization of ethosomes for topical thymoquinone delivery for the treatment of skin acne. J. Drug Deliv. Sci. Technol. 2019, 49, 177–187. [Google Scholar] [CrossRef]
- Md, S.; Alhakamy, N.; Aldawsari, H.; Husain, M.; Khan, N.; Alfaleh, M.; Asfour, H.; Riadi, Y.; Bilgrami, A.; Akhter, M.H. Plumbagin-Loaded Glycerosome Gel as Topical Delivery System for Skin Cancer Therapy. Polymers 2021, 13, 923. [Google Scholar] [CrossRef]
- Akhter, M.H.; Kumar, S.; Nomani, S. Sonication tailored enhance cytotoxicity of naringenin nanoparticle in pancreatic cancer: Design, optimization, and in vitro studies. Drug Dev. Ind. Pharm. 2020, 46, 659–672. [Google Scholar] [CrossRef]
- Soni, K.; Mujtaba, A.; Akhter, H.; Zafar, A.; Kohli, K. Optimisation of ethosomalnanogel for topical nano-CUR and sulphoraphane delivery in effective skin cancer therapy. J. Microencapsul. 2019, 37, 91–108. [Google Scholar] [CrossRef] [PubMed]
- Shkodra, B.; Grune, C.; Traeger, A.; Vollrath, A.; Schubert, S.; Fischer, D. Effect of surfactant on the size and stability of PLGA nanoparticles encapsulating a protein kinase C inhibitor. Int. J. Pharm. 2019, 566, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Rizwan, K.; Muhammad, A.; Sarfaraz, K.; Jiménez, A.N.; Park, D.R.; Yeom, I.T. The influence of ionic and nonionic surfactants on the colloidal stability and removal of CuO nanoparticles from water by chemical coagulation. Int. J. Environ. Res. Pub. Health 2019, 16, 1260. [Google Scholar] [CrossRef] [Green Version]
- Kim, Q.; Jhe, W. Interfacial thermodynamics of spherical nanodroplets: Molecular understanding of surface tension via a hydrogen bond network. Nanoscale 2020, 12, 18701–18709. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.; Hedberg, J.; Blomberg, E.; Wold, S.; Wallinder, I.O. Effect of sonication on particle dispersion, administered dose and metal release of non-functionalized, non-inert metal nanoparticles. J. Nanoparticle Res. 2016, 18, 285. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Giottonini, K.Y.; Rodríguez-Córdova, R.J.; Gutiérrez-Valenzuela, C.A.; Peñuñuri-Miranda, O.; Zavala-Rivera, P.; Guerrero-Germán, P.; Lucero-Acuña, A. PLGA nanoparticle preparations by emulsification and nanoprecipitation techniques: Effects of formulation parameters. RSC Adv. 2020, 10, 4218–4231. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, P.C.; Verma, A.; Al-Abassi, F.A.; Anwar, F.; Kumar, V.; Panda, B.P. Development of surface-engineered PLGA nanoparticulate-delivery system of Tet-1-conjugated nattokinase enzyme for inhibition of Aβ40 plaques in Alzheimer’s disease. Int. J. Nanomed. 2017, 12, 8749–8768. [Google Scholar] [CrossRef] [Green Version]
- Mudalige, T.; Qu, H.; Van Haute, D.; Ansar, S.M.; Paredes, A.; Ingle, T. Characterization of Nanomaterials. In Nanomaterials for Food Applications; Elsevier: Amsterdam, The Netherlands, 2019; pp. 313–353. [Google Scholar] [CrossRef]
- Sharma, N.; Madan, P.; Lin, S. Effect of process and formulation variables on the preparation of parenteral paclitaxel-loaded biodegradable polymeric nanoparticles: A co-surfactant study. Asian J. Pharm. Sci. 2016, 11, 404–416. [Google Scholar] [CrossRef] [Green Version]
