Thiol-Mediated Chemoselective Strategies for In Situ Formation of Hydrogels

Abstract
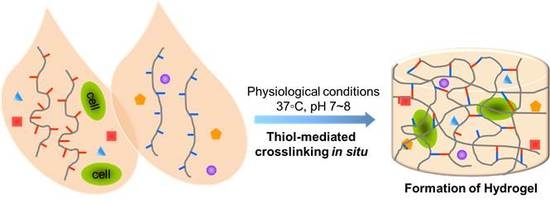
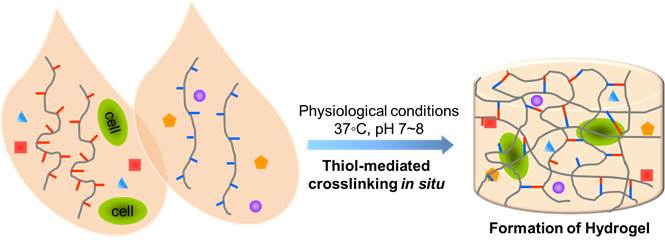
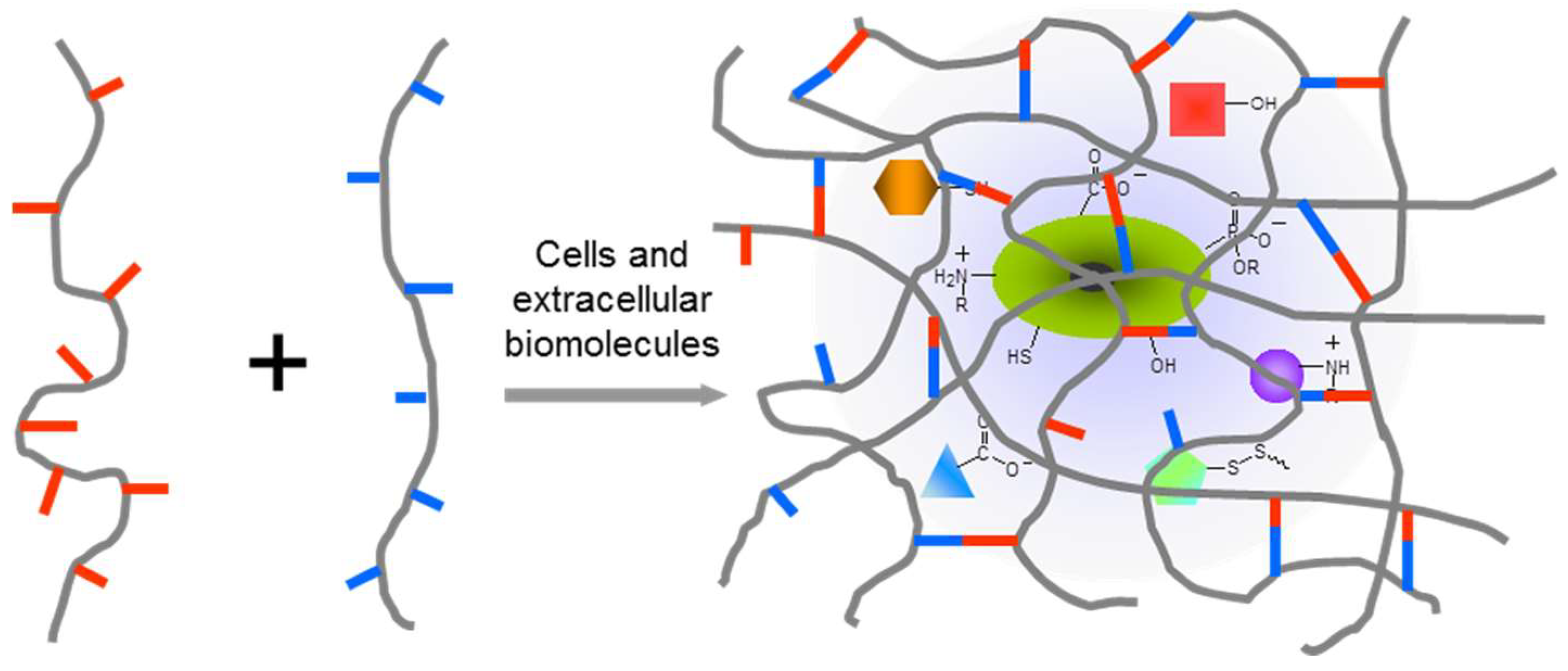
1. Introduction
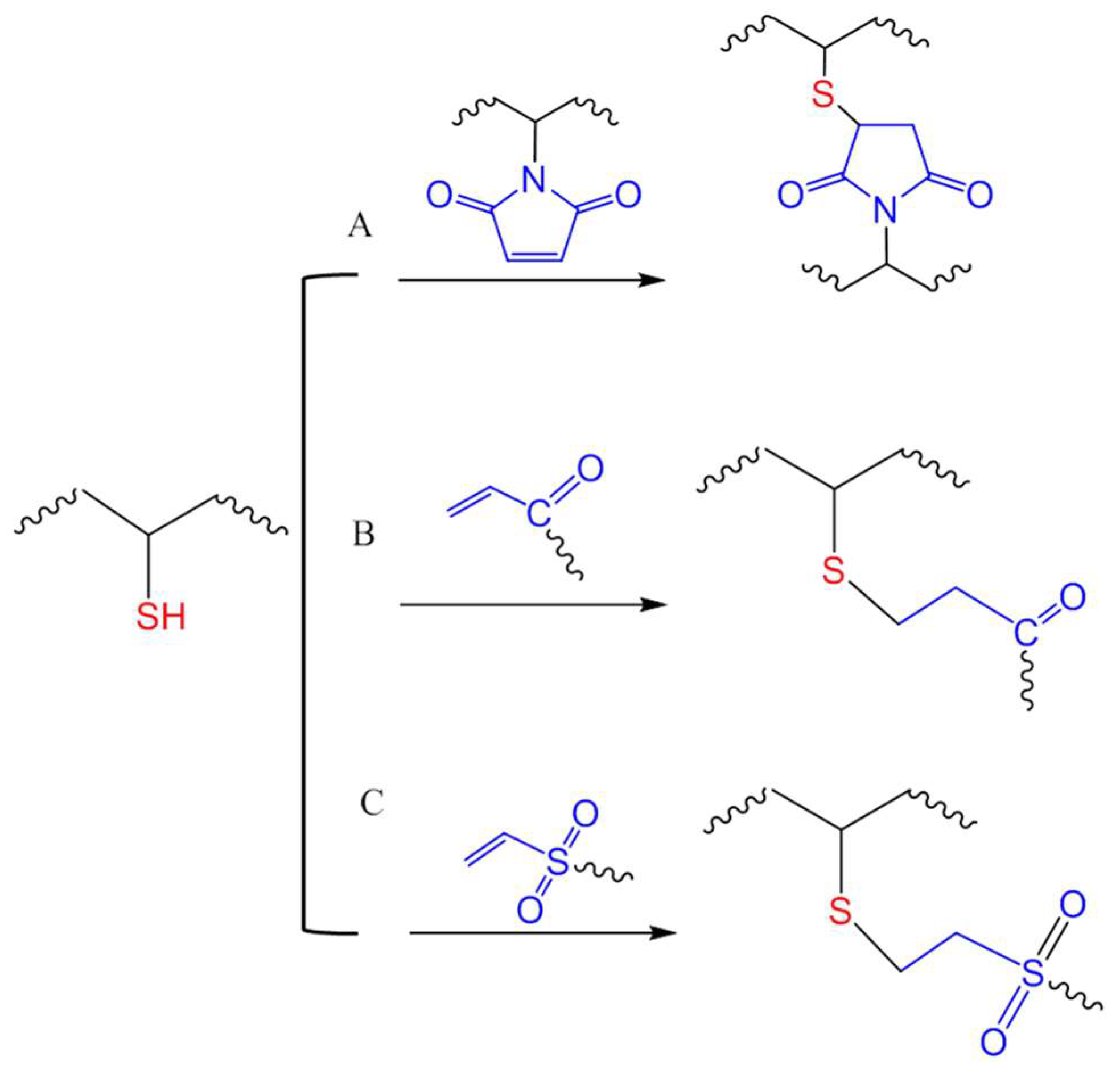
2. Reactivity of the Thiol Group
3. Substitution Reactions of Thiols for Hydrogel Crosslinking
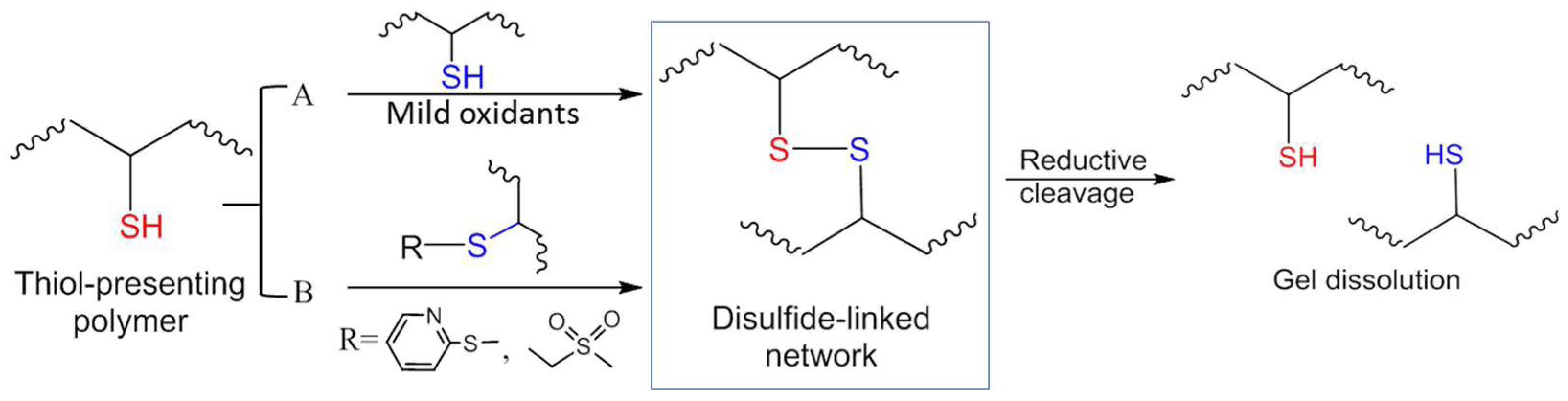
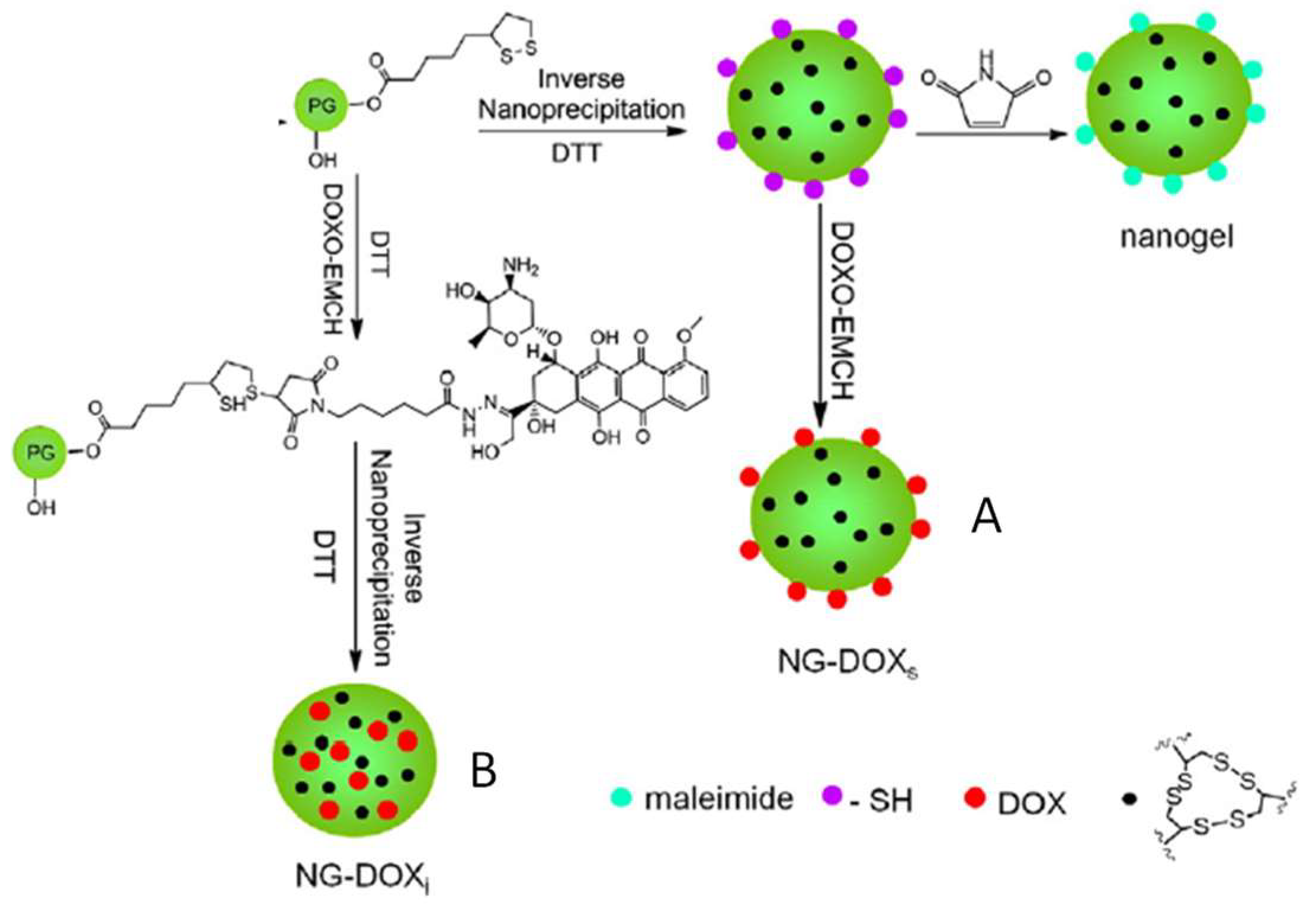
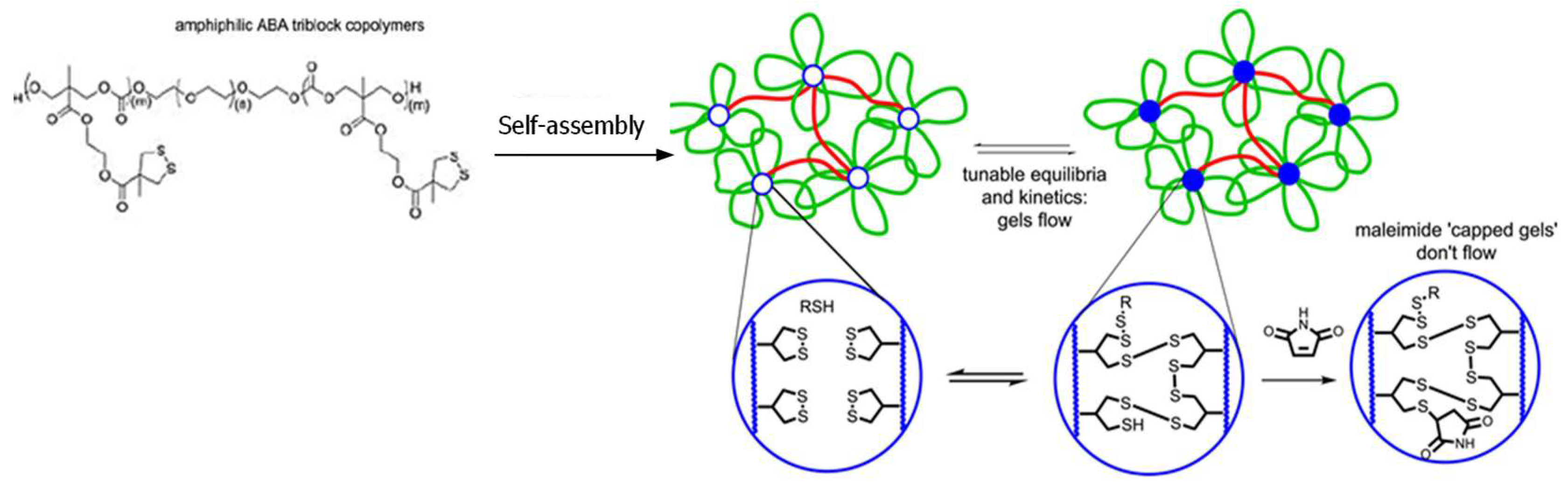
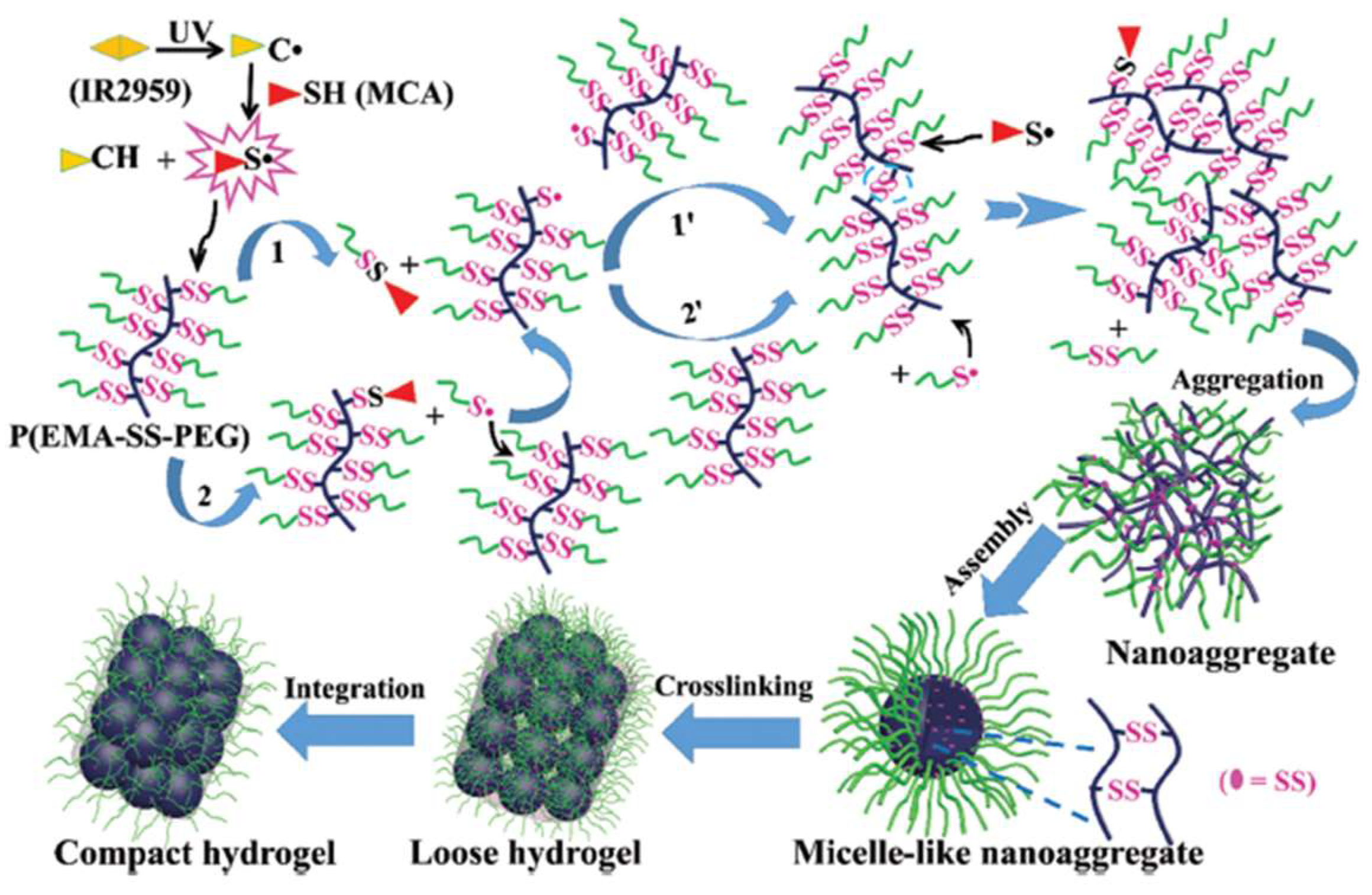
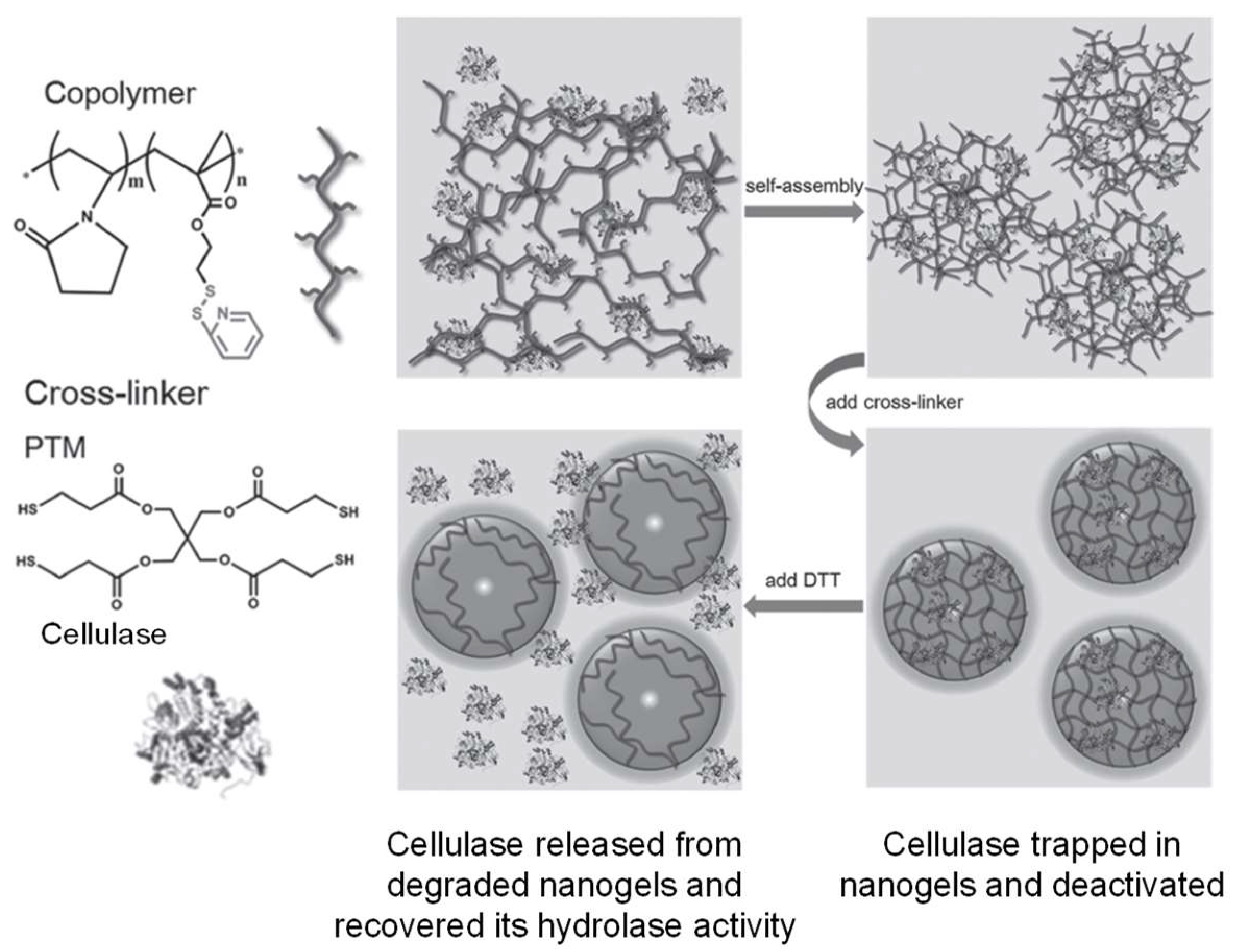
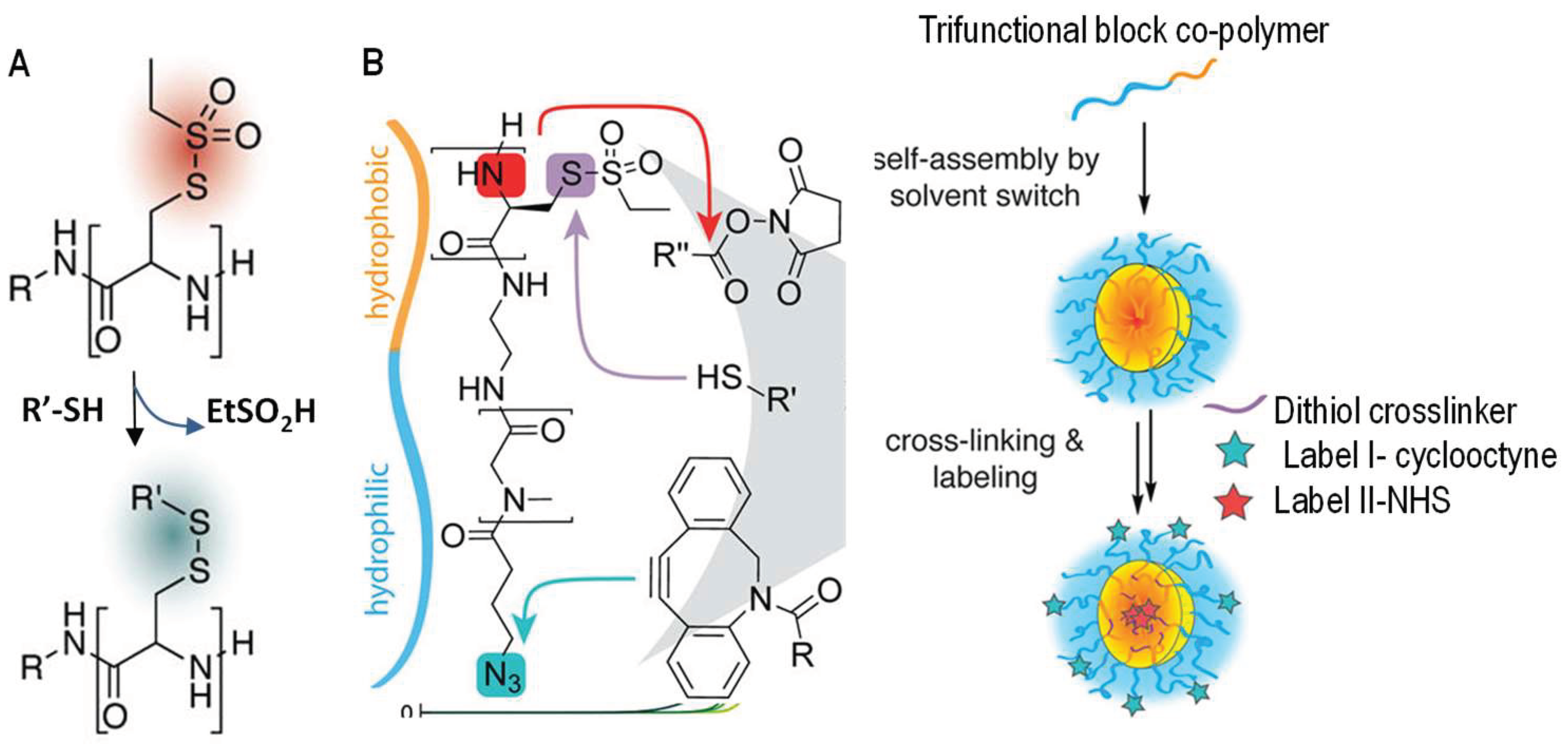
3.1. Formation of Disulfide Bonds
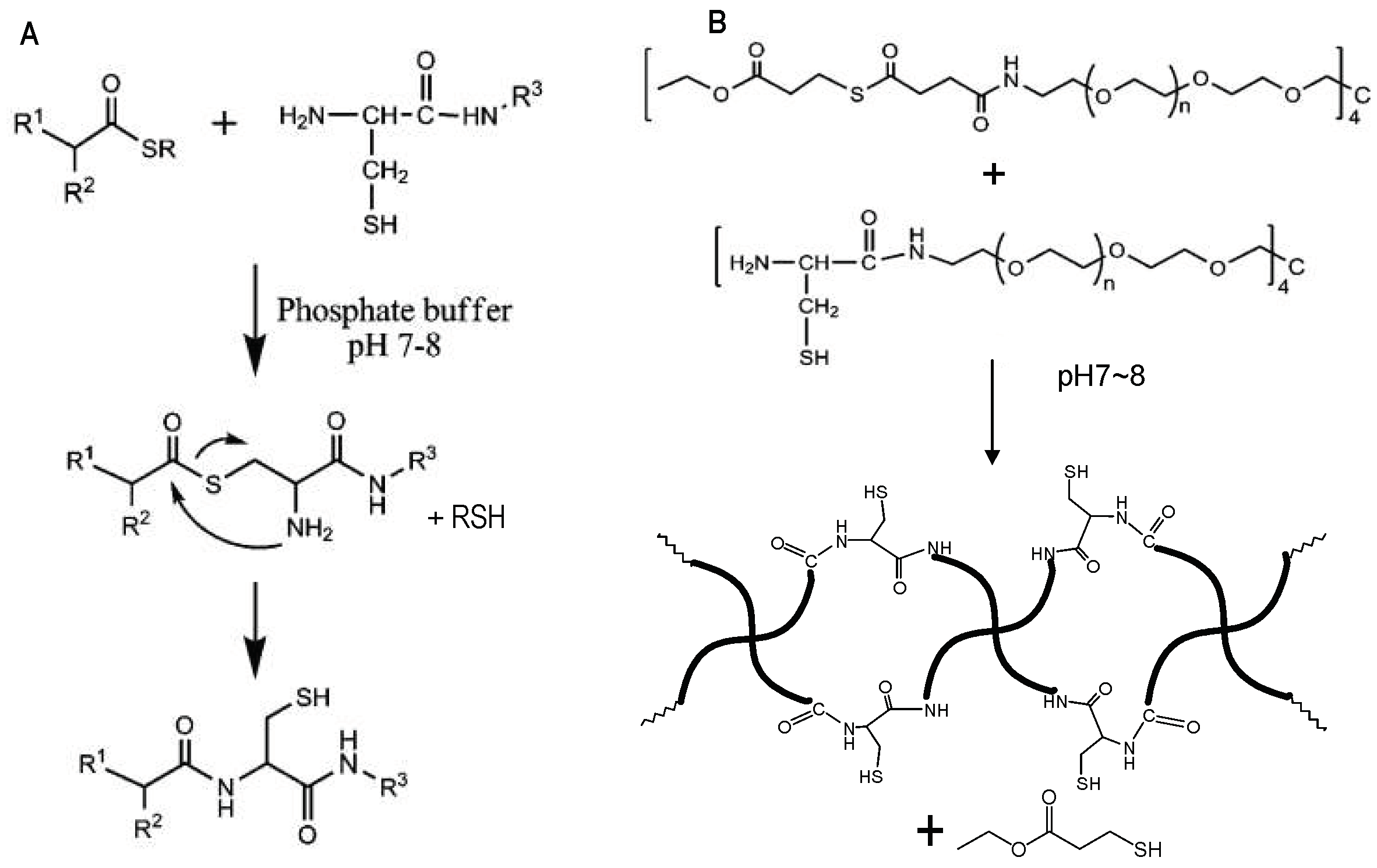
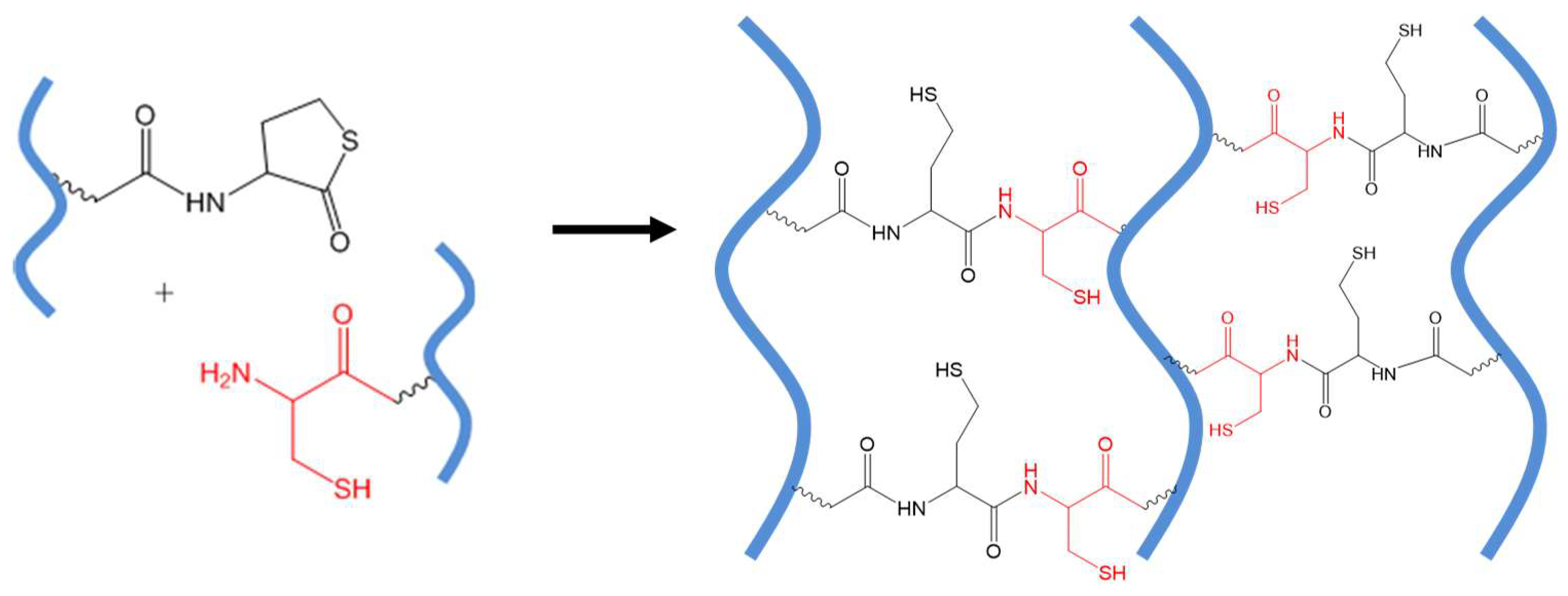
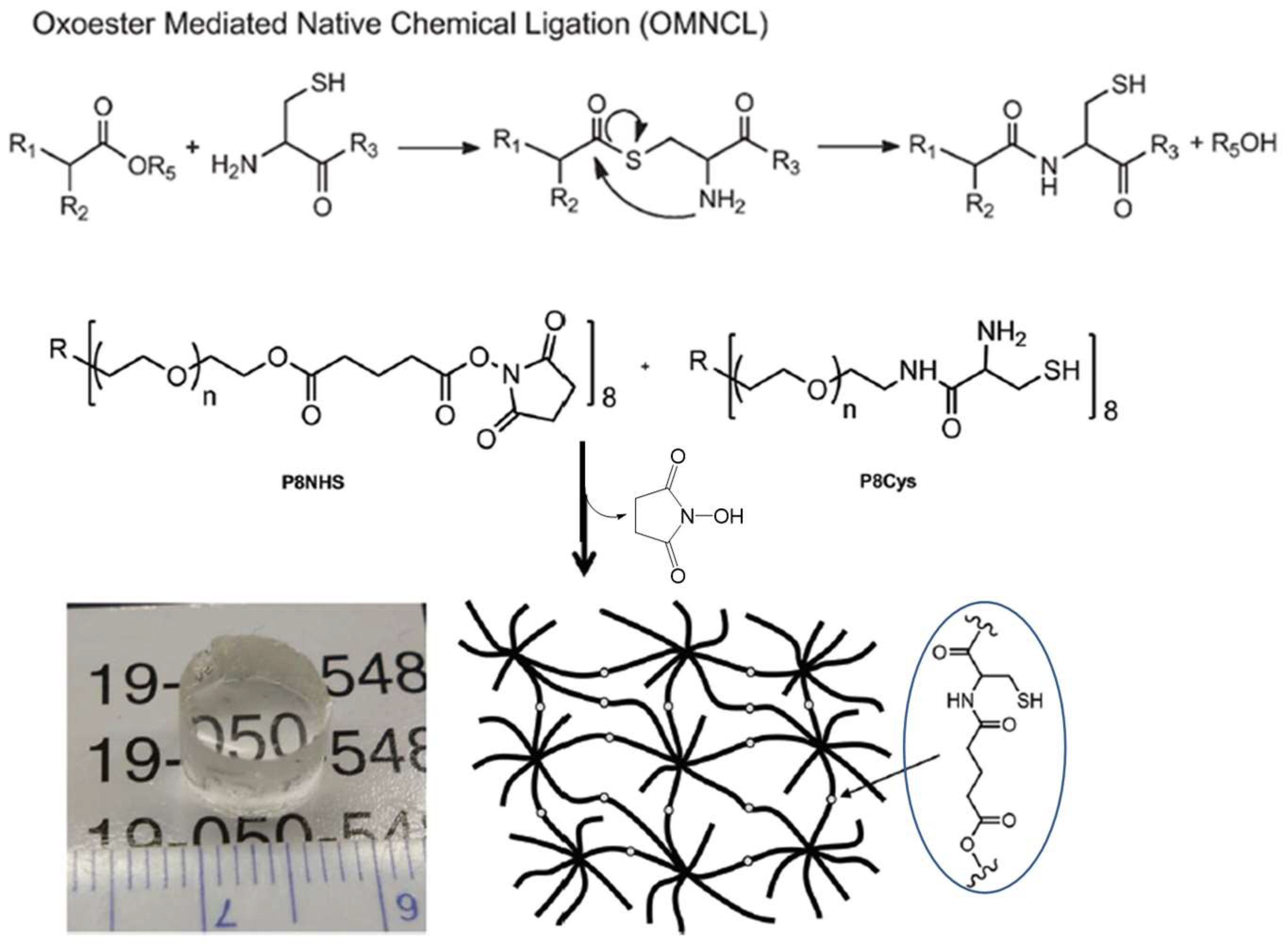
3.2. Native Chemical Ligation
3.3. Thiol-Epoxy Reaction (Ring-Opening Reaction of Epoxides)
4. Addition Reactions Involving Thiols for Hydrogel Crosslinking
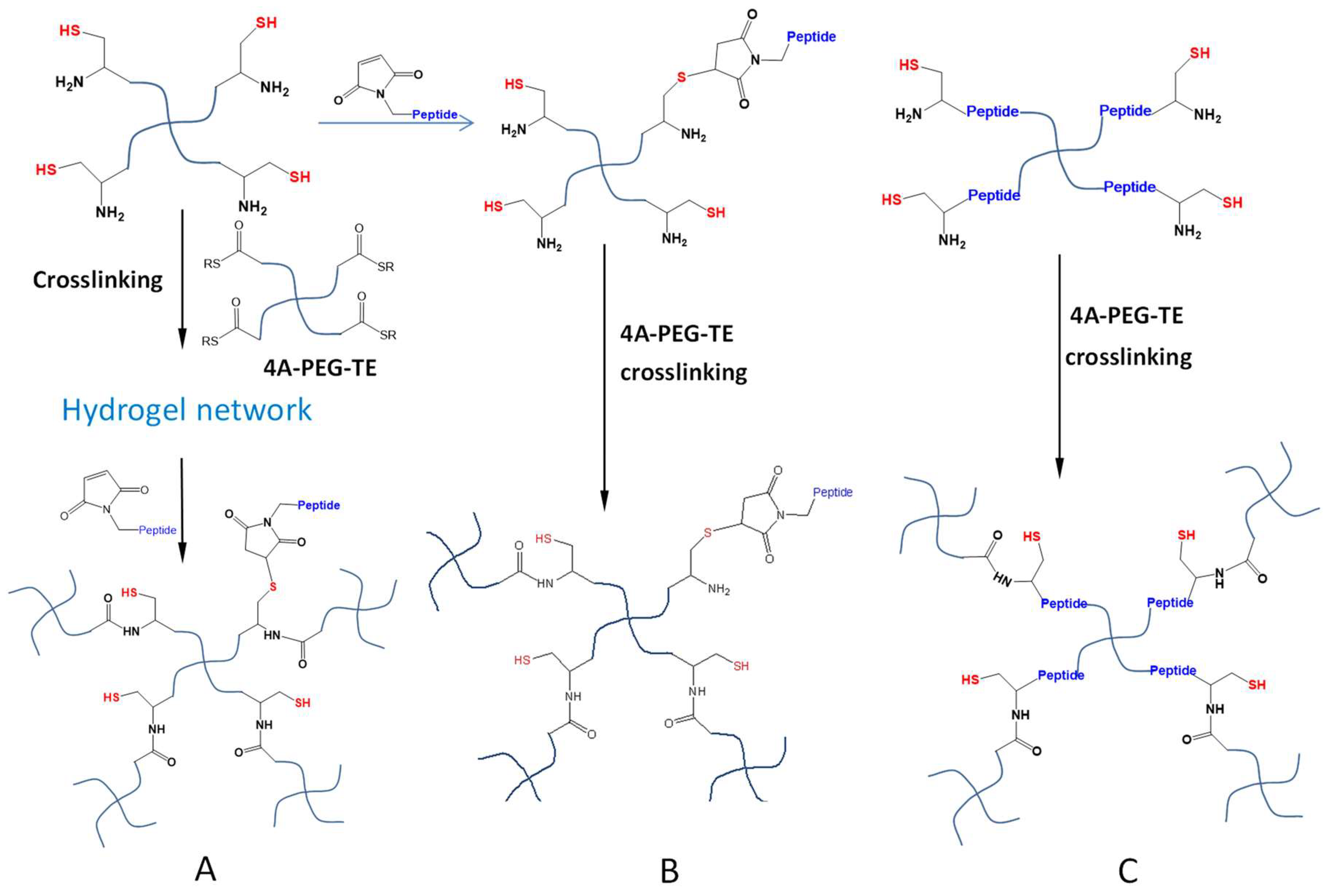
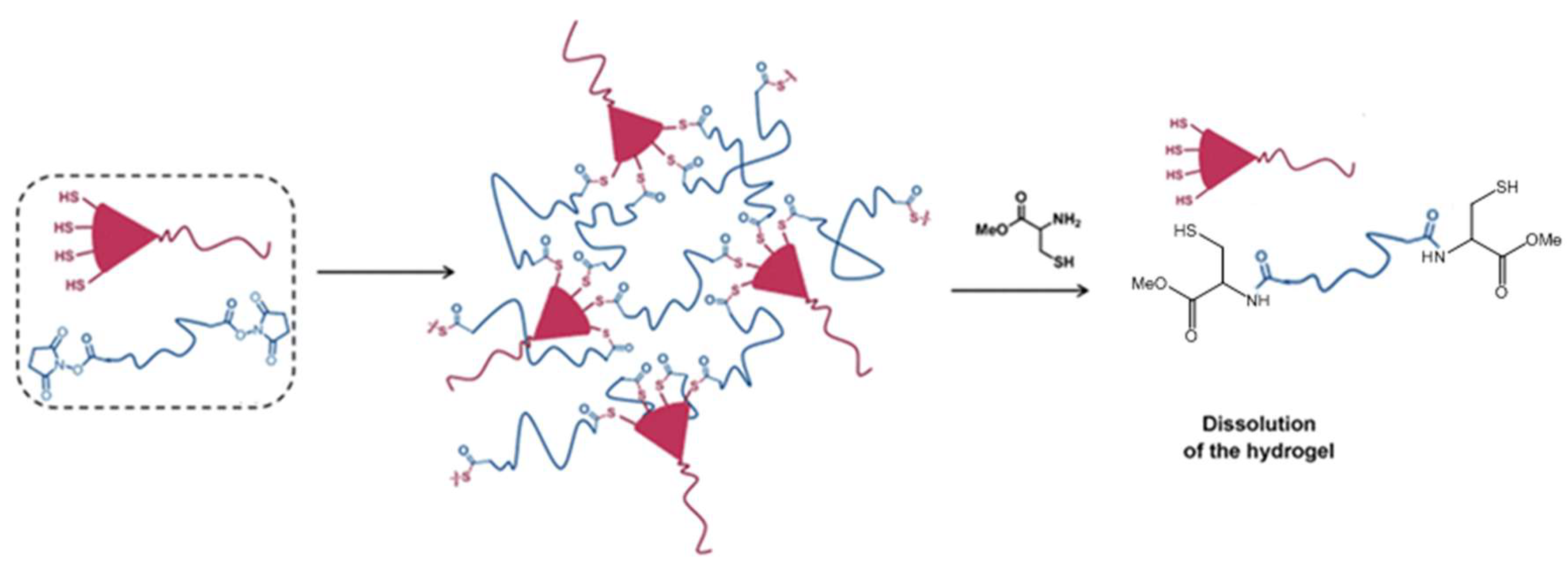
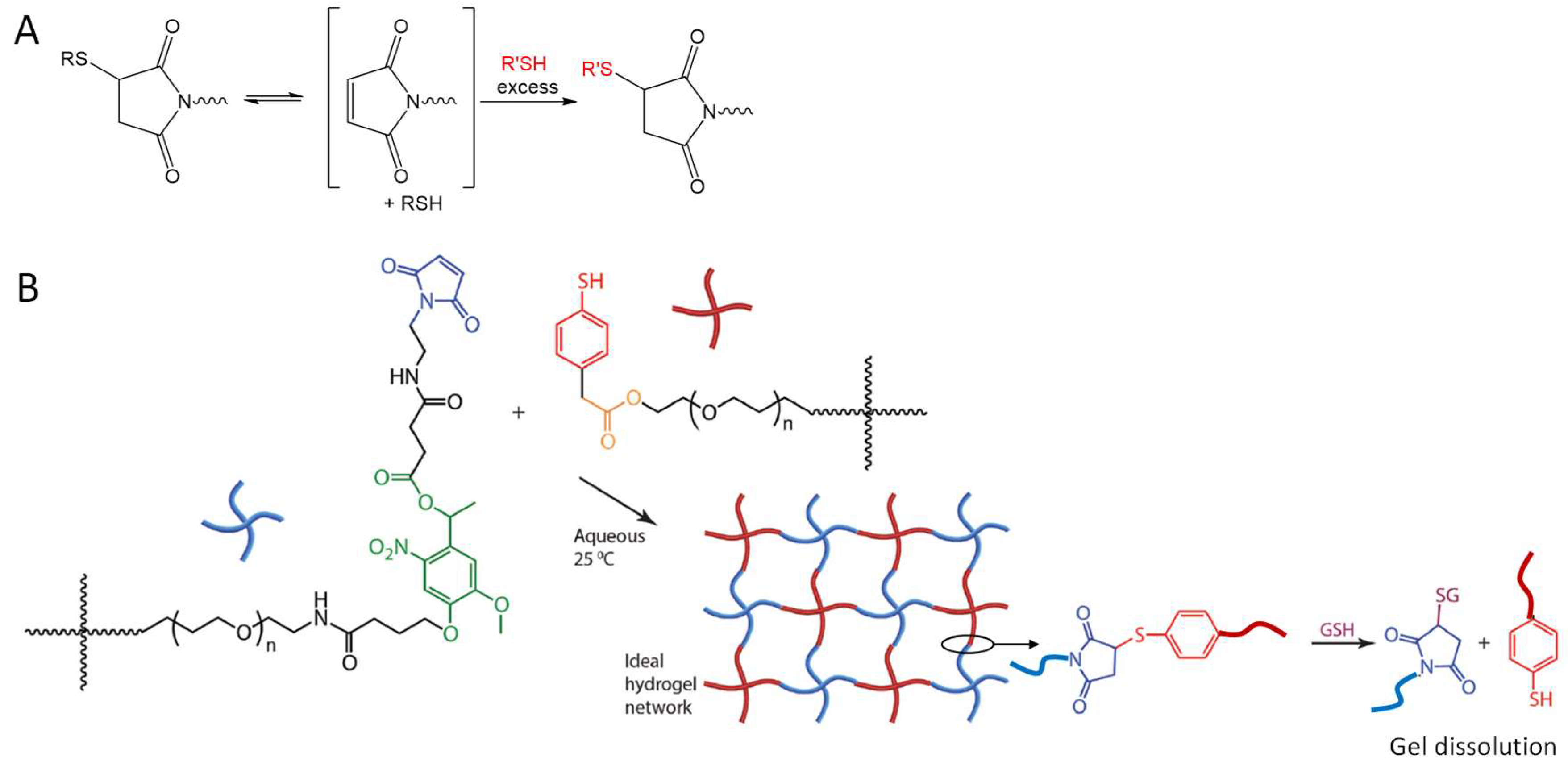
4.1. Michael-Type Additions
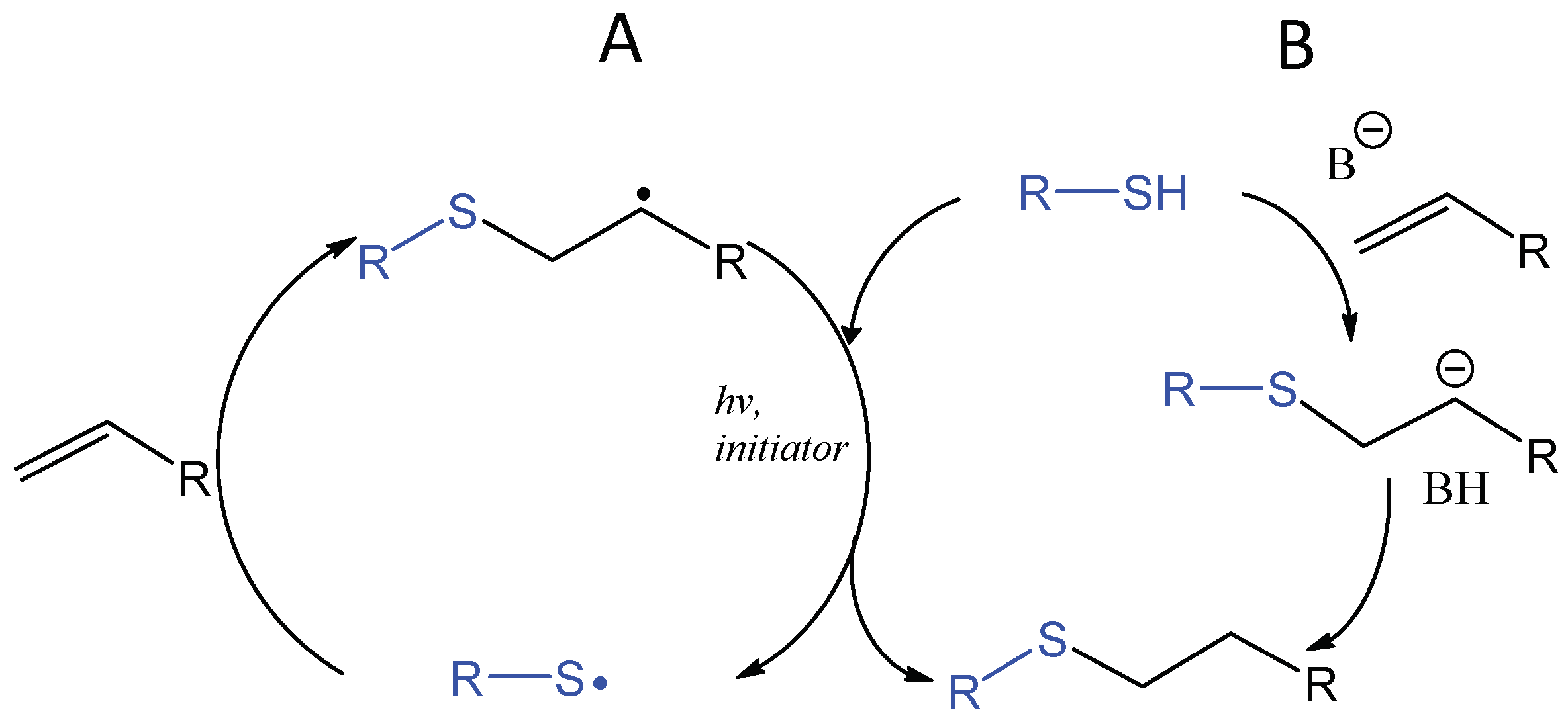
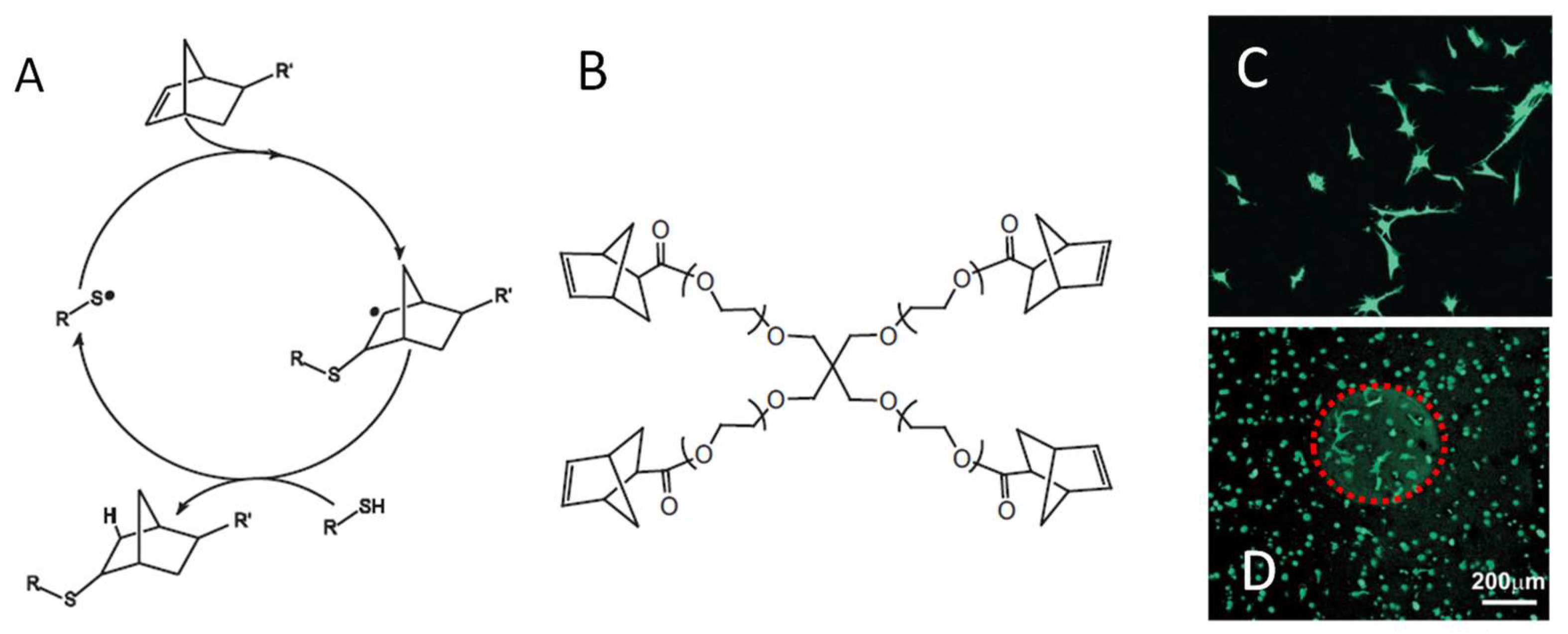
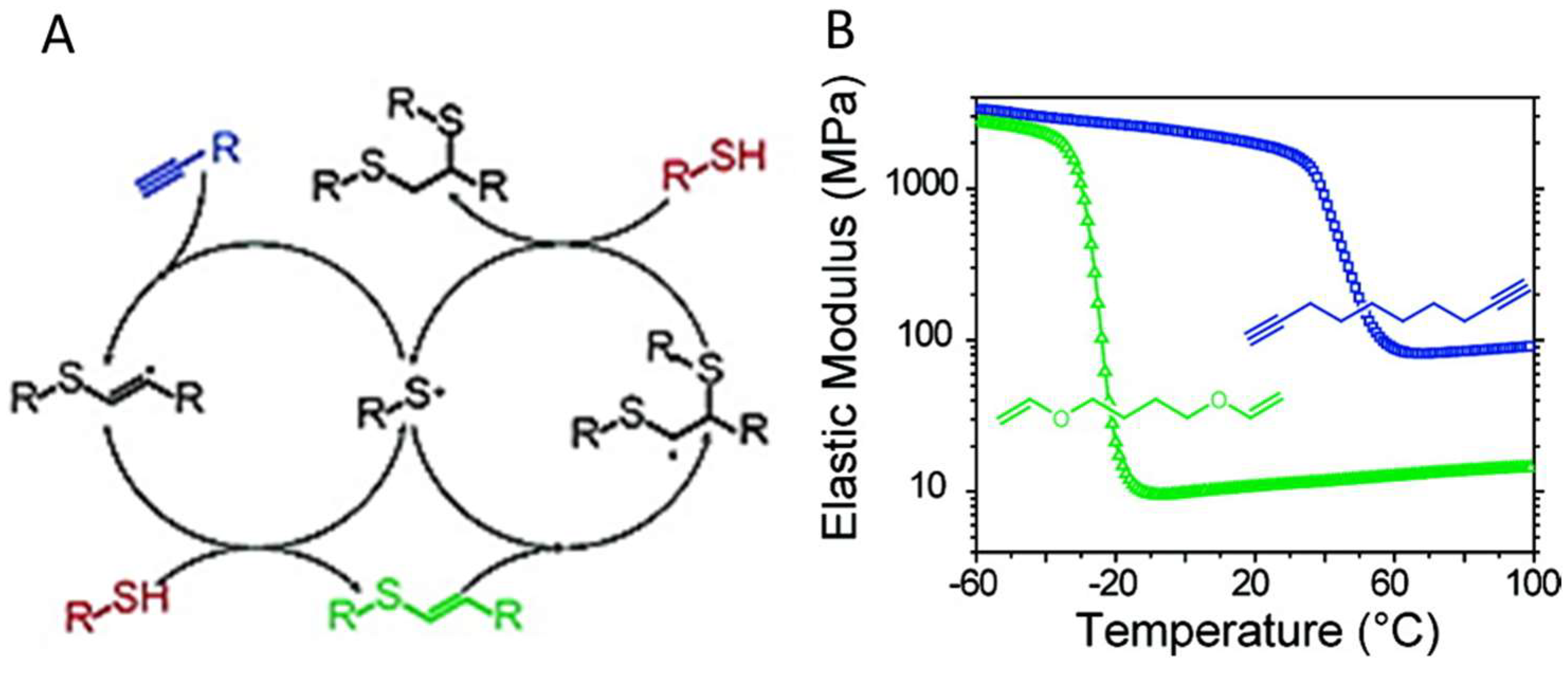
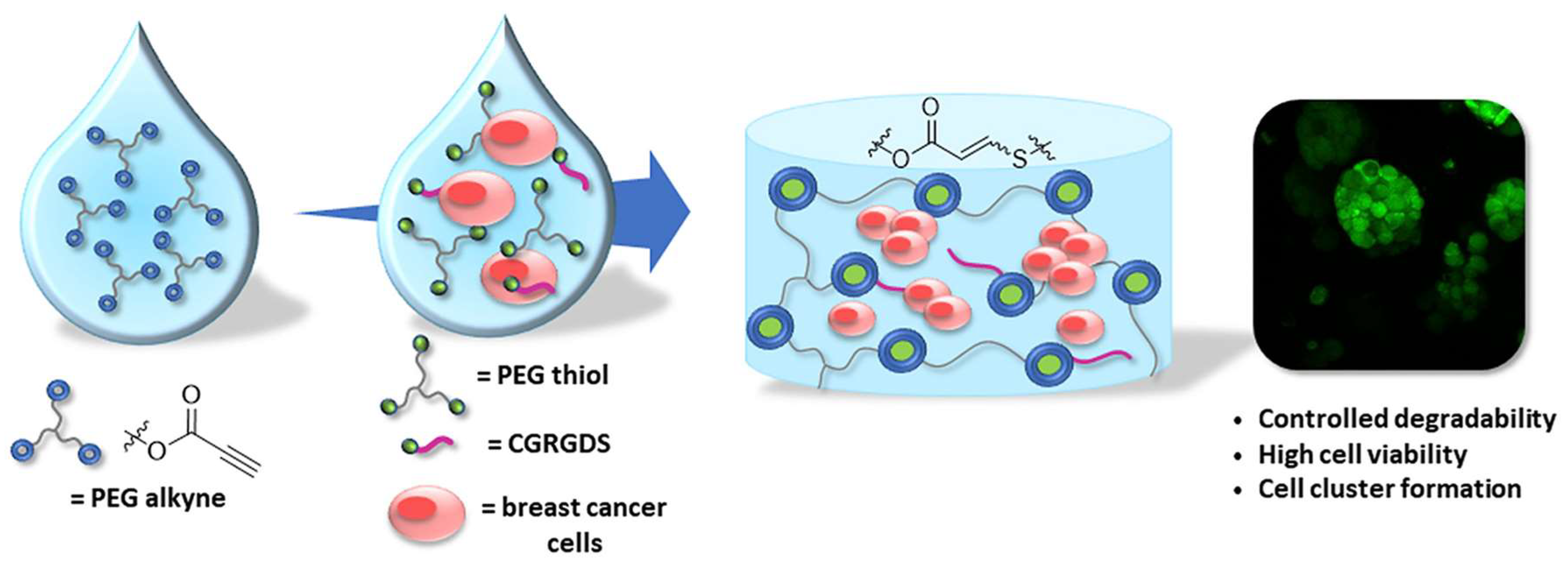
4.2. Photo-Initiated Thiol-Ene and Thiol-Yne Additions
5. Summary and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Peppas, N.A.; Hilt, J.Z.; Khademhosseini, A.; Langer, R. Hydrogels in biology and medicine: From molecular principles to bionanotechnology. Adv. Mater. 2006, 18, 1345–1360. [Google Scholar] [CrossRef]
- Willis, S.L.; Court, J.L.; Redman, R.P.; Wang, J.H.; Leppard, S.W.; O’Byrne, V.J.; Small, S.A.; Lewis, A.L.; Jones, S.A.; Stratford, P.W. A novel phosphorylcholine-coated contact lens for extended wear use. Biomaterials 2001, 22, 3261–3272. [Google Scholar] [CrossRef]
- Miyata, T.; Uragami, T.; Nakamae, K. Biomolecule-sensitive hydrogels. Adv. Drug Del. Rev. 2002, 54, 79–98. [Google Scholar] [CrossRef]
- Chaterji, S.; Kwon, I.K.; Park, K. Smart polymeric gels: Redefining the limits of biomedical devices. Prog. Polym. Sci. 2007, 32, 1083–1122. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.; Paschew, G.; Klatt, S.; Lienig, J.; Arndt, K.F.; Adler, H.J.P. Review on hydrogel-based pH sensors and microsensors. Sensors 2008, 8, 561–581. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, C.E.; Messersmith, P.B. Enzymatically degradable mussel-inspired adhesive hydrogel. Biomacromolecules 2011, 12, 4326–4334. [Google Scholar] [CrossRef] [PubMed]
- Ghobril, C.; Grinstaff, M.W. The chemistry and engineering of polymeric hydrogel adhesives for wound closure: A tutorial. Chem. Soc. Rev. 2015, 44, 1820–1835. [Google Scholar] [CrossRef] [PubMed]
- Li, R.H. Materials for immunoisolated cell transplantation. Adv. Drug Del. Rev. 1998, 33, 87–109. [Google Scholar] [CrossRef]
- Zimmermann, U.; Mimietz, S.; Zimmermann, H.; Hillgartner, M.; Schneider, H.; Ludwig, J.; Hasse, C.; Haase, A.; Rothmund, M.; Fuhr, G. Hydrogel-based non-autologous cell and tissue therapy. BioTechniques 2000, 29, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.K.F.; Matthew, H.W.T. Application of chitosan-based polysaccharide biomaterials in cartilage tissue engineering: A review. Biomaterials 2000, 21, 2589–2598. [Google Scholar] [PubMed]
- Slaughter, B.V.; Khurshid, S.S.; Fisher, O.Z.; Khademhosseini, A.; Peppas, N.A. Hydrogels in regenerative medicine. Adv. Mater. 2009, 21, 3307–3329. [Google Scholar] [CrossRef] [PubMed]
- Sgambato, A.; Cipolla, L.; Russo, L. Bioresponsive hydrogels: Chemical strategies and perspectives in tissue engineering. Gels 2016, 2, 28. [Google Scholar] [CrossRef]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef]
- Kabanov, A.V.; Vinogradov, S.V. Nanogels as pharmaceutical carriers: Finite networks of infinite capabilities. Angew. Chem. Int. Ed. 2009, 48, 5418–5429. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Ding, J.D. Injectable hydrogels as unique biomedical materials. Chem. Soc. Rev. 2008, 37, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Kretlow, J.D.; Young, S.; Klouda, L.; Wong, M.; Mikos, A.G. Injectable biomaterials for regenerating complex craniofacial tissues. Adv. Mater. 2009, 21, 3368–3393. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.V.; Huynh, D.P.; Park, J.H.; Lee, D.S. Injectable polymeric hydrogels for the delivery of therapeutic agents: A review. Eur. Polym. J. 2015, 72, 602–619. [Google Scholar] [CrossRef]
- Petka, W.A.; Harden, J.L.; McGrath, K.P.; Wirtz, D.; Tirrell, D.A. Reversible hydrogels from self-assembling artificial proteins. Science 1998, 281, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, C.M.; Asada, S.; Kitagawa, K.; Koide, T. Artificial collagen gels via self-assembly of de novo designed peptides. Biopolymers 2008, 90, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Mano, J.F.; Silva, G.A.; Azevedo, H.S.; Malafaya, P.B.; Sousa, R.A.; Silva, S.S.; Boesel, L.F.; Oliveira, J.M.; Santos, T.C.; Marques, A.P.; et al. Natural origin biodegradable systems in tissue engineering and regenerative medicine: Present status and some moving trends. J. R. Soc. Interface 2007, 4, 999–1030. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; Andukuri, A.; Lim, D.J.; Jun, H.W. Modulating the gelation properties of self-assembling peptide amphiphiles. ACS Nano 2009, 3, 3447–3454. [Google Scholar] [CrossRef] [PubMed]
- Capito, R.M.; Azevedo, H.S.; Velichko, Y.S.; Mata, A.; Stupp, S.I. Self-assembly of large and small molecules into hierarchically ordered sacs and membranes. Science 2008, 319, 1812–1816. [Google Scholar] [CrossRef] [PubMed]
- Hainline, K.M.; Fries, C.N.; Collier, J.H. Progress toward the clinical translation of bioinspired peptide and protein assemblies. Adv. Healthc. Mater. 2018, 7, 1700930. [Google Scholar] [CrossRef] [PubMed]
- Nimmo, C.M.; Shoichet, M.S. Regenerative biomaterials that “click”: Simple, aqueous-based protocols for hydrogel synthesis, surface immobilization, and 3d patterning. Bioconjug. Chem. 2011, 22, 2199–2209. [Google Scholar] [CrossRef] [PubMed]
- Buwalda, S.J.; Vermonden, T.; Hennink, W.E. Hydrogels for therapeutic delivery: Current developments and future directions. Biomacromolecules 2017, 18, 316–330. [Google Scholar] [CrossRef] [PubMed]
- Anseth, K.S.; Klok, H.-A. Click chemistry in biomaterials, nanomedicine, and drug delivery. Biomacromolecules 2016, 17, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C. Recent advances in crosslinking chemistry of biomimetic poly(ethylene glycol) hydrogels. RSC Adv. 2015, 5, 39844–398583. [Google Scholar] [CrossRef] [PubMed]
- Gruhlke, M.C.H.; Slusarenko, A.J. The biology of reactive sulfur species (rss). Plant Physiol. Biochem. 2012, 59, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yuan, J. Caspases in apoptosis and beyond. Oncogene 2008, 27, 6194–6206. [Google Scholar] [CrossRef] [PubMed]
- Tolbert, B.S.; Tajc, S.G.; Webb, H.; Snyder, J.; Nielsen, J.E.; Miller, B.L.; Basavappa, R. The active site cysteine of ubiquitin-conjugating enzymes has a significantly elevated PKA: Functional implications. Biochemistry 2005, 44, 16385–16391. [Google Scholar] [CrossRef] [PubMed]