- Budhian, A.; Siegel, S.J.; Winey, K.I. Haloperidol-loaded PLGA nanoparticles: Systematic study of particle size and drug content. Int. J. Pharm. 2007, 336, 367–375. [Google Scholar] [CrossRef]
- Español, L.; Larrea, A.; Andreu, V.; Mendoza, G.; Arruebo, M.; Sebastian, V.; Aurora-Prado, M.S.; Kedor-Hackmann, E.R.M.; Santoro, M.I.R.M.; Santamaria, J. Dual encapsulation of hydrophobic and hydrophilic drugs in PLGA nanoparticles by a single-step method: Drug delivery and cytotoxicity assays. RSC Adv. 2016, 6, 111060–111069. [Google Scholar] [CrossRef] [Green Version]
- Kızılbey, K. Optimization of Rutin-Loaded PLGA Nanoparticles Synthesized by Single-Emulsion Solvent Evaporation Method. ACS Omega 2019, 4, 555–562. [Google Scholar] [CrossRef]
- Raya, S.; Mishrab, A.; Mandal, T.K.; Sac, B.; Chakraborty, J. Optimization of the process parameters for fabrication of polymer coated layered double hydroxide-methotrexate nanohybrid for possible treatment of osteosarcoma. RSC Adv. 2015, 5, 102574–102592. [Google Scholar] [CrossRef]
- Liu, D.; Pan, H.; He, F.; Wang, X.; Li, J.; Yang, X.; Pan, W. Effect of particle size on oral absorption of carvedilolnanosuspensions: In vitro and invivoevaluation. Int. J. Nanomed. 2015, 10, 6425–6434. [Google Scholar] [CrossRef] [Green Version]
- Akhter, M.H.; Ahmad, A.; Ali, J.; Mohan, G. Formulation and Development of CoQ10-Loaded s-SNEDDS for Enhancement of Oral Bioavailability. J. Pharm. Innov. 2014, 9, 121–131. [Google Scholar] [CrossRef]
- Wathoni, N.; Rusdin, A.; Febriani, E.; Purnama, D.; Daulay, W.; Azhary, S.Y.; Panatarani, C.; Joni, I.M.; Lesmana, R.; Motoyama, K.; et al. Formulation and characterization of α-mangostin in chitosan nanoparticles coated by sodium alginate, sodium silicate, and polyethylene glycol. J. Pharm. Bioallied Sci. 2019, 11, S619–S627. [Google Scholar] [CrossRef]
- Tejamukti, E.P.; Setyaningsih, W.; Yasir, B.; Alam, G.; Rohman, A. Application of FTIR Spectroscopy and HPLC Combined with Multivariate Calibration for Analysis of Xanthones in Mangosteen Extracts. Sci. Pharm. 2020, 88, 35. [Google Scholar] [CrossRef]
- Cui, Y.-N.; Xu, Q.-X.; Davoodi, P.; Wang, D.-P.; Wang, C.-H. Enhanced intracellular delivery and controlled drug release of magnetic PLGA nanoparticles modified with transferrin. Acta Pharmacol. Sin. 2017, 38, 943–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azadi, A.; Rouini, M.-R.; Hamidi, M. Neuropharmacokinetic evaluation of methotrexate-loaded chitosan nanogels. Int. J. Biol. Macromol. 2015, 79, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Pandit, J.; Sultana, Y.; Aqil, M. Chitosan-coated PLGA nanoparticles of bevacizumab as novel drug delivery to target retina: Optimization, characterization, and in vitro toxicity evaluation. Artif. Cells Nanomed. Biotechnol. 2017, 45, 1397–1407. [Google Scholar] [CrossRef] [Green Version]
- Körber, M. PLGA Erosion: Solubility- or Diffusion-Controlled? Pharm. Res. 2010, 27, 2414–2420. [Google Scholar] [CrossRef]