- Stenzel, M.H. Bioconjugation using thiols: Old chemistry rediscovered to connect polymers with nature’s building blocks. ACS Macro Lett. 2013, 2, 14–18. [Google Scholar] [CrossRef]
- Li, Y.-L.; Zhu, L.; Liu, Z.; Cheng, R.; Meng, F.; Cui, J.-H.; Ji, S.-J.; Zhong, Z. Reversibly stabilized multifunctional dextran nanoparticles efficiently deliver doxorubicin into the nuclei of cancer cells. Angew. Chem. Int. Ed. 2009, 48, 9914–9918. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Hennink, W.E.; Zhong, Z. Reduction-sensitive polymers and bioconjugates for biomedical applications. Biomaterials 2009, 30, 2180–2198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Achazi, K.; Steinhilber, D.; Kratz, F.; Dernedde, J.; Haag, R. A facile approach for dual-responsive prodrug nanogels based on dendritic polyglycerols with minimal leaching. J. Control. Release 2014, 174, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.H.; Su, J.; Messersmith, P.B. Hydrogels cross-linked by native chemical ligation. Biomacromolecules 2009, 10, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Hammer, N.; Brandl, F.P.; Kirchhof, S.; Messmann, V.; Goepferich, A.M. Protein compatibility of selected cross-linking reactions for hydrogels. Macromol. Biosci. 2015, 15, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P. Kinetics and mechanisms of thiol–disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxid. Redox. Signal. 2013, 18, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.A.; Kamada, J.; Koynov, K.; Mohin, J.; Nicolaÿ, R.; Zhang, Y.; Balazs, A.C.; Kowalewski, T.; Matyjaszewski, K. Self-healing polymer films based on thiol–disulfide exchange reactions and self-healing kinetics measured using atomic force microscopy. Macromolecules 2012, 45, 142–149. [Google Scholar] [CrossRef]
- Zhang, X.; Waymouth, R.M. 1,2-Dithiolane-derived dynamic, covalent materials: Cooperative self-assembly and reversible cross-linking. J. Am. Chem. Soc. 2017, 139, 3822–3833. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, L.; Wang, X.; Huang, D.; Yang, F.; Shen, H.; Li, Z.; Wu, D. UV-triggered thiol-disulfide exchange reaction towards tailored biodegradable hydrogels. Polym. Chem. 2016, 7, 1429–1438. [Google Scholar] [CrossRef]
- Ryu, J.-H.; Jiwpanich, S.; Chacko, R.; Bickerton, S.; Thayumanavan, S. Surface-functionalizable polymer nanogels with facile hydrophobic guest encapsulation capabilities. J. Am. Chem. Soc. 2010, 132, 8246–8247. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Rübsam, K.; Jakob, F.; Pazdzior, P.; Schwaneberg, U.; Pich, A. Reversible deactivation of enzymes by redox-responsive nanogel carriers. Macromol. Rapid Commun. 2016, 37, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Schafer, O.; Huesmann, D.; Muhl, C.; Barz, M. Rethinking cysteine protective groups: S-alkylsulfonyl-l-cysteines for chemoselective disulfide formation. Chemistry (Easton) 2016, 22, 18085–18091. [Google Scholar]
- Schäfer, O.; Klinker, K.; Braun, L.; Huesmann, D.; Schultze, J.; Koynov, K.; Barz, M. Combining orthogonal reactive groups in block copolymers for functional nanoparticle synthesis in a single step. ACS Macro Lett. 2017, 1140–1145. [Google Scholar] [CrossRef]
- Johnson, E.C.B.; Kent, S.B.H. Insights into the mechanism and catalysis of the native chemical ligation reaction. J. Am. Chem. Soc. 2006, 128, 6640–6646. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.E.; Kent, S.B.H. Synthesis of native proteins by chemical ligation. Annu. Rev. Biochem. 2000, 69, 923–960. [Google Scholar] [CrossRef] [PubMed]
- Paramonov, S.E.; Gauba, V.; Hartgerink, J.D. Synthesis of collagen-like peptide polymers by native chemical ligation. Macromolecules 2005, 38, 7555–7561. [Google Scholar] [CrossRef]
- Dirksen, A.; Meijer, E.W.; Adriaens, W.; Hackeng, T.M. Strategy for the synthesis of multivalent peptide-based nonsymmetric dendrimers by native chemical ligation. Chem. Commun. 2006, 1667–1669. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.P.; Jones, J.L.; Cronier, S.A.; Collier, J.H. Modulating the mechanical properties of self-assembled peptide hydrogels via native chemical ligation. Biomaterials 2008, 29, 2143–2151. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.P.; Sprangers, A.J.; Byce, J.R.; Su, J.; Squirrell, J.M.; Messersmith, P.B.; Eliceiri, K.W.; Ogle, B.M. Ecm-incorporated hydrogels cross-linked via native chemical ligation to engineer stem cell microenvironments. Biomacromolecules 2013, 14, 3102–3111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, P.; Huangshan, L.; Hu, B.-H.; Messersmith, P.B. Improved method for synthesis of cysteine modified hyaluronic acid for in situ hydrogel formation. Chem. Commun. 2015, 51, 9662–9665. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Cheng, P.; Liu, M.; Li, D.; Liu, G.; Zhao, Y.; Ding, Z.; Chen, F.; Wang, B.; Tan, X.; et al. Poly(glutamic acid) hydrogels crosslinked via native chemical ligation. New J. Chem. 2017, 41, 8656–8662. [Google Scholar] [CrossRef]
- Strehin, I.; Gourevitch, D.; Zhang, Y.; Heber-Katz, E.; Messersmith, P.B. Hydrogels formed by oxo-ester mediated native chemical ligation. Biomater. Sci. 2013, 1, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Boere, K.W.; van den Dikkenberg, J.; Gao, Y.; Visser, J.; Hennink, W.E.; Vermonden, T. Thermogelling and chemoselectively cross-linked hydrogels with controlled mechanical properties and degradation behavior. Biomacromolecules 2015, 16, 2840–2851. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shu, Y.; Hao, T.; Wang, Y.; Qian, Y.; Duan, C.; Sun, H.; Lin, Q.; Wang, C. A chitosan-glutathione based injectable hydrogel for suppression of oxidative stress damage in cardiomyocytes. Biomaterials 2013, 34, 9071–9081. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Hu, B.H.; Lowe, W.L.; Kaufman, D.B.; Messersmith, P.B. Anti-inflammatory peptide-functionalized hydrogels for insulin-secreting cell encapsulation. Biomaterials 2010, 31, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Cynthia, G.; Kristie, C.; Rodriguez, E.K.; Ara, N.; Grinstaff, M.W. A dendritic thioester hydrogel based on thiol–thioester exchange as a dissolvable sealant system for wound closure. Angew. Chem. Int. Ed. 2013, 52, 14070–14074. [Google Scholar]
- Konieczynska, M.D.; Grinstaff, M.W. On-demand dissolution of chemically cross-linked hydrogels. Acc. Chem. Res. 2017, 50, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Brandle, A.; Khan, A. Thiol-epoxy ‘click’ polymerization: Efficient construction of reactive and functional polymers. Polym. Chem. 2012, 3, 3224–3227. [Google Scholar] [CrossRef]
- Gadwal, I.; Stuparu, M.C.; Khan, A. Homopolymer bifunctionalization through sequential thiol-epoxy and esterification reactions: An optimization, quantification, and structural elucidation study. Polym. Chem. 2015, 6, 1393–1404. [Google Scholar] [CrossRef]
- Gao, L.; Li, X.; Wang, Y.; Zhu, W.; Shen, Z.; Li, X. Injectable thiol-epoxy “click” hydrogels. J. Polym. Sci. A Polym. Chem. 2016, 54, 2651–2655. [Google Scholar] [CrossRef]