- Siegel, S.; Kahn, J.B.; Metzger, K.; Winey, K.I.; Werner, K.; Dan, N. Effect of drug type on the degradation rate of PLGA matrices. Eur. J. Pharm. Biopharm. 2006, 64, 287–293. [Google Scholar] [CrossRef]
- Yostawonkul, J.; Surassmo, S.; Namdee, K.; Khongkow, M.; Boonthum, C.; Pagseesing, S.; Saengkrit, N.; Ruktanonchai, U.R.; Chatdarong, K.; Ponglowhapan, S.; et al. Nanocarrier-mediated delivery of α-mangostin for non-surgical castration of male animals. Sci. Rep. 2017, 7, 16234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurman, S.; Yulia, R.; Noor, E.; Sunarti, T.C. IrmayantiThe Optimization of Gel Preparations Using the Active Compounds of Arabica Coffee Ground Nanoparticles. Sci. Pharm. 2019, 87, 32. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, J.; Ahmad, M.Z.; Akhter, H. Surface-Engineered Cancer Nanomedicine: Rational Design and Recent Progress. Curr. Pharm. Des. 2020, 26, 1181–1190. [Google Scholar] [CrossRef]
- Akhter, M.H.; Ahsan, M.J.; Rahman, M.; Anwar, S.; Rizwanullah, M. Advancement in Nanotheranostics for Effective Skin Cancer Therapy: State of the Art. Curr. Nanomed. 2020, 10, 90–104. [Google Scholar] [CrossRef]
- Ahmad, M.Z.; Rizwanullah, M.; Ahmad, J.; Alasmary, M.Y.; Akhter, M.H.; Abdel-Wahab, B.A.; Warsi, M.H.; Haque, A. Progress in nanomedicine-based drug delivery in designing of chitosan nanoparticles for cancer therapy. Int. J. Polym. Mater. 2021, 1–22. [Google Scholar] [CrossRef]
- Zheng, Y.; Ouyang, W.-Q.; Wei, Y.-P.; Syed, S.F.; Hao, C.-S.; Wang, B.-Z.; Shang, Y.-H. Effects of Carbopol® 934 proportion on nanoemulsion gel for topical and transdermal drug delivery: A skin permeation study. Int. J. Nanomed. 2016, 11, 5971–5987. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, C.; Sahoo, S.K. The in vitro stability and in vivo pharmacokinetics of curcumin prepared as an aqueous nanoparticulate formulation. Biomaterials 2010, 31, 6597–6611. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Sanderson, B.J.; Zhang, W. Cytotoxic effect of xanthones from pericarp of the tropical fruit mangosteen (Garciniamangostana Linn.) on human melanoma cells. Food Chem. Toxicol. 2011, 49, 2385–2391. [Google Scholar] [CrossRef]
- Wang, J.J.; Sanderson, B.J.; Zhang, W. Significant anti-invasive activities of α-mangostin from the mangosteenperi-carp on two human skin cancer cell lines. Anticancer. Res. 2012, 32, 3805–3816. [Google Scholar]
- Gan, Q.; Wang, T. Chitosan nanoparticle as protein delivery carrier—Systematic examination of fabrication conditions for efficient loading and release. Colloids Surf. B Biointerfaces 2007, 59, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, A.; Kazmi, I. Dacarbazine nanoparticle topical delivery system for the treatment of melanoma. Sci. Rep. 2017, 7, 16517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loffler, H.; Dreher, F.; Maibach, H. Stratum corneum adhesive tape stripping: Influence of anatomical site, application pressure, duration and removal. Br. J. Dermatol. 2004, 151, 746–752. [Google Scholar] [CrossRef]