- Elbert, D.L.; Pratt, A.B.; Lutolf, M.P.; Halstenberg, S.; Hubbell, J.A. Protein delivery from materials formed by self-selective conjugate addition reactions. J. Control. Release 2001, 76, 11–25. [Google Scholar] [CrossRef]
- Elia, R.; Fuegy, P.W.; van Delden, A.; Firpo, M.A.; Prestwich, G.D.; Peattie, R.A. Stimulation of in vivo angiogenesis by in situ crosslinked, dual growth factor-loaded, glycosaminoglycan hydrogels. Biomaterials 2010, 31, 4630–4638. [Google Scholar] [CrossRef] [PubMed]
- Speidel, A.T.; Stuckey, D.J.; Chow, L.W.; Jackson, L.H.; Noseda, M.; Abreu Paiva, M.; Schneider, M.D.; Stevens, M.M. Multimodal hydrogel-based platform to deliver and monitor cardiac progenitor/stem cell engraftment. ACS Cent. Sci. 2017, 3, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Kao, W.J. In situ forming poly(ethylene glycol)-based hydrogels via thiol-maleimide michael-type addition. J. Biomed. Mater. Res. 2011, 98A, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Phelps, E.A.; Enemchukwu, N.O.; Fiore, V.F.; Sy, J.C.; Murthy, N.; Sulchek, T.A.; Barker, T.H.; García, A.J. Maleimide cross-linked bioactive peg hydrogel exhibits improved reaction kinetics and cross-linking for cell encapsulation and in situ delivery. Adv. Mater. 2012, 24, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Stefanos, S.; Ma, J.; Lalloo, A.; Perry, B.A.; Leibowitz, M.J.; Sinko, P.J.; Stein, S. A hydrogel prepared by in situ cross-linking of a thiol-containing poly(ethylene glycol)-based copolymer: A new biomaterial for protein drug delivery. Biomaterials 2003, 24, 11–18. [Google Scholar] [CrossRef]
- Fontaine, S.D.; Reid, R.; Robinson, L.; Ashley, G.W.; Santi, D.V. Long-term stabilization of maleimide–thiol conjugates. Bioconjug. Chem. 2015, 26, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Darling, N.J.; Hung, Y.-S.; Sharma, S.; Segura, T. Controlling the kinetics of thiol-maleimide michael-type addition gelation kinetics for the generation of homogenous poly(ethylene glycol) hydrogels. Biomaterials 2016, 101, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.D.; Kiick, K.L. Reversible maleimide-thiol adducts yield glutathione-sensitive poly(ethylene glycol)-heparin hydrogels. Polym. Chem. 2013, 4, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Kharkar, P.M.; Kiick, K.L.; Kloxin, A.M. Design of thiol- and light-sensitive degradable hydrogels using michael-type addition reactions. Polym. Chem. 2015, 6, 5565–5574. [Google Scholar] [CrossRef] [PubMed]
- Chakma, P.; Rodrigues Possarle, L.H.; Digby, Z.A.; Zhang, B.; Sparks, J.L.; Konkolewicz, D. Dual stimuli responsive self-healing and malleable materials based on dynamic thiol-michael chemistry. Polym. Chem. 2017, 8, 6534–6543. [Google Scholar] [CrossRef]
- Kuhl, N.; Geitner, R.; Bose, R.K.; Bode, S.; Dietzek, B.; Schmitt, M.; Popp, J.; Garcia, S.J.; van der Zwaag, S.; Schubert, U.S.; et al. Self-healing polymer networks based on reversible michael addition reactions. Macromol. Chem. Phys. 2016, 217, 2541–2550. [Google Scholar] [CrossRef]
- Elisseeff, J.; Anseth, K.; Sims, D.; McIntosh, W.; Randolph, M.; Langer, R. Transdermal photopolymerization for minimally invasive implantation. Proc. Natl. Acad. Sci. USA 1999, 96, 3104–3107. [Google Scholar] [CrossRef] [PubMed]
- Elbert, D.L.; Hubbell, J.A. Conjugate addition reactions combined with free-radical cross-linking for the design of materials for tissue engineering. Biomacromolecules 2001, 2, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Leach, J.B.; Bivens, K.A.; Patrick, C.W.; Schmidt, C.E. Photocrosslinked hyaluronic acid hydrogels: Natural, biodegradable tissue engineering scaffolds. Biotechnol. Bioeng. 2003, 82, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Ki, C.S.; Shih, H. Thiol–norbornene photoclick hydrogels for tissue engineering applications. J. Appl. Polym. Sci. 2015, 132, 41563. [Google Scholar] [CrossRef] [PubMed]
- Bryant, S.J.; Nuttelman, C.R.; Anseth, K.S. Cytocompatibility of uv and visible light photoinitiating systems on cultured nih/3t3 fibroblasts in vitro. J. Biomater. Sci. Polym. Ed. 2000, 11, 439–457. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.G.; Malik, A.N.; Kim, T.K.; Manson, P.N.; Elisseeff, J.H. Variable cytocompatibility of six cell lines with photoinitiators used for polymerizing hydrogels and cell encapsulation. Biomaterials 2005, 26, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.A.; Varghese, S.; Sharma, B.; Strehin, I.; Fermanian, S.; Gorham, J.; Fairbrother, D.H.; Cascio, B.; Elisseeff, J.H. Multifunctional chondroitin sulphate for cartilage tissue-biomaterial integration. Nat. Mater. 2007, 6, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Cramer, N.B.; Reddy, S.K.; O’Brien, A.K.; Bowman, C.N. Thiol−ene photopolymerization mechanism and rate limiting step changes for various vinyl functional group chemistries. Macromolecules 2003, 36, 7964–7969. [Google Scholar] [CrossRef]
- Dondoni, A. The emergence of thiol-ene coupling as a click process for materials and bioorganic chemistry13. Angew. Chem. Int. Ed. 2008, 47, 8995–8997. [Google Scholar] [CrossRef] [PubMed]
- Kade, M.J.; Burke, D.J.; Hawker, C.J. The power of thiol-ene chemistry. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 743–750. [Google Scholar] [CrossRef]
- Lowe, A.B. Thiol-yne ‘click’/coupling chemistry and recent applications in polymer and materials synthesis and modification. Polymer 2014, 55, 5517–5549. [Google Scholar] [CrossRef]
- Rydholm, A.E.; Bowman, C.N.; Anseth, K.S. Degradable thiol-acrylate photopolymers: Polymerization and degradation behavior of an in situ forming biomaterial. Biomaterials 2005, 26, 4495–4506. [Google Scholar] [CrossRef] [PubMed]
- Hoyle, C.E.; Bowman, C.N. Thiol-ene click chemistry. Angew. Chem. Int. Ed. 2010, 49, 1540–1573. [Google Scholar] [CrossRef] [PubMed]
- McCall, J.D.; Anseth, K.S. Thiol–ene photopolymerizations provide a facile method to encapsulate proteins and maintain their bioactivity. Biomacromolecules 2012, 13, 2410–2417. [Google Scholar] [CrossRef] [PubMed]
- Killops, K.L.; Campos, L.M.; Hawker, C.J. Robust, efficient, and orthogonal synthesis of dendrimers via thiol-ene “click” chemistry. J. Am. Chem. Soc. 2008, 130, 5062–5064. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Discher, D.E.; Justynska, J.; Schlaad, H. Grafting short peptides onto polybutadiene-block-poly(ethylene oxide): A platform for self-assembling hybrid amphiphiles. Angew. Chem. Int. Ed. 2006, 45, 7578–7581. [Google Scholar] [CrossRef] [PubMed]
- Wittrock, S.; Becker, T.; Kunz, H. Synthetic vaccines of tumor-associated glycopeptide antigens by immune-compatible thioether linkage to bovine serum albumin. Angew. Chem. Int. Ed. 2007, 46, 5226–5230. [Google Scholar] [CrossRef] [PubMed]