- Olesen, C.M.; Fuchs, C.S.K.; Philipsen, P.A.; Hædersdal, M.; Agner, T.; Clausen, M.-L. Advancement through epidermis using tape stripping technique and Reflectance Confocal Microscopy. Sci. Rep. 2019, 9, 12217. [Google Scholar] [CrossRef]
- Lee, H.N.; Jang, H.Y.; Kim, H.J.; Shin, S.A.; Choo, G.S.; Park, Y.S.; Kim, S.K.; Jung, J.Y. Antitumor and apoptosis-inducing effects of α-mangostin extracted from the pericarp of the mangosteen fruit (Garciniamangostana L.)in YD-15 tongue mucoepidermoid carcinoma cells. Int. J. Mol. Med. 2016, 37, 939–948. [Google Scholar] [CrossRef] [Green Version]
- Krishnakumar, N.; Sulfikkarali, N.; Prasad, R.; Karthikeyan, S. Enhanced anticancer activity of naringenin-loaded nanoparticles in human cervical (HeLa) cancer cells. Biomed. Prev. Nutr. 2011, 1, 223–231. [Google Scholar] [CrossRef]
- Zeng, X.; Luo, M.; Liu, G.; Wang, X.; Tao, W.; Lin, Y.; Ji, X.; Nie, L.; Mei, L. Polydopamine-Modified Black Phosphorous Nanocapsule with Enhanced Stability and Photothermal Performance for Tumor Multimodal Treatments. Adv. Sci. 2018, 5, 1800510. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, N.A.; Zaidel, D.N.A.; Muhamad, I.I.; Hamid, M.A.; Yaakob, H.; Jusoh, Y.M.M. Optimization of the antioxidant-rich xanthone extract from mangosteen (Garciniamangostana L.) pericarp via microwave-assisted extraction. Heliyon 2019, 5, e02571. [Google Scholar] [CrossRef] [Green Version]
- Phan, T.K.T.; Tran, T.Q.; Pham, D.T.N.; Nguyen, D.T. Characterization, Release Pattern, and Cytotoxicity of Liposomes Loaded With α-Mangostin Isolated From Pericarp of Mangosteen (Garciniamangostana L.). Nat. Prod. Commun. 2020, 15, 1–8. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Variables | Level Used (Coded) | ||
|---|---|---|---|
| Low (−1) | Medium (0) | High (+1) | |
| X1: Polymer concentration (% w/v) | 2.5 | 3.00 | 3.50 |
| X2: Surfactant concentration (% w/v) | 1.00 | 1.75 | 2.50 |
| X3: Sonication time (min) | 4 | 8 | 12 |
| Dependent variables | |||
| Y1: Particle size (nm) | Minimize | ||
| Y2: PDI | Minimize | ||
| Y3: Entrapment efficiency (%) | Maximize | ||
| Formulation Code | Independent Variables | Responses | ||||
|---|---|---|---|---|---|---|
| X1 (% w/v) | X2 (% w/v) | X3 (min) | Y1 (nm) | Y2 | Y3 (%) | |
| * NP1 | 3.00 | 1.75 | 8.00 | 176 ± 19.04 | 0.205 ± 0.02 | 78 ± 8.67 |
| NP2 | 2.50 | 1.00 | 8.00 | 187 ± 24.12 | 0.302 ± 0.03 | 68 ± 6.12 |
| * NP3 | 3.00 | 1.75 | 8.00 | 171 ± 15.67 | 0.210 ± 0.04 | 79 ± 9.34 |
| NP4 | 3.50 | 1.75 | 12.00 | 167 ± 13.45 | 0.233 ± 0.02 | 83 ± 7.98 |
| * NP5 | 3.00 | 1.75 | 8.00 | 172 ± 21.81 | 0.206 ± 0.01 | 74.9 ± 6.80 |
| NP6 | 3.00 | 2.50 | 12.00 | 125 ± 10.01 | 0.276 ± 0.05 | 75 ± 10.07 |
| NP7 | 2.50 | 1.75 | 4.00 | 230 ± 33.76 | 0.281 ± 0.01 | 51 ± 9.23 |
| NP8 | 3.50 | 1.00 | 8.00 | 172 ± 11.93 | 0.215 ± 0.03 | 83 ± 9.65 |
| NP9 | 3.00 | 2.50 | 4.00 | 198 ± 19.62 | 0.306 ±0.04 | 72 ± 11.92 |
| NP10 | 2.50 | 2.50 | 8.00 | 167 ± 17.12 | 0.321 ± 0.02 | 53 ± 4.47 |
| * NP11 | 3.00 | 1.75 | 8.00 | 173 ± 16.02 | 0.199 ± 0.01 | 76 ± 11.21 |