- Kharkar, P.M.; Rehmann, M.S.; Skeens, K.M.; Maverakis, E.; Kloxin, A.M. Thiol–ene click hydrogels for therapeutic delivery. ACS Biomater. Sci. Eng. 2016, 2, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Fairbanks, B.D.; Schwartz, M.P.; Halevi, A.E.; Nuttelman, C.R.; Bowman, C.N.; Anseth, K.S. A versatile synthetic extracellular matrix mimic via thiol-norbornene photopolymerization. Adv. Mater. 2009, 21, 5005–5010. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.; Lin, C.C. Cross-linking and degradation of step-growth hydrogels formed by thiol-ene photoclick chemistry. Biomacromolecules 2012, 13, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Xin, S.; Wyman, O.M.; Alge, D.L. Assembly of peg microgels into porous cell-instructive 3d scaffolds via thiol-ene click chemistry. Adv. Healthc. Mater. 2018, 7, 1800160. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, R.R.; Kwandou, G.; Spicer, P.T.; Stenzel, M.H. (-)-riboflavin (vitamin b2) and flavin mononucleotide as visible light photo initiators in the thiol-ene polymerisation of peg-based hydrogels. Polym. Chem. 2017, 8, 980–984. [Google Scholar] [CrossRef]
- Yue, K.; Liu, Y.; Byambaa, B.; Singh, V.; Liu, W.; Li, X.; Sun, Y.; Zhang, Y.S.; Tamayol, A.; Zhang, P.; et al. Visible light crosslinkable human hair keratin hydrogels. Bioeng. Transl. Med. 2018, 3, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Le, N.N.T.; Zorn, S.; Schmitt, S.K.; Gopalan, P.; Murphy, W.L. Hydrogel arrays formed via differential wettability patterning enable combinatorial screening of stem cell behavior. Acta Biomater. 2016, 34, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Jivan, F.; Yegappan, R.; Pearce, H.; Carrow, J.K.; McShane, M.; Gaharwar, A.K.; Alge, D.L. Sequential thiol–ene and tetrazine click reactions for the polymerization and functionalization of hydrogel microparticles. Biomacromolecules 2016, 17, 3516–3523. [Google Scholar] [CrossRef] [PubMed]
- Mabry, K.M.; Schroeder, M.E.; Payne, S.Z.; Anseth, K.S. Three-dimensional high-throughput cell encapsulation platform to study changes in cell-matrix interactions. ACS Appl. Mater. Interfaces 2016, 8, 21914–21922. [Google Scholar] [CrossRef] [PubMed]
- Richard, H. Thiol–yne chemistry: A powerful tool for creating highly functional materials. Angew. Chem. Int. Ed. 2010, 49, 3415–3417. [Google Scholar]
- Fairbanks, B.D.; Scott, T.F.; Kloxin, C.J.; Anseth, K.S.; Bowman, C.N. Thiol−yne photopolymerizations: Novel mechanism, kinetics, and step-growth formation of highly cross-linked networks. Macromolecules 2009, 42, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.W.; Shin, J.; Hoyle, C.E.; Bowman, C.N.; Lowe, A.B. Synthesis, thiol-yne “click” photopolymerization, and physical properties of networks derived from novel multifunctional alkynes. Macromolecules 2010, 43, 4937–4942. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, H.; Jiang, X.; Yin, J. Versatile functionalization of the micropatterned hydrogel of hyperbranched poly(ether amine) based on “thiol-yne” chemistry. Adv. Funct. Mater. 2014, 24, 1679–1686. [Google Scholar] [CrossRef]
- Cai, X.Y.; Li, J.Z.; Li, N.N.; Chen, J.C.; Kang, E.-T.; Xu, L.Q. Peg-based hydrogels prepared by catalyst-free thiol–yne addition and their post-antibacterial modification. Biomater. Sci. 2016, 4, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Truong, V.X.; Dove, A.P. Organocatalytic, regioselective nucleophilic “click” addition of thiols to propiolic acid esters for polymer–polymer coupling. Angew. Chem. Int. Ed. 2013, 52, 4132–4136. [Google Scholar] [CrossRef] [PubMed]
- Macdougall, L.J.; Truong, V.X.; Dove, A.P. Efficient in situ nucleophilic thiol-yne click chemistry for the synthesis of strong hydrogel materials with tunable properties. ACS Macro Lett. 2017, 6, 93–97. [Google Scholar] [CrossRef]
- Macdougall, L.J.; Wiley, K.L.; Kloxin, A.M.; Dove, A.P. Design of synthetic extracellular matrices for probing breast cancer cell growth using robust cyctocompatible nucleophilic thiol-yne addition chemistry. Biomaterials 2018, 178, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Ifkovits, J.L.; Burdick, J.A. Review: Photopolymerizable and degradable biomaterials for tissue engineering applications. Tissue Eng. 2007, 13, 2369–2385. [Google Scholar] [CrossRef] [PubMed]
- Azagarsamy, M.A.; Anseth, K.S. Bioorthogonal click chemistry: An indispensable tool to create multifaceted cell culture scaffolds. ACS Macro Lett. 2013, 2, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.S.; Khademhosseini, A. Advances in engineering hydrogels. Science 2017, 356, eaaf3627. [Google Scholar] [CrossRef] [PubMed]
- Khire, V.S.; Harant, A.W.; Watkins, A.W.; Anseth, K.S.; Bowman, C.N. Ultrathin patterned polymer films on surfaces using thiol-ene polymerizations. Macromolecules 2006, 39, 5081–5086. [Google Scholar] [CrossRef]
- Khire, V.S.; Benoit, D.S.W.; Anseth, K.S.; Bowman, C.N. Ultrathin gradient films using thiol-ene polymerizations. J. Polym. Sci. A Polym. Chem. 2006, 44, 7027–7039. [Google Scholar] [CrossRef]
- Dadoo, N.; Landry, S.B.; Bomar, J.D.; Gramlich, W.M. Synthesis and spatiotemporal modification of biocompatible and stimuli-responsive carboxymethyl cellulose hydrogels using thiol-norbornene chemistry. Macromol. Biosci. 2017, 17, 1700107. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, L.A.; Kloxin, A.M. Design of thiol–ene photoclick hydrogels using facile techniques for cell culture applications. Biomater. Sci. 2014, 2, 1612–1626. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Chu, C.C. Visible light induced dextran-methacrylate hydrogel formation using (-)-riboflavin vitamin b2 as a photoinitiator and l-arginine as a co-initiator. Fibers Polym. 2009, 10, 14–20. [Google Scholar] [CrossRef]
- Fairbanks, B.D.; Schwartz, M.P.; Bowman, C.N.; Anseth, K.S. Photoinitiated polymerization of peg-diacrylate with lithium phenyl-2,4,6-trimethylbenzoylphosphinate: Polymerization rate and cytocompatibility. Biomaterials 2009, 30, 6702–6707. [Google Scholar] [CrossRef] [PubMed]
- Holmes, R.; Yang, X.-B.; Dunne, A.; Florea, L.; Wood, D.; Tronci, G. Thiol-ene photo-click collagen-peg hydrogels: Impact of water-soluble photoinitiators on cell viability, gelation kinetics and rheological properties. Polymers 2017, 9, 226. [Google Scholar] [CrossRef]
- Perera, M.M.; Ayres, N. Gelatin based dynamic hydrogels via thiol–norbornene reactions. Polym. Chem. 2017, 8, 6741–6749. [Google Scholar] [CrossRef]
- Schweller, R.M.; West, J.L. Encoding hydrogel mechanics via network cross-linking structure. ACS Biomater. Sci. Eng. 2015, 1, 335–344. [Google Scholar] [CrossRef] [PubMed]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | Advantages | Limitations |
|---|---|---|
| Disulfide Formation |
|
|
| Native Chemical Ligation |
| Low-molecular-weight thiol by-product released |
| Thiol-Epoxy Reaction |
| Relatively slow gelation, high concentrations of reactants needed |
| Michael-Type Additions |
|
|
| Photo-initiated Thiol-Ene and Thiol-Yne Additions |
|
|
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, J. Thiol-Mediated Chemoselective Strategies for In Situ Formation of Hydrogels. Gels 2018, 4, 72. https://doi.org/10.3390/gels4030072
Su J. Thiol-Mediated Chemoselective Strategies for In Situ Formation of Hydrogels. Gels. 2018; 4(3):72. https://doi.org/10.3390/gels4030072
Chicago/Turabian StyleSu, Jing. 2018. "Thiol-Mediated Chemoselective Strategies for In Situ Formation of Hydrogels" Gels 4, no. 3: 72. https://doi.org/10.3390/gels4030072
APA StyleSu, J. (2018). Thiol-Mediated Chemoselective Strategies for In Situ Formation of Hydrogels. Gels, 4(3), 72. https://doi.org/10.3390/gels4030072