| NP12 | 3.50 | 2.50 | 8.00 | 162 ± 14.03 | 0.268 ± 0.02 | 90 ± 12.76 |
| NP13 | 3.00 | 1.00 | 4.00 | 194 ± 24.98 | 0.203 ± 0.07 | 74 ± 9.67 |
| NP14 | 3.50 | 1.75 | 4.00 | 174 ± 17.54 | 0.222 ± 0.09 | 81 ± 8.78 |
| NP15 | 3.00 | 1.00 | 12.00 | 166 ± 19.32 | 0.312 ± 0.06 | 84 ± 7.43 |
| * NP16 | 3.00 | 1.75 | 8.00 | 172 ± 21.78 | 0.212 ± 0.03 | 77 ± 9.56 |
| NP17 | 2.50 | 1.75 | 12.00 | 132 ± 12.67 | 0.321 ± 0.05 | 61 ± 7.12 |
| Response Surface Quadratic Model | R-Squared | Adj R-Squared | Pred R-Squared | Adeq Precision | PRESS | % CV | Mean | SD |
|---|---|---|---|---|---|---|---|---|
| Response 1 (Y1) | 0.9970 | 0.9940 | 0.9853 | 79.146 | 128.59 | 1.04 | 172.82 | 1.81 |
| Response 2 (Y2) | 0.9942 | 0.9867 | 0.9476 | 28.134 | 1.874 × 10−3 | 2.16 | 0.25 | 5.457 × 10−3 |
| Response 3 (Y3) | 0.9935 | 0.9852 | 0.9799 | 39.150 | 36.26 | 1.74 | 74.11 | 1.29 |
| PS = +173.16 – 5.12 × X1 – 8.37 × X2 – 25.75 × X3 + 2.50 × X1 × X2 + 22.75 × X1 × X3 – 11.25 × X2 × X3 + 2.14 × X12 – 2.86 × X22 | ||||||||
| PDI = +0.21 – 0.036 × X1 + 0.017 × X2 + 0.016 × X3 + 8.500E − 0.03 × X1 × X2 – 7.250 – 0.03 × X1 × X3 – 0.035 × X2 × X3 + 0.03 × X12 + 0.040 × X22 + 0.028 × X32 | ||||||||
| EE = +76.98 + 13.00 × X1 − 2.37 × X2 + 3.13 × X3 + 5.50 × X1 × X2 − 2.00 × X1 × X3 – 1.75 × X2 × X3 − 5.37 × X12 + 1.88 × X22 − 2.61 × X32 | ||||||||
| Variables | Optimum Composition | Response | Observed Value of Response | Predicted Value of Response | Percentage Error |
|---|---|---|---|---|---|
| X1 | 3.39 % w/v | Y1 | 168.06 ± 17.02 | 150.87 | 11.39 |
| X2 | 1.82 % w/v | Y2 | 0.201 ± 0.01 | 0.214 | −6.07 |
| X3 | 8.79 min | Y3 | 84.26 ± 8.23 | 79.16 | 6.44 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Md, S.; Alhakamy, N.A.; Neamatallah, T.; Alshehri, S.; Mujtaba, M.A.; Riadi, Y.; Radhakrishnan, A.K.; Khalilullah, H.; Gupta, M.; Akhter, M.H. Development, Characterization, and Evaluation of α-Mangostin-Loaded Polymeric Nanoparticle Gel for Topical Therapy in Skin Cancer. Gels 2021, 7, 230. https://doi.org/10.3390/gels7040230
Md S, Alhakamy NA, Neamatallah T, Alshehri S, Mujtaba MA, Riadi Y, Radhakrishnan AK, Khalilullah H, Gupta M, Akhter MH. Development, Characterization, and Evaluation of α-Mangostin-Loaded Polymeric Nanoparticle Gel for Topical Therapy in Skin Cancer. Gels. 2021; 7(4):230. https://doi.org/10.3390/gels7040230
Chicago/Turabian StyleMd, Shadab, Nabil A. Alhakamy, Thikryat Neamatallah, Samah Alshehri, Md Ali Mujtaba, Yassine Riadi, Ammu K. Radhakrishnan, Habibullah Khalilullah, Manish Gupta, and Md Habban Akhter. 2021. "Development, Characterization, and Evaluation of α-Mangostin-Loaded Polymeric Nanoparticle Gel for Topical Therapy in Skin Cancer" Gels 7, no. 4: 230. https://doi.org/10.3390/gels7040230
APA StyleMd, S., Alhakamy, N. A., Neamatallah, T., Alshehri, S., Mujtaba, M. A., Riadi, Y., Radhakrishnan, A. K., Khalilullah, H., Gupta, M., & Akhter, M. H. (2021). Development, Characterization, and Evaluation of α-Mangostin-Loaded Polymeric Nanoparticle Gel for Topical Therapy in Skin Cancer. Gels, 7(4), 230. https://doi.org/10.3390/gels7040230